
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning energetic properties through co-crystallisation – a high-pressure experimental and computational study of nitrotriazolone: 4,4′-bipyridine†
Imogen L.
Christopher
a,
Xiaojiao
Liu
b,
Hayleigh J.
Lloyd
a,
Craig L.
Bull
 c,
Nicholas P.
Funnell
c,
Peter
Portius
d,
Adam A. L.
Michalchuk
e,
Stuart R.
Kennedy
a,
Colin R.
Pulham
*a and
Carole A.
Morrison
*a
c,
Nicholas P.
Funnell
c,
Peter
Portius
d,
Adam A. L.
Michalchuk
e,
Stuart R.
Kennedy
a,
Colin R.
Pulham
*a and
Carole A.
Morrison
*a
aEaSTChem School of Chemistry, The University of Edinburgh, King's Buildings, West Mains Road, Edinburgh, EH9 3FJ, UK. E-mail: c.r.pulham@ed.ac.uk; c.morrison@ed.ac.uk
bDiamond Light Source, Harwell Science and Innovation Campus, Fermi Avenue, Didcot, OX11 0DE, UK
cISIS Neutron and Muon Source, Science and Technology Facilities Council, Rutherford Appleton Laboratory, Harwell Campus, Didcot, OX11 0QX, UK
dDepartment of Chemistry, University of Sheffield, S3 7HF, UK
eSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, West Midlands B15 2TT, UK
First published on 28th May 2024
Abstract
We report the preparation of a co-crystal formed between the energetic molecule 3-nitro-1,2,4-triazol-5-one (NTO) and 4,4′-bipyridine (BIPY), that has been structurally characterised by high-pressure single crystal and neutron powder diffraction data up to 5.93 GPa. No phase transitions or proton transfer were observed up to this pressure. At higher pressures the crystal quality degraded and the X-ray diffraction patterns showed severe twinning, with the appearance of multiple crystalline domains. Computational modelling indicates that the colour changes observed on application of pressure can be attributed to compression of the unit cell that cause heightened band dispersion and band gap narrowing that coincides with a shortening of the BIPY π⋯π stacking distance. Modelling also suggests that the application of pressure induces proton migration along an N–H⋯N intermolecular hydrogen bond. Impact-sensitivity measurements show that the co-crystal is less sensitive to initiation than NTO, whereas computational modelling suggests that the impact sensitivities of NTO and the co-crystal are broadly similar.
Introduction
An active topic of research into energetic materials (EMs) is the use of co-crystallisation to alter the properties of already known and well-characterised energetic compounds.1 The potential of this approach has been demonstrated by successful attempts to tune energetic performance and environmental impact. Examples of properties that can be altered through co-crystallisation include: mechanical impact sensitivities (IS), aqueous solubilities, acidities, thermal decomposition temperatures, and energetic performance.2 Co-crystallisation also reduces the requirement to synthesise and test very novel EMs, which is a costly undertaking due to stringent safety requirements. Thus, co-crystallisation studies that re-employ existing EMs and associated manufacturing infrastructure offer a new and facile design space to explore new materials that are likely to exhibit different properties.One of the first reports of an EM co-crystal concerned TNT (trinitrotoluene) with CL-20 (hexanitrohexaazaisowurtzitane).3 CL-20 has a high energy density and high explosive power but has thus far not been widely used due to its high mechanical sensitivity.4 In contrast, its 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 co-crystal with TNT was reported to have a significantly reduced impact sensitivity. Heating this material above 136 °C resulted in phase separation of the two energetic components, and the authors suggested that the co-crystal form provides a safer form for transportation. CL-20 has also been crystallised with 1-methyl-3,4,5-trinitropyrazole (MTNP) to give a 1:1 co-crystal that has a high crystal density and superior (calculated) detonation performance.5 Moreover, measured impact and friction sensitivities show that it is less sensitive than CL-20, with values close to those for RDX and HMX.
:1 co-crystal with TNT was reported to have a significantly reduced impact sensitivity. Heating this material above 136 °C resulted in phase separation of the two energetic components, and the authors suggested that the co-crystal form provides a safer form for transportation. CL-20 has also been crystallised with 1-methyl-3,4,5-trinitropyrazole (MTNP) to give a 1:1 co-crystal that has a high crystal density and superior (calculated) detonation performance.5 Moreover, measured impact and friction sensitivities show that it is less sensitive than CL-20, with values close to those for RDX and HMX.
More recently, a co-crystal was reported that combined the oxidiser ammonium dinitramide (ADN) with pyrazine-1,4-dioxide to give a system with a balanced oxidiser/fuel ratio, i.e. complete combustion to H2O, N2, and CO.6 Moreover, the IS of the material was reported to be similar to ADN while the detonation performance was superior. An appreciable number of energetic co-crystalline materials have now been reported for azoles,6–9 nitrobenzenes,10–13 and acetone peroxides.12,14
Although many reports concern the co-crystallisation of two EM components, work has also been undertaken on the co-crystallisation of an EM with a non-energetic component. While the obvious drawback of this approach is a loss of stored energy density and performance, several advantages can arise. For example, it provides an accessible route to tuneable intermolecular hydrogen bonds, which is a commonly employed approach in creating pharmaceutically relevant co-crystals.15 EMs predominantly feature nitro groups, which can act as hydrogen bond acceptors, but often lack other functional groups that can act as hydrogen bond donors. Limiting co-crystallisation studies solely to EMs narrows the scope to explore variations in intermolecular interactions that can be employed to form stable co-crystals. Replacing one component with a non-EM hydrogen bond donor moiety lifts this limitation, and the opportunity to explore structure–property relationships is increased.
This work concerns 3-nitro-1,2,4-triazol-5-one (NTO), an insensitive EM with a detonation velocity comparable to that of RDX (1,3,5-trinitro-1,3,5-triazine).16 Its relatively high solubility in water is a potential disadvantage because of the adverse effects of organic nitro-compounds on aquatic life.17 Somewhat unusually, it contains both hydrogen bond donor (N–H) and acceptor (NO2) functional groups. The former renders it acidic in nature (pKa = 3.67),16 and consequently NTO readily forms salts with numerous metals and amines.18 In attempts to obtain a co-crystal, we investigated its propensity to co-crystallise with a relatively strong base, 4,4′-bipyridine (BIPY, pKa of conjugate acid = 4.82).19 According to the empirical ΔpKa rule,20 ΔpKa ≈ 1 gives an approximately equal likelihood for a two-component system to crystallise as either a co-crystal or a salt.
It is now widely recognised that it is important to study the structural response of EMs over a range of pressures and temperatures, as polymorphic phase transitions have often been reported within P–T ranges that are relevant to initiation conditions.21 Different polymorphs of EMs have been demonstrated to have different sensitivities to mechanical impact, e.g. HMX22 and CL-20.23 Moreover, recent modelling studies have also indicated that predicted IS values for HMX,24 CL-20,24 1,1-diamino-2,2-dinitroethane (FOX-7),25 and RDX26 are polymorph-dependent. Exploration of the structural response to pressure is particularly pertinent for a crystal formed between NTO and BIPY given the potential for ambiguity between the co-crystal and salt forms, and the concomitant modification of material properties this may cause.
Herein, the synthesis and characterisation of a two-component crystal of NTO and 4,4′-bipyridine (NTO·BIPY) is described. Ambient and high-pressure single-crystal X-ray and powder neutron diffraction studies at pressures up to ca. 6 GPa have been undertaken, supplemented with computational modelling to track the intermolecular hydrogen bond structural parameters and account for the change in colour of the molecular crystal at high pressure. In addition, the IS of NTO·BIPY is measured relative to that of α-NTO and explored using a vibrational up-pumping model that has been successfully applied across a range of single-component EMs,27 making the first step towards its validation for multicomponent crystals.
Results and discussion
Ambient-pressure crystal structure of NTO·BIPY (single crystal X-ray diffraction)
Pale-cream coloured crystals of NTO·BIPY were obtained by dissolving equimolar amounts of NTO and BIPY (on a 10 mg scale) of NTO in a few mls of ethanol followed by slow evaporation (see Experimental section). Qualitative solubility tests in a selection of solvents showed that NTO·BIPY is much less soluble in water than NTO.The crystal structure of NTO·BIPY at ambient pressure was solved and refined in the monoclinic crystal system (space group P21/n) with unit-cell parameters a = 7.9440(7) Å, b = 5.7979(5) Å, c = 27.874(2) Å, and β = 95.810(3)° (see ESI†). The asymmetric unit comprises a single NTO and BIPY moiety, connected via two hydrogen bonds. The first, N1–H⋯N3, is associated with the acidic proton on NTO (Fig. 1(a)). Difference Fourier maps unambiguously assigned this hydrogen atom to the NTO (see ESI†), thereby defining this structure as a co-crystal rather than a salt. The second, N2–H⋯N4, leads to the formation of parallel stacks of alternating herringbone planes (Fig. 1(b)).
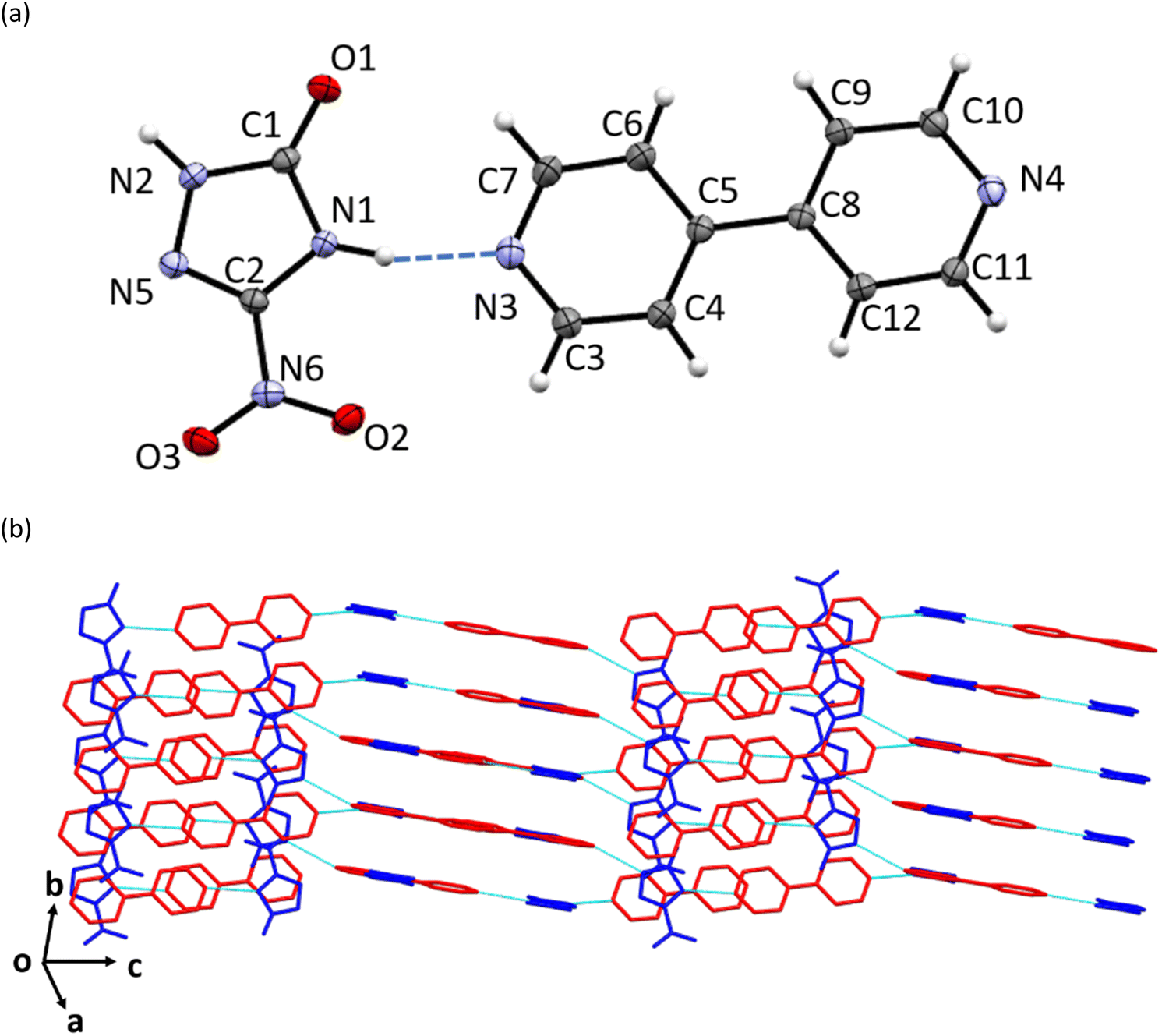 | ||
| Fig. 1 (a) The molecular structure of the asymmetric unit, (b) crystal packing of NTO·BIPY as determined at ambient pressure by single crystal X-ray diffraction (shown without H atoms for clarity, NTO in blue, BIPY in red, hydrogen bonds shown as light blue dash-lines). | ||
Response of NTO·BIPY to high pressure (single crystal X-ray diffraction)
A search for either potential pressure-induced phase transitions or proton transfer to produce a salt was conducted up to 7.74 GPa using a Merrill–Bassett diamond-anvil cell. Up to 5.93 GPa, it was found that all crystallographic unit-cell axes compressed monotonically without any obvious change in space group (Fig. 2(a)). Over the studied pressure range, there was a gradual change in crystal colour, from colourless at ambient pressure, to bright yellow at 2.85 GPa, and finally to orange at 7.74 GPa (Fig. 3). At just above 6 GPa striations were observed on the crystal surface, and by 7.74 GPa the X-ray diffraction pattern shows severe twinning, with multiple crystalline domains orientated along the 0kl plane (see ESI†); characterisation of this crystal form was unfortunately not possible due to the complexity and poor quality of the diffraction patterns. We note that the limit of the pressure transmitting media (PTM) employed (a 1:1 pentane/isopentane mixture) is around 7.4 GPa,28i.e. above the pressure range where the crystal striations started to appear. It is therefore unlikely that any potential solidification of the PTM is responsible for the changes in crystal morphology. Upon decompression to ambient pressure, the crystal reverted to colourless, but the striations remained (Fig. 3(d)). Despite the limited quality of the diffraction data, it was possible to index the crystal to the ambient-pressure structure.
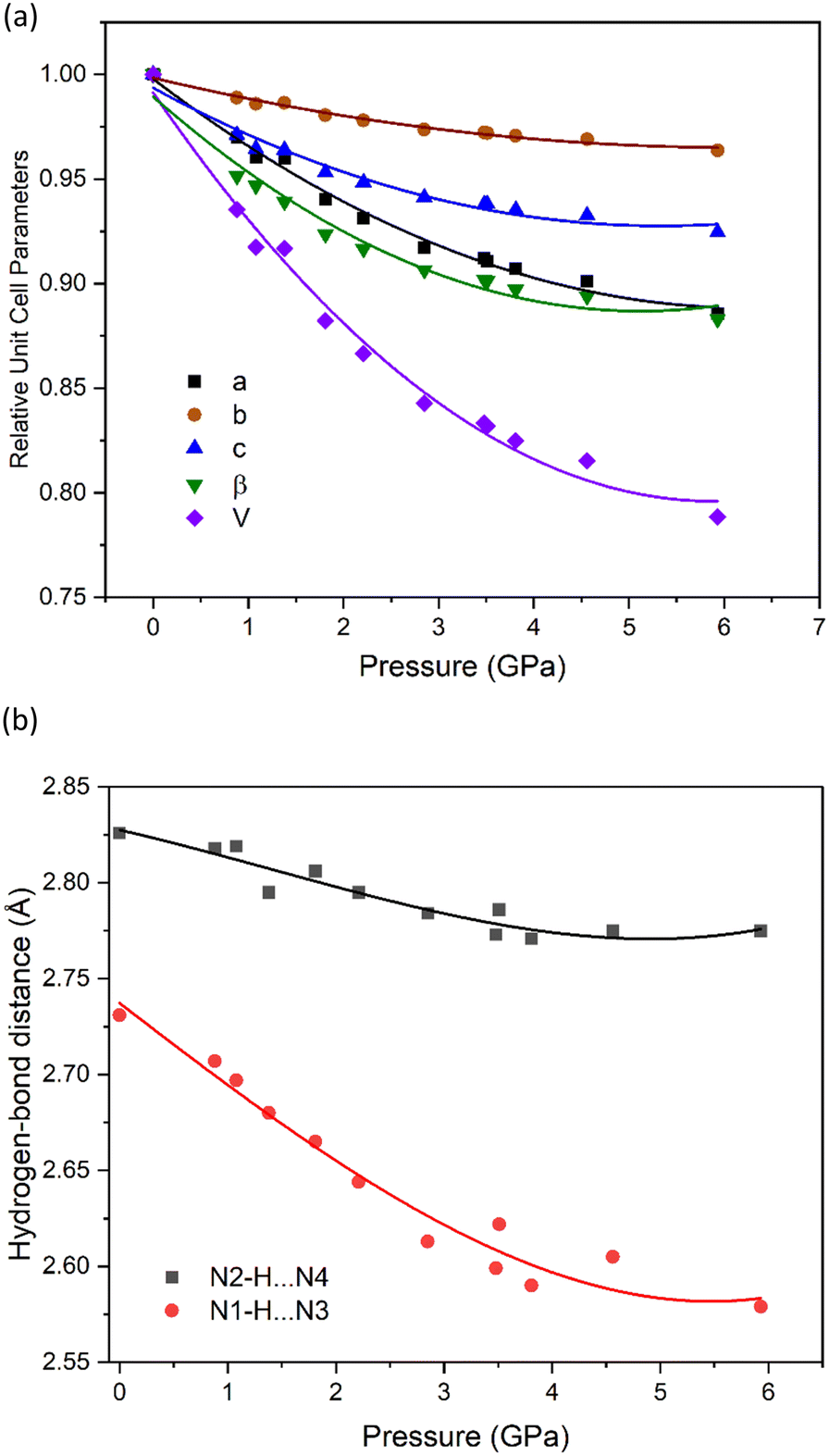 | ||
| Fig. 2 Plots from the single-crystal X-ray high-pressure diffraction study showing (a) the relative change in unit-cell parameters for NTO·BIPY, alongside (b) changes in the N⋯N hydrogen bond distances. Curves are derived from 2nd order polynomial fits. | ||
 | ||
| Fig. 3 Photographs showing colour changes in a crystal of NTO·BIPY in the DAC at different pressure conditions: (a) ambient (colourless, without PTM and ruby spheres), (b) 2.85 GPa (yellow), (c) 6.04 GPa (yellow, with striations), (d) 7.74 GPa (orange), and (e) ambient pressure after decompression (colourless). The ruby spheres, used for pressure calibration, are visible top right. | ||
Even though the high-pressure single crystal X-ray diffraction study was unable to locate the position of the hydrogen atom within the hydrogen bond, it was able to map the bond compression with respect to the heavy-atom positions. This is shown for the two hydrogen bonds in NTO·BIPY in Fig. 2(b), where N1–H⋯N3 decreases significantly more than N2–H⋯N4, despite being the shorter of the two interactions. No discontinuities were observed in either trend, suggesting that the colour change observed with increasing pressure is not associated with significant changes in hydrogen bond interactions. This is important, as it would otherwise be tempting to attribute the colour changes to single and double proton transfer to BIPY, as similar pressure-induced colour changes have been observed in for the co-crystal formed between BIPY and squaric acid.29 In an attempt to explore further the positions of hydrogen atoms in NTO·BIPY, we turned to computational modelling and high-pressure powder neutron diffraction measurements.
Computational modelling
Geometry optimisations were performed on the ambient-pressure and three high-pressure structures (at 1.81, 3.48 and 5.93 GPa, i.e. the maximum pressure for which structural data is available). The input models were derived from the experimental single crystal X-ray diffraction crystal structures. All unit-cell parameters returned optimised values well within the expected deviation from the experimental values (ca. 5%), indicating that reliable outputs had been obtained (see ESI†). For the ambient-pressure structure, the hydrogen atom in the N1–H⋯N3 hydrogen bond remained on the NTO molecule, which confirms the conclusion drawn from the X-ray diffraction study that this is a co-crystal. Similarly, optimisation of the three high-pressure structures also showed no proton transfer, suggesting that NTO·BIPY remains as a co-crystal over the investigated pressure range.In an effort to understand the changes in the hydrogen bond potential energy surface in more detail, we performed a series of potential energy surface (PES) scans for the four structures, with the outputs shown in Fig. 4(a). At lower pressures (0.00 to 3.48 GPa), the PES appears as broad minima at ca. 1.1 Å, which confirms that proton attachment to NTO gives rise to the lowest energy structure. A second metastable minimum is predicted at ca. 1.55 Å (some 100 kJ mol−1 higher in energy), which corresponds to proton attachment to BIPY. At 5.93 GPa the PES appears as a single well. In order to portray more accurately how these PES scans impact on the predicted N–H bond length, the calculated single point energies for each surface were fitted to a sixth-order polynomial function and subsequently solved within the one-dimensional Schrödinger equation formalism to locate the ground-state wavefunction. This also provides the associated expectation value for a deuterium atom located on each potential energy surface (note: in this analysis deuterium, instead of hydrogen, was modelled, to allow for a more direct comparison against the deuterated sample explored using high-pressure powder neutron diffraction). The resulting predicted N1–D bond lengths, alongside the calculated N1⋯N3 distances, are shown as a function of pressure in Fig. 4(b) (see also ESI†). With a predicted bond lengthening of ca. 0.07 Å to occupy a position just 0.11 Å away from the mid-point of the N1⋯N3 bond, the calculated PES suggests that NTO·BIPY should be classed as a proton migration system. It is therefore unlikely to form a salt, unless a phase transition (at high temperatures, or pressures beyond 5.93 GPa) could induce a more significant change in the crystal packing. Comparable changes to those seen are reported for the well-investigated proton migration system of urea-phosphoric acid, in which the proton position moves along a N⋯O hydrogen bond by 0.03–0.05 Å at elevated temperatures.30,31
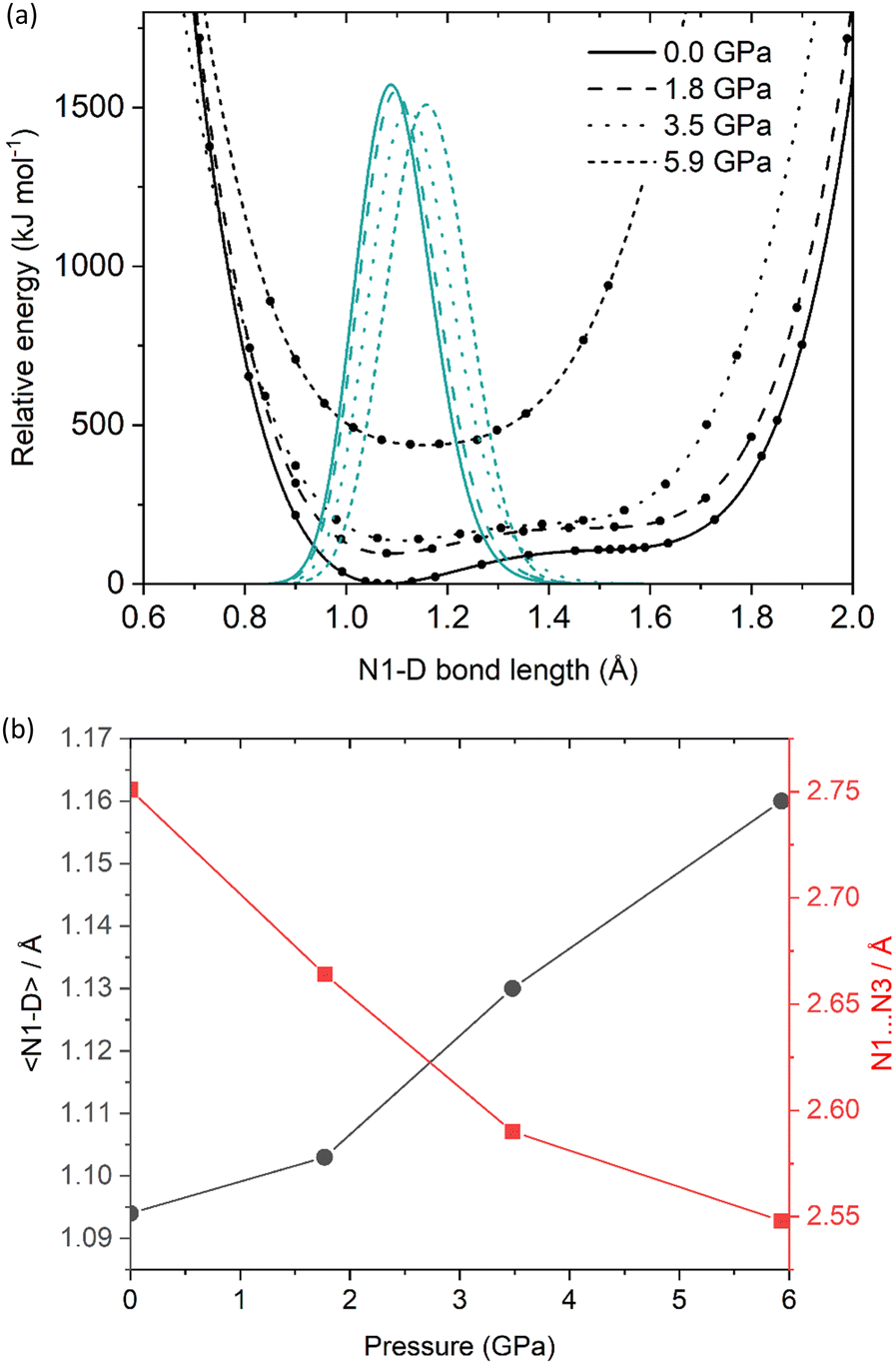 | ||
| Fig. 4 (a) Calculated potential energy surfaces for the N1–D⋯N3 hydrogen bond in NTO·BIPY as a function of pressure. The data points are fitted to a sixth-order polynomial function (black curves), alongside the vibrational ground state expectation values for a deuterium atom located on each PES (green curves); (b) N1–D expectation values and calculated N1⋯N3 distances are plotted as a function of pressure. | ||
With the computational modelling therefore appearing to suggest proton migration rather than transfer, another factor must be responsible for the colour changes observed at high pressure. In order to determine if the colour change can simply be attributed to unit-cell compression, electronic band structures were calculated for the structures at ambient pressure and 5.93 GPa. The results are presented in Fig. 5 alongside the partial density of states; associated Brillouin zone k-point paths are given in the ESI.†
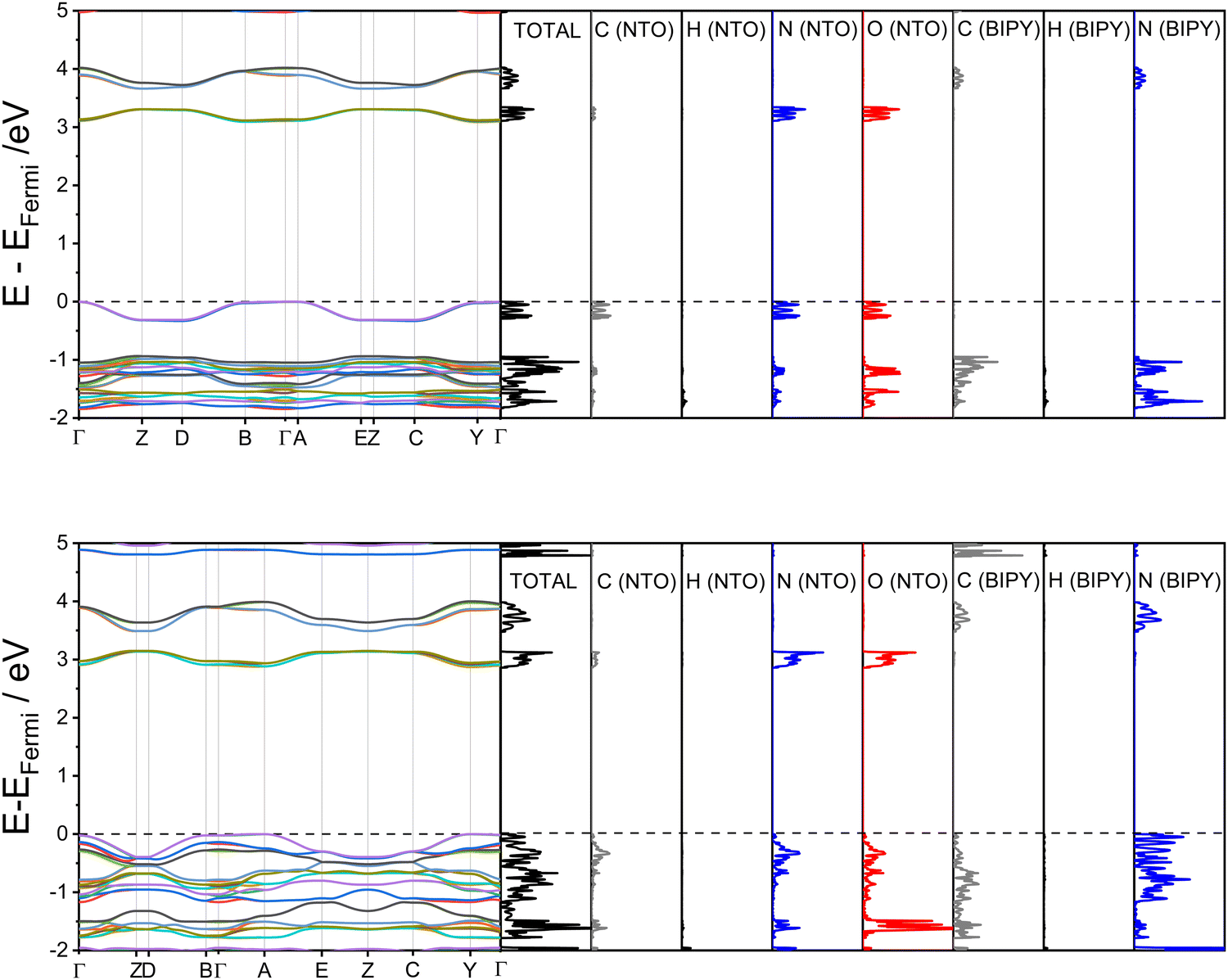 | ||
| Fig. 5 Calculated electronic band structures for NTO·BIPY, alongside partial density of states (top: 0 GPa, bottom 5.93 GPa). The dotted black line indicates the Fermi level, set to 0 eV in each case (absolute EFermi values: 0 GPa = −6.02 eV; 5.93 GPa = −5.61 eV). | ||
The first point to note from the electronic band structure plots are the values of the direct (i.e. vertical) band gaps measured at the Γ-point, which reduces from 3.09 eV for the ambient-pressure structure to 2.87 eV for the 5.93 GPa structure. The ambient-pressure band gap for NTO·BIPY is similar to that found for the white pigment ZnO (3.3 eV),32 while the high-pressure band gap size is in line with values reported for yellow molecular crystals which were obtained using the same hybrid DFT functional (2.5–2.8 eV for the yellow forms of the highly polymorphic organic molecule known as ‘ROY’).33 Thus, the pressure-induced colour change can be directly attributed to a compression of the unit cell, and an associated electronic transition between the frontier highest occupied crystalline orbitals (HOCOs) and the lowest unoccupied crystalline orbitals (LUCOs). Partial density of states plots (see Fig. 5) demonstrate that both of these crystalline orbitals are located on NTO. In the high-pressure structure the HOCOs are dominated by contributions from the nitrogen atoms on the BIPY, whereas the LUCOs remain unchanged from their ambient-pressure character on NTO. Based on the electronic band structure diagrams it is apparent that the narrowing of the electronic band gap for the high-pressure structure arises from an increase in energy dispersion with respect to the k-point paths Γ→ Z, D → B, A → E and C → Y. These k-point vectors all coincide with the unit cell b-vector (see ESI†), which partially aligns with the herringbone π⋯π stacking direction in both the ambient-pressure and high-pressure structures of NTO·BIPY; this distance shortens from 4.37(1) to 3.31(2) Å in the single-crystal diffraction study. While the electronic band structure calculations therefore account for the clear to yellow colour change observed from ambient pressure to 5.9 GPa, the reason for the further colour change (to orange, at 7.7 GPa) remains unknown. However, given that the striated crystal recovered at ambient pressure (Fig. 3(d)) could be successfully indexed to the ambient pressure structure suggests either further unit cell compression, or the appearance of a new phase only stable at high pressure, accounts for the yellow to orange colour change.
High-pressure powder neutron diffraction study
In order to explore experimentally the structural changes in NTO·BIPY with pressure, we conducted high-pressure powder neutron diffraction measurements. The resulting data sets sample more of the diffraction patterns than those collected in the X-ray single crystal measurements, which are limited by the absorbing steel body of the diamond-anvil cell. High-pressure neutron diffraction measurements were obtained in ca. 0.2–0.5 GPa steps from ambient pressure to ca. 5.4 GPa using perdeutero-NTOb7BIPY (NTO·BIPY-d10) to avoid incoherent scattering that would otherwise arise from 1H. The variation in unit-cell volume for the 26 data points is plotted in Fig. 6, where the lack of any discontinuous behaviour confirms that no phase transition occurs. The bulk modulus (B0) was found to be 7.1(3) GPa, and its derivative (B′) 11.0(5). Compression behaviour can only be understood directly from the principal axes (Xi) of the strain tensor. We have determined the compressibilities (Ki) of these principal axes, as well as their spatial relationship with respect to the (non-orthogonal) unit cell axes; both are shown Table 1 and Fig. 6, along with the equation of state fit to the volume data. Of particular interest is the finding that the three principal axes undergo highly anisotropic compression, as well as a small degree of negative linear compression along the X3 direction. Further investigation shows this latter effect only persists up to ca. 0.7 GPa (see ESI†).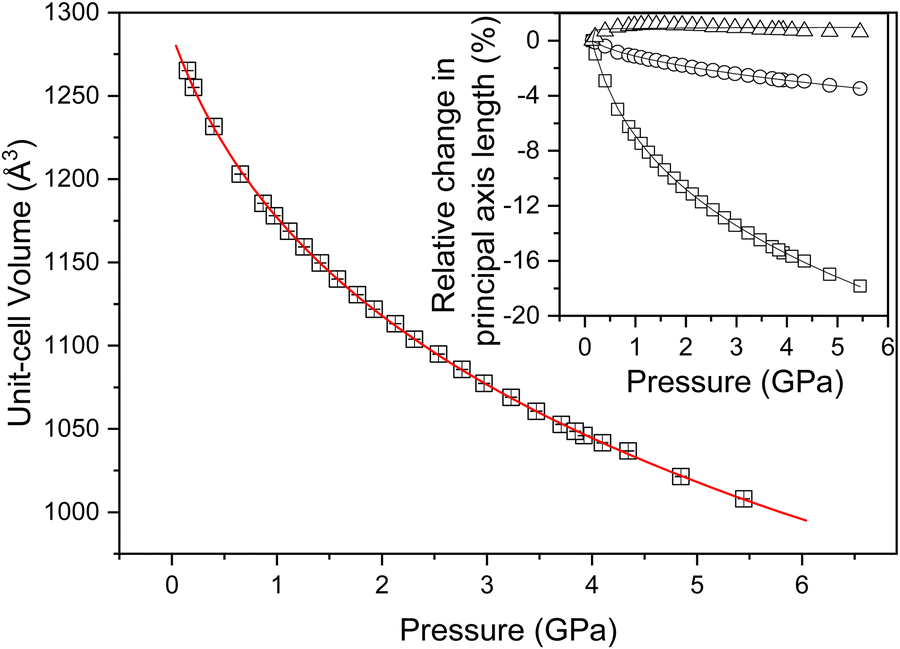 | ||
| Fig. 6 Unit cell volume data as a function of pressure. The red line indicates the third-order Birch–Murnaghan fit. The inset graphic shows the relative axis compressibilities, with triangles, circles, and squares indicating the X3, X2, and X1 principal directions, respectively. | ||
| Axes | K i (TPa−1) | Direction |
|---|---|---|
| X 1 | 29.43(9) | ∼a − c/4 |
| X 2 | 6.01(5) | +b |
| X 3 | −0.2(4) | ∼a + c/3 |
Structural models were refined against all neutron powder data using the Rietveld method within Topas Academic v6.34 Due to the limited d-spacing data range imposed by the Paris–Edinburgh press, it was necessary to minimise the number of independent parameters. Both BIPY and NTO were parameterised as separate rigid bodies, encoded as a Z-matrix description of bond distances and angles, alongside parameters describing whole-molecule translations and rotations. Intramolecular, non-D, bond lengths and angles were constrained to average values derived from the X-ray crystal structure at ambient pressure. The N–D bond lengths in NTO were allowed to refine, but were restrained against the ambient pressure DFT-calculated distances. All C–D bonds on the BIPY molecule were constrained to be the same length, which in turn was restrained against a literature-determined value.35 Each molecule was permitted some torsional flexibility, through rotation of the NTO nitro group and the relative orientation of the pyridine rings in BIPY. Lastly, common elements within the same molecule were constrained to have identical isotropic displacement parameters and refined with Marquardt-style damping. Contaminant alumina, zirconia (both anvil components), and lead (pressure marker) phases were also included in the refinement, which together accounted for all non-sample diffraction intensity. The pressure inside the press was determined using the relationship between the lead equation of state, and its refined unit cell volume.36 The Rietveld fits to the lowest and highest pressures measured is shown in Fig. 7.
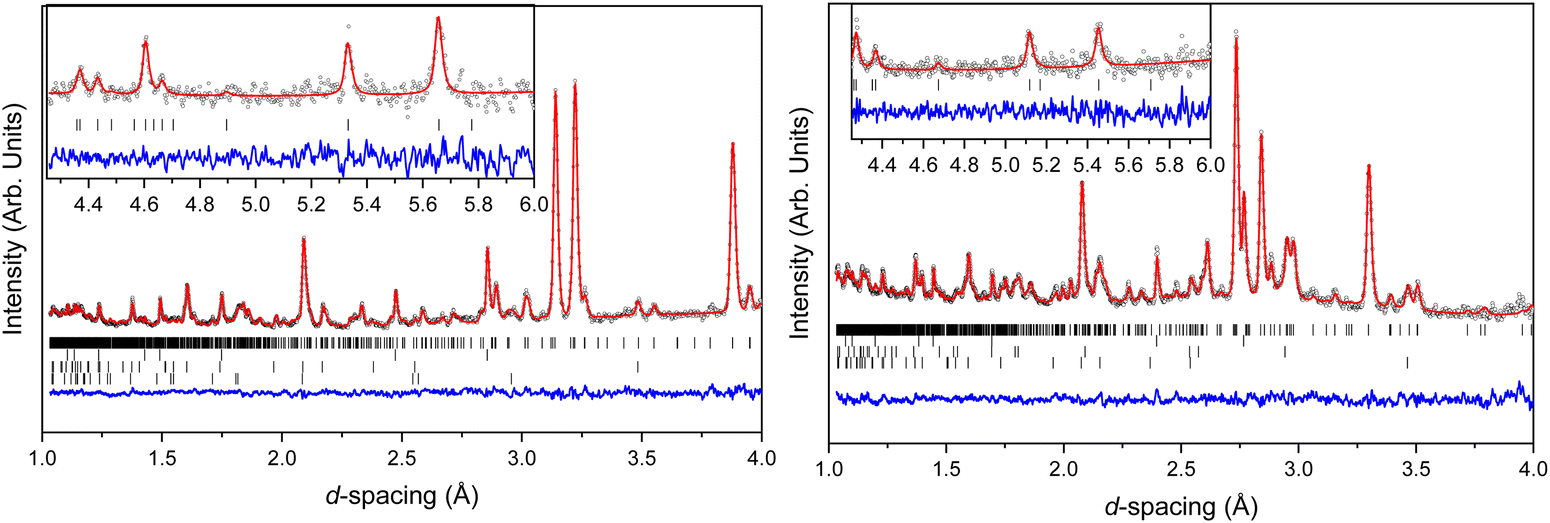 | ||
| Fig. 7 Neutron powder diffraction patterns of NTO·BIPY (left at 0.1 GPa, right at 5.4 GPa). Experimental data are shown as open spheres, Rietveld refinement fits as solid red lines, and residual data as the blue traces. Vertical tick marks indicate the determined peak positions of the four phases, which are (from top to bottom) NTO·BIPY, lead, Al2O3 and ZrO2. The insert in both panels shows the data collected in the long frame providing access to the high d-spacing range in the diffraction pattern. | ||
In an attempt to determine whether the scattering data indicated any migration of the deuterium atom along the N1⋯N3 distance, initial refinements allowed free variation of the N–D bond distances. However, this frequently resulted in physically unrealistic bond geometries so the covalent bond lengths were instead restrained against the ambient-pressure values observed in the DFT-calculations. The refined structures suggest that the two N–D⋯N hydrogen bonds compress to similar extents (N4⋯D and N3⋯D by 0.14 and 0.18 Å, respectively). However, the magnitude of the bond length uncertainties render these changes as insignificant (Δ/σ significance tests return values of 2.2 and 2.3, respectively). The N⋯N distances are more revealing, and show a marked difference in compression: N2⋯N4 and N1⋯N3 decrease by 0.061 and 0.21 Å, respectively, which are similar to those observed in the single-crystal diffraction study (see Fig. 2). Interaction distances from the neutron diffraction study are plotted in Fig. 8.
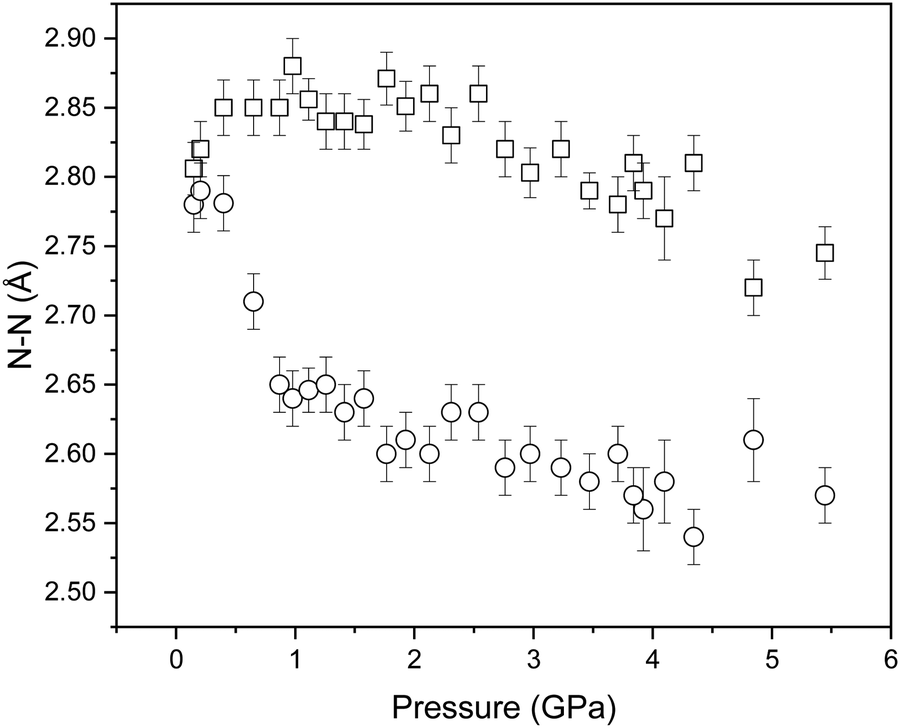 | ||
| Fig. 8 Variation in the determined N⋯N distances of NTO·BIPY with increasing pressure by Rietveld profile fitting of the neutron powder diffraction patterns. | ||
Impact-sensitivity measurements of NTO·BIPY
In order to explore what effect co-crystallising NTO with BIPY has on its sensitivity to mechanical impact, we performed a series of drop-weight measurements using the Sheffield drop-weight apparatus (see ESI† for details). For the purpose of ranking, NTO·BIPY was compared with synthesis-grade NTO, synthesis-grade RDX and NaCl (i.e. a non-energetic). Since mechanical impact-induced reactions are not always clearly identifiable by direct audio-visual observations, results were also evaluated using video and sound recordings as well as mass balances. Using NaCl as a baseline, the data (see Table S5, ESI†) clearly confirms that RDX is initiated at the maximum drop height available (equivalent to 8.3 J), as noted by visible dust clouds, an increase in sound intensity (factors 0.5 to 1.3 above the background signal), and a mass loss ratio of 1.6 to 1.7 (see ESI†) compared to NaCl (−0.06 to 0.05 and 0.05 to 0.22, respectively). NTO samples initiated two out of three times (sound intensity factors −0.1, 0.8, 1.0 above the background). As with RDX, all NTO samples underwent mass loss at a level far above the baseline, ranging from 1.4 to 2.4, which correlates with the sound intensities produced. Finally, mechanical impact testing on NTO·BIPY shows no increase in the recorded sound intensity, but mass loss similar to that recorded for RDX and NTO (normalised ratios range from 1.0 to 2.0). A comparison of the infrared spectrum of recovered NTO·BIPY material after drop weight impact-induced compression revealed no sign of chemical reaction. Thus, unlike the reference materials RDX and pure NTO, NTO·BIPY fails to give either audible reports or signs of chemical reaction, apart from a degree of material ejection upon impact from maximum drop height. The order of impact sensitivity is therefore RDX > NTO > NTO·BIPY. Note: a recent reassessment of experimentally determined impact sensitivites for a range of common energetic materials has highlighted once more the variability of results depending on method and apparatus (10 J, RDX, and 13 J, NTO, as average over ten trials by Muravyev et al., vs. 3.5 to 7.5 J and 25 to >120 J, respectively, for the BAM tester.37 It is therefore not surprising that the initiation of synthesis-grade NTO is within the range of the Sheffield drop-weight apparatus (5.5 J, RDX; extrapolated 11 J, NTO, assuming an unchanged curvature of the probability to initiation – impact energy function).In an effort to understand this experimental observation, we have also studied NTO·BIPY by vibrational up-pumping. Full details on our IS prediction model are available elsewhere.25–27 Briefly, the model is based on the principles of vibrational up-pumping, whereby the energy produced by mechanical impact or shock is deposited into the acoustic lattice vibrations. The energy is then rapidly equilibrated across the phonon bath modes, defined by an upper limit of Ωmax, which is typically around 200 cm−1. These low energy modes reach temperatures on the order of thousands of kelvin (denoted by Tshock). Through a number of phonon scattering pathways, their increased vibrational excitation creates new higher-energy vibrational states, termed the two-phonon density of states, Ω(2). As a result, the mechanical energy is vibrationally pumped up into the molecular modes occupying the 1–3 Ωmax region. Increased population of these vibrational modes introduces molecular strain, which subsequently leads to the initiation of the material. Thus, our IS prediction model takes the phonon density of states (termed g(ω), calculated for the optimised crystal structure using plane-wave density functional theory), as input data, and generates its corresponding Ω(2). Integrating the projection of Ω(2) onto the underlying g(ω) provides a quantitative prediction of the ability of the vibrational modes inherent to the crystal lattice to transfer mechanical energy into the molecular vibrations. This is the metric we report for relative predicted IS.
Prior to performing the up-pumping analysis, the calculated g(ω) for NTO·BIPY was investigated in more detail in order to better understand the nature of the vibrational motions in the IS prediction manifold. For instance, while it is reasonable to expect that the phonon bath modes will be delocalised across both the NTO and BIPY molecules (i.e. inseparable mixed modes, and therefore all included in the up-pumping process), the same may not be true across the up-pumping window. To that end, partial g(ω) plots were constructed, where the relative movements of the atoms in NTO and BIPY for each eigenvector were interrogated in turn. The outcome is shown in Fig. 9, which shows that some modes (marked with an asterisk) that fall under the Ω(2) envelope have very small NTO character, and thus should not be included in the Ω(2) onto g(ω) projection and integration. For direct comparison with NTO·BIPY, Fig. 9 also shows the corresponding g(ω) and Ω(2) for α-NTO,27 which allows some important factors to be considered. As expected, the molecular vibrational modes for NTO above Ωmax are broadly consistent in both the co-crystal and in pure NTO, but the phonon bath modes below Ωmax are noticeably different, which reflects differences in the unit cells for the two crystal structures. The phonon bath region for the co-crystal, as well as containing more modes of vibration to capture and up-pump the mechanical shock wave energy, also extends slightly further (210 cm−1 for the co-crystal vs. 200 cm−1 for pure NTO, see ESI†). Consequently, the Ω(2) envelope for NTO·BIPY is slightly broader and more pronounced than for α-NTO. However, integrating the projection of Ω(2) onto g(ω) (and excluding the peaks marked with asterisks which are BIPY molecular modes) produces a broadly similar outcome in which NTO·BIPY has only a slightly higher IS value than pure NTO (see Fig. 10). Also shown in Fig. 10 are predicted IS values for a range of EMs, using the same vibrational up-pumping model,26 against literature BAM Fall–Hammer experimental IS values, which reveals a correlation between up-pumping intensity and experimental IS values. From this analysis it is clear that while a clear ranking of predicted IS values for highly sensitive EMs are obtained by this model, our prediction curve is essentially flat for the less sensitive EMs, with α-FOX-7, α-NTO and TATB having near parity. This most likely arises due to the computed g(ω) plots sampling the Γ-point only in the Brillouin zone, as full Brillouin zone sampling has previously demonstrated greater differentiation between lower sensitive EMs.38 Within the accuracy of Γ-point g(ω) vibrational-up pumping, the predicted IS value for NTO·BIPY is broadly similar to α-NTO; while this is at odds with the experimental data reported in the previous section using the Sheffield apparatus, it is nonetheless encouraging that the up-pumping model does not predict a drastic change in material response behaviour for the co-crystal.
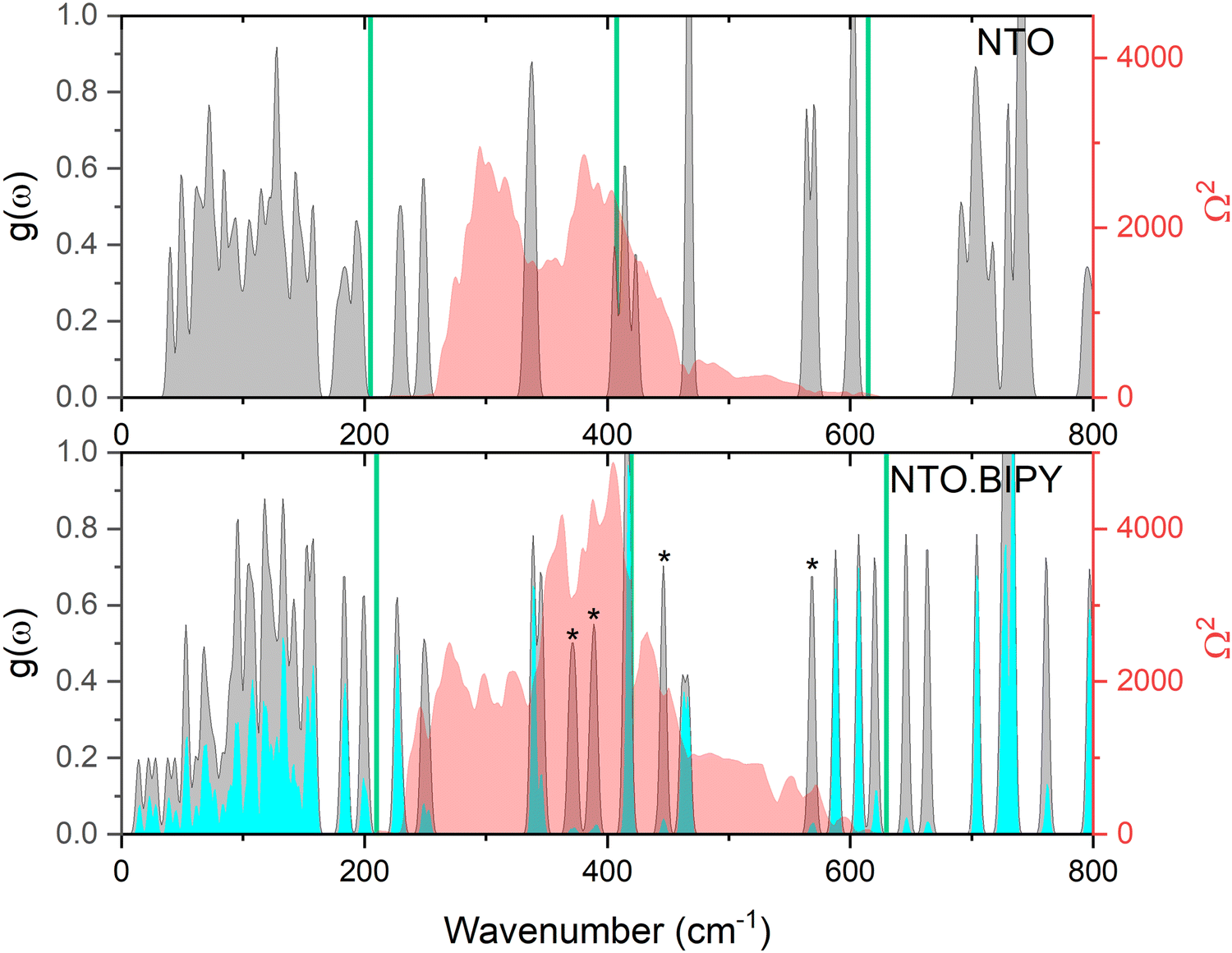 | ||
| Fig. 9 Simulated g(ω) (grey), and corresponding Ω(2) (red) for NTO (top)27 and NTO·BIPY (bottom) at ambient conditions. The partial g(ω) for NTO in the co-crystal is shown in cyan, and peaks with negligble NTO character are marked with an asterisk. The green vertical lines represent sequential multiples of Ωmax, within an 1–3 Ωmax up-pumping window. | ||
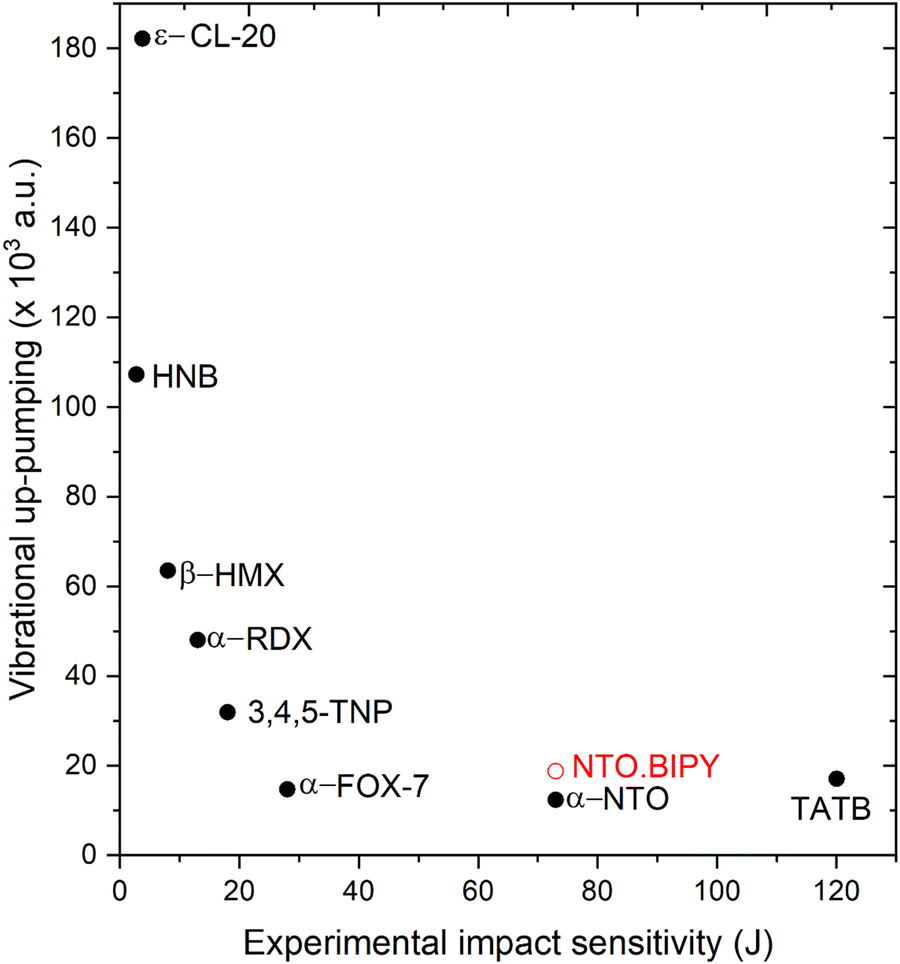 | ||
| Fig. 10 Vibrational up-pumping densities vs. experimental IS values for a range of previously reported EMs26,27,39 alongside the prediction for NTO·BIPY (shown in red). Filled symbols correspond to experimentally determined data, unfilled to predicted values only. | ||
Experimental and computational modelling methods
Synthesis
Safety warning: NTO and its co-crystals are highly energetic and may explode in response to certain external stimuli. It is strongly recommended to carry out small-scale reactions and take appropriate precautions such as the use of leather gloves, protective coats, face shields, and explosion-proof baffles.4,4′-BIPY was obtained from Alfa Aesar, semicarbazide hydrochloride, ethanol (absolute, HPLC grade), formic acid (pure, ca. 85%) and nitric acid (ca. 70%) from Fisher Scientific. Water was purified by ion exchange. All chemicals were used without further purification. The synthesis of NTO is based on Coburn's report40 and was performed in a simple two-step process: (i) the condensation of semicarbazide hydrochloride and formic acid producing triazolone (TO), and (ii) the nitration of TO to form nitrotriazolone. Solid semicarbazide hydrochloride (45.02 g, 0.404 mol) was suspended in 85% formic acid (48 ml, 1.08 mol). The stirred reaction mixture was then heated to 60 °C and kept at this temperature for a period of 1.5 h. After this time, the residual formic acid and water were removed by vacuum distillation. The solid residue was then suspended in ice cold water for 1 h, and the crude TO collected by vacuum filtration. The crude TO was purified by washing with ice-cold water followed by drying in air over night to give a white, odourless, crystalline material (16.63 g, 0.195 mol, 48% yield). While maintaining a temperature of 20 °C, the purified intermediate (TO, 6.01 g, 70.7 mmol) was slowly added to 60 ml of nitric acid (69–70%). The resulting mixture was heated to a temperature of 60–70 °C, which was maintained for approximately 2 h under constant stirring. During this time a precipitate formed. After the heating period, the reaction vessel was immersed in an ice-bath (0–5 °C) which completed the product precipitation. The precipitate was then collected by filtration, and the filter residue washed with water to remove any adsorbed or co-precipitated nitric acid. After air-drying, white, odorless, crystalline NTO was obtained (5.84 g, 44.9 mmol, 64% yield).
A colourless mixture of NTO (118 mg, 907 μmol) and BIPY (142 mg, 909 μmol) was prepared in a glass vial. The addition of ethanol (10 ml) produced an intense yellow colour (NB: both NTO and BIPY produce colourless solutions in EtOH, which, upon combination also produce a yellow colour). The mixture was heated to the boiling point of the solvent which dissolved nearly all solids and resulted in a pale yellow-green solution. The decanted solution (a 3 mg decantation residue was discarded) was allowed to slowly cool to r.t., which spontaneously produced yellow crystals. After 18 h the crystals were collected by filtration and then washed with cold EtOH (2 × 2 ml) and dried in air to give 188 mg of the NTO·BIPY adduct (C12H10N6O3, 286.25 g mol−1, 671 μmol, 66% yield with respect to NTO). Additional characterisation data are available in the ESI† (FTIR spectra of NTO, BIPY and NTO·BIPY, PXRD of NTO·BIPY). The deutero adduct NTO-d2·BIPY-d8 was obtained following the method stated above using NTO-d2 and BIPY-d8. The latter were synthesised by H/D exchange reactions of the starting components NTO-h2 and BIPY-h8, followed by recrystallisation in D2O.
Ambient-pressure X-ray single crystal diffraction
A suitable single crystal was selected and mounted on a Bruker D8 Venture diffractometer (Mo-Kα radiation, λ = 0.71073 Å). The crystal was maintained at 293 K using an Oxford Cryostream Plus during the data collection. Unit-cell parameters were obtained using the Bruker APEX III software package, while Olex241 was employed for structure determination. The structure was solved with ShelXT42 using instrinsic phasing methods. The positions of hydrogen atoms involved in hydrogen bonds were determined by difference Fourier maps (see ESI†); all other hydrogen atoms were located at calculated positions. Structure refinements were performed using ShelXL43 using least squares minimisation. Full details of the collected diffraction data are given in the ESI.†High-pressure X-ray single crystal diffraction
A suitable single crystal was loaded into a Merrill–Bassett diamond anvil cell (DAC) with a 1:1 mixture of n-pentane and iso-pentane as the pressure-transmitting medium. Ruby spheres were used for pressure calibration. Diffraction data were collected on a Bruker D8 Venture X-ray diffractometer (Mo-Kα radiation, λ = 0.71073 Å) at room temperature condition (ca. 295 K). Data reduction and application of Lorentz polarization and absorption corrections were performed using the Bruker APEX III software package. Olex241 was employed for structure determination. The structure was solved and refined with ShelXT using instrinsic phasing methods and least squares minimisation.42 Full details of the collected diffraction data are given in the ESI.†
High-pressure neutron diffraction study
Neutron powder diffraction patterns were collected on the PEARL instrument based at the ISIS Neutron and Muon Source, UK.44 A V3 Paris–Edinburgh press was used for the data collection. The perdeuterated powdered sample of NTO·BIPY was loaded into an encapsulated Ti–Zr alloy gasket45 and placed between single-toroidal (ZTA) anvils for measurements in the 0–5.4GPa pressure region. Pb was included in the sample volume as pressure marker.36 A 1:1 mixture of perdeuterated iso-pentane and n-pentane was used as the pressure-transmitting medium.28 The experimental patterns were normalised and corrected for attenuation effects using Mantid software,46 and Rietveld refinement of the data was performed using the Topas suite.34
Conclusions
The formation of co-crystals in energetic materials research continues to present an attractive design space for the tuning of material properties of known compounds. We have presented the synthesis and characterisation of a co-crystal of the energetic compound NTO with the non-energetic compound 4,4′-bipyridine (BIPY), which mitigates the undesirable water-solubility properties of NTO. While differences in the pKa values between NTO and protonated BIPY would suggest a fine balance between salt or co-crystal formation, we have demonstrated that the material exists as a co-crystal. This form persists at high pressure, and no evidence of a phase transition up to 5.93 GPa was observed. At higher pressures the crystal quality degraded and the X-ray diffraction patterns showed severe twinning, with the appearance of multiple crystalline domains.Computational modelling suggests that the application of pressure induces proton migration along an N–H⋯N intermolecular hydrogen bond. Although a pressure-induced phase transition was not observed, the crystals were observed to change colour with increasing pressure. Electronic band structure calculations revealed that this colour change could be attributed to straightforward compression effects of the the unit cell that cause heightened band dispersion and band gap narrowing along the k-point paths that are associated with the crystallographic b-axis. This partially aligns with the herringbone π⋯π stacking direction which shortens on application of pressure.
Finally, we have measured and modelled the response to mechanical impact stimulation for NTO·BIPY. Experimental measurements indicated a small reduction in impact sensitivity compared to NTO, whereas computational modelling suggests a broadly similar response, but this is likely at the limits of using a Γ-point-only simulated density of states for vibrational up-pumping. As a whole, this study shows that co-crystallisation of EMs continues to present an attractive pathway to tune the properties of energetic materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr B. Westwater for the synthesis of additional samples of NTO and collection of PXRD data for NTO·BIPY. This work was supported by Dstl and the Air Force Office of Scientific Research under award no. FA8655-20-1-7000. We are grateful for the computational support from the United Kingdom Materials and Molecular Modeling Hub, which is partly funded by EPSRC (grant no. EP/PO20194 and EP/TO22213), for which access was obtained via the UKCP consortium and funded by EPSRC grant no. ref EP/PO22561/1. I. L. C. thanks the University of Edinburgh, School of Chemistry for the award of a PhD scholarship.References
- J. C. Bennion and A. J. Matzger, Development and Evolution of Energetic Cocrystals, Acc. Chem. Res., 2021, 54, 1699–1710 CrossRef CAS PubMed.
- S. R. Kennedy and C. R. Pulham, in Co-crystals: Preparation, Characterization and Applications Edited, ed. B. Aakeroy and A. Sinha, The Royal Society of Chemistry, 2018, pp. 231–264 Search PubMed.
- O. Bolton and A. J. Matzger, Improved Stability and Smart-Material Functionality Realized in an Energetic Cocrystal, Angew. Chem., Int. Ed., 2011, 123, 9122–9125 CrossRef.
- U. R. Nair, R. Sivabalan, G. M. Gore, M. Geetha, S. N. Asthana and H. Singh, Hexanitrohexaazaisowurtzitane (CL-20) and CL-20-Based Formulations (Review), Compust. Explos. Shock Waves, 2005, 41, 3–16 Search PubMed.
- Q. Ma, T. Jiang, Y. Chi, Y. Chen, J. Wang, J. Huang and F. Nie, A novel multi-nitrogen 2,4,6,8,10,12-hexanitrohexaazaisowurtzitane-based energetic co-crystal with 1-methyl-3,4,5-trinitropyrazole as a donor: experimental and theoretical investigations of intermolecular interactions, New J. Chem., 2017, 41, 4165 RSC.
- M. K. Bellas and A. J. Matzger, Achieving Balanced Energetics through Cocrystallization, Angew. Chem., Int. Ed., 2019, 58, 17185–17188 CrossRef CAS PubMed.
- J. C. Bennion, A. McBain, S. F. Son and A. J. Matzger, Design and synthesis of a series of nitrogen-rich energetic cocrystals of 5,5′-dinitro-2H,2H′-3,3′-bi-1,2,4-triazole (DNBT), Cryst. Growth Des., 2015, 15, 2545–2549 CrossRef CAS.
- S. R. Anderson, P. Dubé, M. Krawiec, J. S. Salan, D. J. A. Ende and P. Samuels, Promising CL-20-Based Energetic Material by Cocrystallization, Propellants, Explos. Pyrotech., 2016, 41, 783–788 CrossRef CAS.
- J. C. Bennion, Z. R. Siddiqi and A. J. Matzger, A melt castable energetic cocrystal, Chem. Commun., 2017, 53, 6065–6068 RSC.
- Y. Wang, Z. Yang, H. Li, X. Zhou, Q. Zhang, J. Wang and Y. Liu, A Novel Cocrystal Explosive of HNIW with Good Comprehensive Properties, Propellants, Explos. Pyrotech., 2014, 39, 590–596 CrossRef CAS.
- J. C. Bennion, L. Vogt, M. E. Tuckerman and A. J. Matzger, Isostructural Cocrystals of 1,3,5-Trinitrobenzene Assembled by Halogen Bonding, Cryst. Growth Des., 2016, 16, 4688–4693 CrossRef CAS.
- K. B. Landenberger, O. Bolton and A. J. Matzger, Two Isostructural Explosive Cocrystals with Significantly Different Thermodynamic Stabilities, Angew. Chem., Int. Ed., 2013, 52, 6468–6471 CrossRef CAS PubMed.
- S. I. Bozkuş, K. S. Hope, B. Yüksel, N. Atceken, H. Nazır, O. Atakol and N. Şen, Characterization and properties of a novel energetic Co-crystal formed between 2,4,6-Trinitrophenol and 9-Bromoanthracene, J. Mol. Struct., 2019, 1192, 145–153 CrossRef.
- K. B. Landenberger, O. Bolton and A. J. Matzger, Energetic-energetic cocrystals of diacetone diperoxide (DADP): Dramatic and divergent sensitivity modifications via cocrystallization, J. Am. Chem. Soc., 2015, 137, 5074–5079 CrossRef CAS PubMed.
- N. Qiao, M. Li, W. Schlindwein, N. Malek, A. Davies and G. Trappitt, Pharmaceutical cocrystals: An overview, Int. J. Pharm., 2011, 419, 1–11 CrossRef CAS PubMed.
- K. Lee, L. B. Chapman and M. D. Cobura, 3-Nitro-1,2,4-triazol-5-one, a less sensitive explosive, J. Energ. Mater., 1987, 5, 27–33 CrossRef CAS.
- D. A. Pillard, W. S. Eck, M. S. Johnson and S. Packard, Effects of 3-Nitro-1,2,4-triazol-5-one on Survival, Growth and Metamorphosis in the Northern Leopard Frog, Lithobates pipiens, Ecotoxicology, 2017, 26, 1170–1180 CrossRef CAS PubMed.
- G. Singh and S. P. Felix, Studies on energetic compounds: 25. An overview of preparation, thermolysis and applications of the salts of 5-nitro-2,4-dihydro-3H-1,2,4-triazol-3-one (NTO), J. Hazard. Mater., 2002, 90, 1–17 CrossRef CAS PubMed.
- J. A. Dean and N. York St Louis San Francisco Auckland Bogotá Caracus Lisbon London Madrid Mexico Milan Montreal New Delhi Paris San Juan São Paulo, LANGE’S HANDBOOK OF CHEMISTRY.
- A. J. Cruz-Cabeza, Acid–base crystalline complexes and the pKa rule, CrystEngComm, 2012, 14, 6362–6365 RSC.
- F. P. A. Fabbiani and C. R. Pulham, High-pressure studies of pharmaceutical compounds and energetic materials, Chem. Soc. Rev., 2006, 35, 932–942 RSC.
- H. H. Cady and L. C. Smith, Studies on the polymorphs of HMX, Los Alamos National Laboratory, Los Alamos, 1962, pp. 1–50 Search PubMed.
- S. Zeman, M. Jungovà and Q.-L. Yan, Impact sensitivity in respect of the crystal lattice free volume and the characteristics of plasticity of some nitramine explosives, Chin. J. Energetic. Mater., 2015, 13, 124–127 Search PubMed.
- X. Bidault and S. Chaudhuri, Can a shock-induced phonon up-pumping model relate to impact sensitivity of molecular crystals, polymorphs and cocrystals, RSC Adv., 2022, 12, 31282 RSC.
- A. A. L. Michalchuk, S. Rudić, C. R. Pulham and C. A. Morrison, Predicting the impact sensitivity of a polymorphic explosive: the curious case of FOX-7, Chem. Commun., 2021, 57, 11213–11216 RSC.
- I. L. Christopher, C. R. Pulham, A. A. L. Michalchuk and C. A. Morrison, J. Chem. Phys., 2023, 158, 124115 CrossRef CAS PubMed.
- A. A. L. Michalchuk, J. Hemingway and C. A. Morrison, Predicting the impact sensitivities of energetic materials through zone-center phonon up-pumping, J. Chem. Phys., 2021, 154, 1–11 CrossRef PubMed.
- S. Klotz, J.-C. Chervin, P. Munsch and G. Le Marchand, Hydrostatic limits of 11 pressure transmitting media, J. Phys. D, 2009, 42, 075413 CrossRef.
- D. M. S. Martins, D. S. Middlemiss, C. R. Pulham, C. C. Wilson, M. T. Weller, P. F. Henry, N. Shankland, K. Shankland, W. G. Marshall, R. M. Ibberson, K. Knight, S. Moggach, M. Brunelli and C. A. Morrison, Temperature- and pressure-induced proton transfer in the 1:1 adduct formed between squaric acid and 4,4-bipyridine, J. Am. Chem. Soc., 2009, 131, 3884–3893 CrossRef CAS PubMed.
- C. C. Wilson, Migration of the proton in the strong O–H⋯O hydrogen bond in urea-phosphoric acid (1/1), Acta Crystallogr., Sect. B, 2001, 57, 435–439 CrossRef CAS PubMed.
- C. A. Morrison, M. M. Siddick, P. J. Camp and C. C. Wilson, Toward Understanding Mobile Proton Behavior from First Principles Calculation: The Short Hydrogen Bond in Crystalline Urea-Phosphoric Acid, J. Am. Chem. Soc., 2005, 127, 4042–4048 CrossRef CAS PubMed.
- V. Srikant and D. R. Clarke, On the optical band gap of zinc oxide, J. Appl. Phys., 1998, 83, 5447 CrossRef CAS.
- L. R. Warren, E. McGowan, M. Renton, C. A. Morrison and N. P. Funnell, Direct evidence for distinct colour origins in ROY polymorphs, Chem. Sci., 2021, 12, 12711–12718 RSC.
- A. Coelho, TOPAS-Academic V4, Coelho Software, Brisbane, Australia, 2007 Search PubMed.
- I. Heo, J. C. Lee, B. Rukiye Özer and T. Schultz, Mass-correlated high-resolution spectra and the structure of benzene, J. Phys. Chem. Lett., 2022, 13, 8278–8283 CrossRef CAS PubMed.
- A. D. Fortes, RAL Technical Report, RAL-TR-2019-002, 2019.
- N. V. Muravyev, D. B. Meerov, K. A. Monogarov, I. N. Melnikov, E. K. Kosareva, L. L. Fershtat, A. B. Sheremetev, I. L. Dalinger, I. V. Fomenkov and A. N. Pivkina, Sensitivity of energetic materials: Evidence of thermodynamic factor on a large array of CHNOFCl compounds, Chem. Eng. J., 2021, 421, 129804 CrossRef CAS.
- A. A. L. Michalchuk, M. Trestman, S. Rudić, P. Portius, P. T. Fincham, C. R. Pulham and C. A. Morrison, Predicting the reactivity of energetic materials: An: ab initio multi-phonon approach, J. Mater. Chem. A, 2019, 7, 19539–19553 RSC.
- N. Atceken, J. Hemingway, C. L. Bull, X. Liu, A. A. L. Michalchuk, S. Konar, C. A. Morrison and C. R. Pulham, Phys. Chem. Chem. Phys., 2023, 25, 31646–31654 RSC.
- L. Kien-Yin, L. B. Chapman and M. D. Coburn, J. Energ. Mater., 1987, 5, 27–33 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: A complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, SHELXT – Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. L. Bull, N. P. Funnell, M. G. Tucker, S. Hull, D. J. Francis and W. G. Marshall, High Press. Res., 2016, 36(4), 493–511 CrossRef.
- W. G. Marshall and D. J. Francis, J. Appl. Cryst., 2002, 35, 122–125 CrossRef CAS.
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. F. Leal, M. A. Gigg, V. E. Lynch, A. Markvardsen, D. J. Mikkelson, R. L. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. G. Perring, P. F. Peterson, S. Ren, M. A. Reuter, A. T. Savici, J. W. Taylor, R. J. Taylor, R. Tolchenov, W. Zhou and J. Zikovski, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764156 Search PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, Quantum-mechanical condensed matter simulations with CRYSTAL, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1–36 CAS.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D’Arco, M. Llunell, M. Causà, Y. Noël, L. Maschio, A. Erba, M. Rerat and S. Casassa, CRYSTAL17 User's Manual, University of Torino, Torino, 2017 Search PubMed.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 121, 1187–1192 CrossRef CAS PubMed.
- K. Refson, P. R. Tulip and S. J. Clark, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 155114 CrossRef.
- Jmol development team, 2016. Jmol, Available at: https://jmol.sourceforge.net/.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2170530, 2170531, 2170532, 2170533, 2170534, 2170535, 2170536, 2170537, 2170538, 2170539, 2170540, 2170541 and 2256439. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cp01595a |
| This journal is © the Owner Societies 2024 |