
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A case study using spectroscopy and computational modelling for Co speciation in a deep eutectic solvent†
Isuri N.
Perera
 a,
Garima S.
Dobhal
b,
Jennifer M.
Pringle
a,
Luke A.
O’Dell
b,
Sherif Abdulkader
Tawfik
b,
Tiffany R.
Walsh
*b and
Cristina
Pozo-Gonzalo
*acd
a,
Garima S.
Dobhal
b,
Jennifer M.
Pringle
a,
Luke A.
O’Dell
b,
Sherif Abdulkader
Tawfik
b,
Tiffany R.
Walsh
*b and
Cristina
Pozo-Gonzalo
*acd
aInstitute for Frontier Materials, Deakin University, Melbourne, Victoria 3125, Australia. E-mail: cpg@deakin.edu.au; cpozo@icb.csic.es
bInstitute for Frontier Materials, Deakin University, Geelong, VIC 3216, Australia
cAragonese Foundation for Research and Development (ARAID), Av. de Ranillas 1-D, 50018 Zaragoza, Spain
dInstituto de Carboquímica (ICB-CSIC), C/Miguel Luesma Castán, 4, 50018, Zaragoza, Spain
First published on 26th July 2024
Abstract
Cobalt has a vital role in the manufacturing of reliable and sustainable clean energy technologies. However, the forecasted supply deficit for cobalt is likely to reach values of 150 kT by 2030. Therefore, it is paramount to consider end-of-life devices as secondary resources for cobalt. Electrorecovery of cobalt from leached solutions has attracted attention due to the sustainability of the recovery process over solvent extraction followed by chemical precipitation. Recently, we reported Co electrorecovery from two different cobalt sources (CoCl2·6H2O and CoSO4·7H2O) using ethylene glycol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :choline chloride (EG:ChCl) in a 4.5:1 molar ratio, leading to higher purity and easier electrodeposition when sulfate was present as an additive. Here, Co2+ speciation is reported for the two EG:ChCl systems depending on the cobalt source using several spectroscopic techniques (e.g. NMR, EPR, FTIR) in combination with molecular dynamics simulations. Monodentate coordination of SO42− to Co2+, forming the tetrahedral [CoCl3(SO4)]3− was observed as the dominant structure in the system containing CoSO4·7H2O, whereas the system comprising CoCl2·6H2O shows a homoleptic tetrahedral [CoCl4]2− as the dominant structure. This resulted in knowledge being gained regarding Co2+ speciation and the correlation with electrochemistry will contribute to the science required for designing safe electrolytes for efficient electrorecovery.
:choline chloride (EG:ChCl) in a 4.5:1 molar ratio, leading to higher purity and easier electrodeposition when sulfate was present as an additive. Here, Co2+ speciation is reported for the two EG:ChCl systems depending on the cobalt source using several spectroscopic techniques (e.g. NMR, EPR, FTIR) in combination with molecular dynamics simulations. Monodentate coordination of SO42− to Co2+, forming the tetrahedral [CoCl3(SO4)]3− was observed as the dominant structure in the system containing CoSO4·7H2O, whereas the system comprising CoCl2·6H2O shows a homoleptic tetrahedral [CoCl4]2− as the dominant structure. This resulted in knowledge being gained regarding Co2+ speciation and the correlation with electrochemistry will contribute to the science required for designing safe electrolytes for efficient electrorecovery.
Introduction
Cobalt is listed as a critical metal due to its scarcity and important role in developing clean energy, as it is one of the key metals in Li-ion battery (LIB) cathodes. The demand for LIBs is expanding due to the ever-increasing demand for consumer electronics and electric vehicles, thereby leading to a large volume of spent LIBs as waste. Recent research publications have projected that by 2030, more than 11 million tonnes of spent LIBs will reach their end of life.1,2 This is a significant problem as spent LIBs contain toxic and flammable chemicals as well as metals, including cobalt that can create a serious impact on human health and the ecosystems.3 The current recovery process of cobalt from spent LIB cathodes is mainly based on hydrometallurgical methods involving leaching (using traditional concentrated acids or alkali), separation steps (e.g., solvent extraction, ion exchange resins), and product recovery steps (e.g., precipitation, electrodeposition).4,5The use of deep eutectic solvents (DESs) for metal recovery has received a great deal of attention since they can be safer and more environmentally friendly compared to conventional solvents such as H2SO4, HNO3, and HCl, which are currently used in metal recovery.6–9 DESs can be easily tuned by mixing different hydrogen bond donors (HBD) and hydrogen bond acceptors (HBA) to form a liquid at room temperature.10 Moreover, the suitability of DESs for the electrochemical deposition of cobalt has been illustrated using several DESs in the recovery process.10–16
Recently, we have investigated the electrochemical recovery of Co using two model Co sources (CoCl2·6H2O and CoSO4·7H2O) in three solvent systems containing different EG amounts (67 molar%, 82 molar%, and 100 molar%) in a mixture with choline chloride to study the role of EG. 82 molar% EG with ChCl (EG:ChCl in 4.5:1 molar ratio) led to an easier reduction of Co2+ compared to the 2:1 (67 molar% EG) system.17 These Co sources were chosen based on the possible anions in the solution after leaching the spent LIBs with the most common acids (HCl and H2SO4) in hydrometallurgy. This previous study showed clear differences in the electrochemical behaviour of the two systems, indicating the likely existence of different speciation for Co2+. The sulfate system (0.1 mol L−1 CoSO4·7H2O in EG:ChCl (4.5:1)) showed more positive Co2+ reduction potential, stable electrodeposition for 45 minutes (constant current density with time), and higher metallic Co content in the electrodeposit in comparison to the chloride system (0.1 mol L−1 CoCl2·6H2O in EG:ChCl (4.5:1)).17 Both systems showed similar physicochemical properties such as viscosity and ionic conductivity even though the water contents differed, with a higher content for the sulfate system (chloride system: <4000 ppm and sulfate system: <7500 ppm), which interestingly did not translate into a higher content of side products (Co(OH)2) in the electrodeposits. Therefore, we hypothesised that the above-mentioned differences in the electrochemistry of the two systems could be due to the differences in Co2+ speciation in the bulk. Hence, in-depth speciation studies for Co2+ are required to understand the differences in the bulk of the two systems and, thus, their impact on the electrochemistry. The correlation between the electrochemistry and speciation of metal ions such as Nd, Na, Cu, and Co in ILs,18–23 and Zn, Te, I2, Ni, and U in DESs,24–31 have been previously reported.
Speciation can be investigated using analysis techniques to identify or quantify a chemical species in a sample. Ultraviolet-visible (UV-vis) and Fourier transform infrared (FTIR) spectroscopies are the most reported analytical techniques used to gain knowledge on Co2+ speciation in the literature.11,32,33 UV-vis analysis of Co2+ in ChCl:EG (1:2), ChCl:citric acid (2:1) and ChCl:organic acid (e.g. lactic, malonic, succinic, glutaric, glycolic acids) in a 1:1 and 1:2 molar ratio have shown three signature bands of the tetrachlorocobaltate(II) anion (tetrahedral) between 600–700 nm, irrespective of the chemical nature of the HBD.32 However, Co2+ in ChCl:urea (1:2) and aqueous dilutions of p-tolunesulfonic acid:ChCl (1:1) has shown only one broad band (at λmax 638 nm and ∼515 nm respectively) which corresponds to an octahedral Co2+.34,35 On the other hand, FTIR spectroscopic analysis of Co2+ in ChCl:lactic acid (1:1) and ChCl:urea (1:2) has been used to understand the coordination of Co2+ in DESs. A new band at 2208 cm−1 has been identified upon adding Co2+ in ChCl:urea (1:2) compared to the neat DES. This new absorption band has been attributed to the Co–O bond,35 confirming the coordination of urea with Co2+.
Speciation can be affected by factors such as the presence of additives, e.g., water. A study was carried out by Amphlett et al. on the effect of increasing water content on the speciation of Co2+ in three DESs (EG:ChCl in 2:1, urea:ChCl in 2:1, and malonic acid:ChCl in 1:1).36 They have shown that with less than 20 weight% of water, Co2+ in all three DESs presents tetrahedral geometry. Once the water content was increased up to 50 wt%, the speciation of Co2+ changed from tetrahedral to octahedral geometry.
Molecular dynamics (MD) simulations can provide complementary atomic-scale insights into the coordination of Co2+ in these complex liquid environments. Computational studies on neat EG:ChCl (2:1) have been reported previously.37–39 Bezerra et al. reported outcomes of force-field MD (FFMD) simulations of CoCl2·6H2O in EG:ChCl (2:1) and found that the Co2+ interacted strongly with Cl− independent of temperature,40 as expected due to the chlorophilic nature of Co2+.41,42 It has also been reported that the Co–Cl coordination number increased with higher temperatures (333 and 353 K).40 However, to date, no simulations of the EG:ChCl (4.5:1) composition have been reported, nor have any simulations of this DES containing both Co2+, chloride and the additives present in our system (e.g. sulfate ions, ethylene glycol, water).
Therefore, this work investigates the speciation of Co2+ in two systems (0.1 mol L−1 CoCl2·6H2O in EG:ChCl (4.5:1) and 0.1 mol L−1 CoSO4·7H2O in EG:ChCl (4.5:1)) using UV-vis spectroscopy, FTIR spectroscopy, electron paramagnetic resonance (EPR) spectroscopy, nuclear magnetic resonance (NMR) spectroscopy and MD simulations. Our results reveal that even when SO42− (Co2+:SO42−, 1:1) is present in a significantly lower concentration compared with Cl− (Co2+:Cl−, 1:26), it could still be coordinated to Co2+ together with 3 Cl−, forming a mixed ligand coordinated tetrahedral complex that eases the Co electrodeposition. This mono-coordination of SO42− to Co2+ in the sulfate system has been corroborated using FTIR spectroscopy and MD simulations, whereas 1H NMR spectroscopy has clearly shown a significant difference in the chemical environment between sulfate and chloride systems. Hence, this study demonstrates new insights into the tunability of the electrochemical behaviour of Co2+ based on the chemical composition of the electrolyte.
Experimental
Materials and solution mixtures
Cobalt chloride hexahydrate, CoCl2·6H2O with 98% purity, cobalt sulfate heptahydrate, CoSO4·7H2O with ≥98% purity, anhydrous ethylene glycol with 99.8% purity, and choline chloride, with ≥98% purity were purchased from Sigma-Aldrich. All chemicals were used as received.The solvent system was prepared by mixing ethylene glycol (EG) and choline chloride (ChCl) in a 4.5:1 molar ratio and stirring at 500 rpm for 2–3 h at room temperature in a fume hood until a clear and homogeneous solution was obtained. Two solution mixtures were prepared by adding CoCl2·6H2O and CoSO4·7H2O salts to obtain 0.1 mol L−1 in EG:ChCl (4.5:1). (0.0242 g of CoCl2·6H2O and 0.0281 g of CoSO4·7H2O were dissolved in 1.0 mL of solvent system). Solutions were stirred for 1 h at 50 °C in an Ar-filled glovebox (Kiyon, H2O, and O2 <0.1 ppm) until homogeneous solutions were obtained.
Ultraviolet-visible (UV-vis) spectroscopy
A UV-vis spectrophotometer (UV-2600, SHIMADZU) was used to identify the coordination geometry of Co2+ in each solution mixture. UV spectra were obtained before and after diluting the solution mixtures (0.001 mol L−1, 100 times to lower concentrations) with the solvent system consisting of EG:ChCl (4.5:1) to avoid saturation of the absorbance and performing a wavelength scan.
Fourier transform infrared (FTIR) spectroscopy
The FTIR spectra of the samples were recorded using a PerkinElmer Frontier spectrometer, with the background being measured under identical conditions (4000–500 cm−1, 4 cm−1 resolution, and 32 scans) at room temperature. The spectrometer was equipped with a Golden Gate Diamond ATR stage with a heating option. The detachable top plate of the stage's specially designed anvil attachment prevented the sample from being exposed to atmospheric moisture after loading the samples inside a glovebox. The spectra were ATR-corrected, and baseline corrected.Electron paramagnetic resonance (EPR) spectroscopy
The EPR measurements were performed at room temperature using an ADANI CMS 8400 X-band bench-top EPR spectrometer. Samples were placed into quartz glass capillary tubes with a 4 mm diameter. Typical instrument settings were center field: 334 mT, sweep width: 250 mT, modulation amplitude: 200 μT, power attenuation: 12 dB, and sweep time: 50 s. The average signal after 50 scans was plotted for each sample. The background was corrected using Origin.Nuclear magnetic resonance (NMR) spectroscopy
1H NMR spectra of the samples were measured using a Bruker Avance III 500 MHz spectrometer at room temperature. Samples were filled into 3 mm capillary tubes and inserted into 5 mm NMR tubes filled with D2O after heat sealing.Force-field molecular dynamics (FFMD) simulations
Force-field molecular dynamics (FFMD) simulations of Co2+ in the DES were conducted using GROMACS software.43 The optimized potentials for liquid simulations – all atom (OPLS–AA) force field with Doherty et al.'s scaled parameters for the EG:ChCl (2:1) were used.44,45 Sulfate ion parameters were taken from Cannon et al. and were also scaled.46 Three liquid compositions were modelled in total, all with a 4.5:1 molar ratio: two compositions were modelled with CoSO4 salt (at two different concentrations: one at a higher concentration (Co2+:EG, 1:45) and one at the lower (Co2+:EG, 1:120) similar to the experimental concentration) and one with CoCl2 salt (at a higher concentration only), summarised in the ESI,† Table S1. These systems were subjected to equilibration under an isothermal-isobaric ensemble (NPT) at 1 bar for 10–20 ns until a stable density was achieved. Following this step, the liquid was thoroughly mixed using simulated annealing (SA). The complexity of the mixture led to several adjustments of this computational mixing protocol (details are given in the ESI†). Convergence of the relevant radial distribution functions was used to determine the appropriate number of SA cycles required to ensure thorough mixing. Production runs were performed in triplicate, with different initial atomic coordinates in each case, and comprised 100 ns at 300 K in the NVT ensemble at the equilibrium density. Frames were saved every 10 ps. Analysis of coordination numbers and hydrogen bonding used in-house python codes were then used in MD analysis.47,48 Full details, including liquid compositions, are provided in the ESI.†
Ab initio molecular dynamics (AIMD) simulations
Ab initio molecular dynamics (AIMD) simulations were also used to investigate these liquids. Due to their basis in density functional theory (DFT), AIMD is more computationally intensive than FFMD and therefore is constrained by relatively smaller system sizes to ensure appropriate simulation timescales can be achieved. To generate the initial configurations for AIMD, a small system comprising 1 Co2+ atom, and the respective number of molecules (703 atoms, compared with ∼22000 atoms for the FFMD simulations) was prepared using FFMD simulations to ensure these smaller samples matched the liquid density obtained from the larger-sized FFMD simulations (details in the ESI,† Table S2). Spin-polarized AIMD simulations were performed using the Vienna Ab Initio Simulation Package (VASP),49 with the PBE functional,50 along with projector augmented wave potentials.49,51–53 The temperature was controlled using the Nosé–Hoover54,55 thermostat at 300 K. The two systems were run for 21 ps of dynamics with a timestep of 1 fs.
Results and discussion
UV-vis analysis was conducted to investigate the coordination geometry of Co2+. The blue colour of both systems indicates a tetrahedral coordination geometry, which was also corroborated by UV-vis spectra showing three signature bands between 600–700 nm32,33,56 (Fig. 1). Tran et al. reported [CoCl4]2− is indicated by the three signature bands at 630, 667, and 696 nm in the EG:ChCl (2:1) solvent after leaching LiCoO2.33 In our study, the two systems had to be diluted 100 times with EG:ChCl (4.5:1) before the analysis to avoid the saturation of the absorbance,17 and no colour change was observed upon dilution, indicating that the tetrahedral geometry of Co2+ was maintained. However, the UV-vis spectra of 0.1 mol L−1 Co2+, prior to dilution, shows a small proportion of octahedral Co2+ (λmax = 531 nm) in both systems together with the dominant tetrahedral Co2+ as shown in Fig. S1 (ESI†). Recently an octahedral structure [Co(EG)6]2+ was reported at λmax = 534 nm, which is slightly different from the value that we observe.57 This difference may be related to a different octahedral speciation in our system, which may combine EG and other ligands available in the DES mixture.
 | ||
| Fig. 1 UV-vis spectra of 0.001 mol L−1 (a) CoCl2·6H2O in EG:ChCl (4.5:1) and (b) CoSO4·7H2O in EG:ChCl (4.5:1), the wavelength of each band, colour in the original solution mixture (0.1 mol L−1) and hypothesised Co2+ tetrahedral coordination in the sulfate system. | ||
Moreover, the blue colour was maintained in both systems when heated at 50 °C, in which the electrochemical experiments were performed, indicating that Co2+ tetrahedral coordination remained at 50 °C.
[CoCl4]2− is the most commonly reported tetrahedral structure for Co2+ in chloride-containing media; however, it has been reported to be electrochemically inactive.20,23 As we have reported the successful reduction of Co2+ to cobalt metal in a chloride rich environment that contained other additives (e.g. EG, water, and sulfate anion),17 it is plausible that those additives or other molecules such as sulfate and/or EG could be part of the solvation sphere in addition to chloride while maintaining the tetrahedral coordination geometry in the undiluted systems.17 To investigate this, several spectroscopic techniques were used, as described below, to investigate the chemical nature of the coordinated ligands and the speciation of Co2+ in the two systems.
EPR spectroscopy
EPR spectroscopy is a valuable analytical technique to detect different chemical environments in paramagnetic transition metal complexes (i.e., cobalt), including its coordination geometry.58 Unfortunately, there is a lack of EPR spectroscopy studies of Co2+ chemical environments in DESs. The EPR spectra, as shown in Fig. S2(a) (ESI†), arise from the interaction between the external magnetic field and magnetic moments of unpaired electrons in the systems, according to the Zeeman interaction.58Paramagnetic metal ions can exhibit different g values depending on the orientation of the metal complex in the applied magnetic field.58 Therefore, the g factor was calculated for the chloride and sulfate systems using eqn (S2), and plotted as shown in Fig. S2(b) (ESI†).
Both systems showed similar g values (2.4) which lie in the region expected for tetrahedral Co2+ complexes (g = 2.2–2.4) in agreement with the UV-vis spectra.59,60 However, there is no significant difference between the spectra of the two systems that can provide information regarding the chemical nature of the ligands that are coordinated to Co2+. This may be due to the very fast spin lattice relaxation of Co2+ leading to line broadening,60 and the lower sensitivity of this technique when using a high dielectric solvent (dielectric constant of EG = 37) at room temperature. However, we also need to consider that the changes in speciation between the two systems may be too small to be detected accurately by EPR spectroscopy.
FTIR spectroscopy
FTIR spectroscopy is a useful technique to investigate the solvation environment of metals in the solvent system under study. First, the solvent system, EG:ChCl (4.5:1) and its individual constituents (EG as the hydrogen bond donor (HBD) and ChCl as the hydrogen bond acceptor (HBA)) were analysed using FTIR as shown in Fig. S3 (ESI†). Details of the spectral bands are provided in the ESI.†
Subsequently, the cobalt-containing systems were analysed using FTIR spectroscopy, as shown in Fig. 2, and compared with the neat DES. The spectra showed no significant changes in the band's position compared to the neat solvent systems.
 | ||
| Fig. 2 FTIR spectra of EG:ChCl (4.5:1) (bottom), 0.1 mol L−1 CoCl2·6H2O in EG:ChCl (4.5:1) (middle) and 0.1 mol L−1 CoSO4·7H2O in EG:ChCl (4.5:1) (top) analysed at room temperature. | ||
In our previous study, we observed that the mixtures containing a small concentration of sulfate anions, in conjunction with chloride anions, favoured the reduction of Co2+.17 Therefore, here we focused our attention on the S–O vibration region to understand the role of sulfate in cobalt electrochemistry. According to the literature, the free sulfate anion has tetrahedral symmetry with four internal fundamental vibrational modes: symmetric S–O stretching, asymmetric S–O stretching, symmetric O–S–O bending, and asymmetric O–S–O bending. However, only the bands corresponding to asymmetric S–O stretching (1102 cm−1) and asymmetric O–S–O bending (611 cm−1) are active and within the range of the FTIR spectrometer.61–64
On the other hand, a coordinated sulfate anion shows a splitting of the asymmetric S–O stretching band to two or three bands depending on the coordination denticity, allowing the weak symmetric S–O stretching band to become IR active. Therefore, three bands in total exemplify the monodentate coordination, whereas four bands are present for bidentate coordination with an additional peak at ∼1200 cm−1.65,66 Several authors reported the three bands for monodentate coordinated sulfate in different complexes; [Co(NH3)5SO4]·Br, [Cr(OH2)5SO4Cl]·0.5H2O and FeSO4+ in the range 970–980 cm−1, 1040–1059 cm−1 and 1122–1130 cm−1 for symmetric S–O stretching, and two asymmetric S–O stretching bands respectively.61,65 However, here, the FTIR spectrum of the sulfate system was not clear as an overlapping with the bands of the neat solvent system occurred. Therefore, a higher concentration of CoSO4·7H2O was added to EG:ChCl (4.5:1), and Fig. 3 depicts the sulfate region of interest.
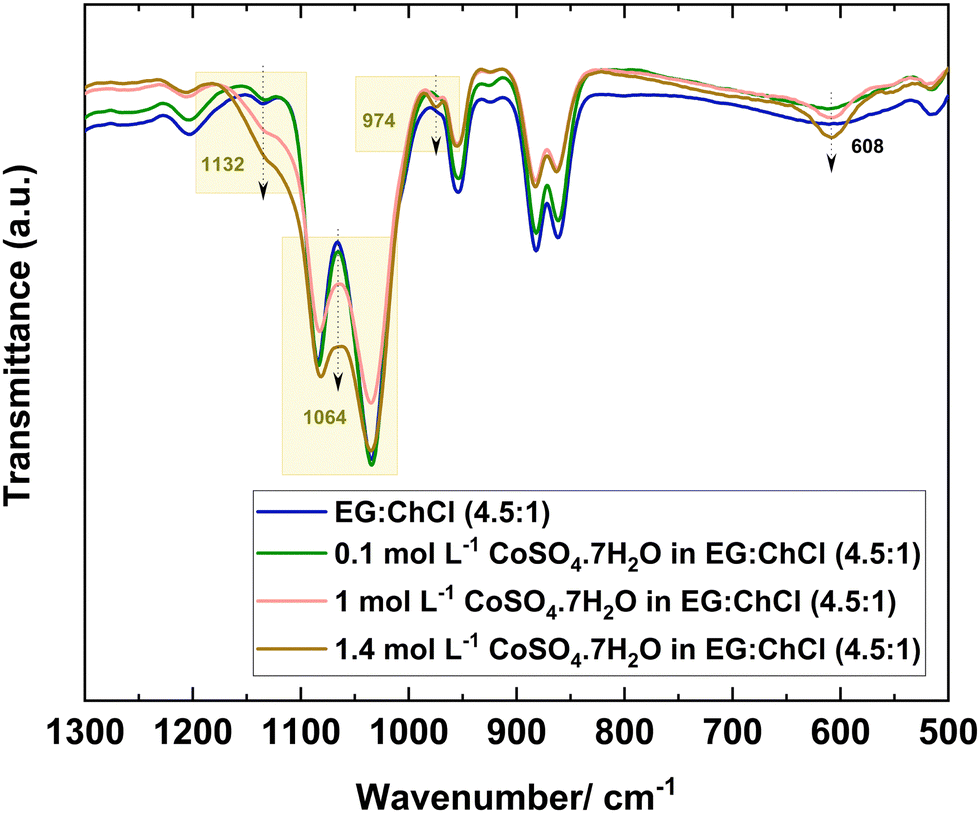 | ||
| Fig. 3 FTIR spectra of EG:ChCl (4.5:1), 0.1 mol L−1, 1 mol L−1, and 1.4 mol L−1 of CoSO4·7H2O in EG:ChCl (4.5:1) analysed at room temperature. | ||
The concentration of CoSO4·7H2O was increased by more than 10 times while maintaining the Co:sulfate ratio at 1:1. An increase in the relative intensity of the band at 1132 cm−1, a small new band at 974 cm−1 and a change at 1064 cm−1, were observed upon increasing the sulfate salt in the DES mixture (Fig. 3). Interestingly, there was no increase in the relative intensity of the band at 1200 cm−1, which originates from the solvent system, EG:ChCl (4.5:1). All these observations suggest a possible monodentate coordination of the sulfate anion with Co2+. Moreover, the relative intensity of the asymmetric O–S–O bending vibration band at 608 cm−1 also increased with the increase in CoSO4·7H2O concentration, suggesting the presence of free sulfate in the sulfate system.67,68 This can be related to the large excess concentration of CoSO4·7H2O in the DES mixture. FTIR spectroscopy was also performed for the 1.4 mol L−1 of CoSO4·7H2O in EG:ChCl (4.5:1) at 50 °C, at the same temperature as the electrochemical experiments were performed (Fig. S4, ESI†). No changes were observed between the spectra at room temperature and at 50 °C, indicating similar Co2+ speciation at those temperatures.
1H NMR spectroscopy
1H NMR spectroscopic analysis was conducted to further investigate the possible interaction of Co2+ with the EG:ChCl (4.5:1) under study. Similar to the FTIR spectroscopic analysis, the individual constituents of the solvent system, EG and ChCl, and the solvent system, EG:ChCl (4.5:1), were analysed using 1H NMR spectroscopy at room temperature, as shown in Fig. S5 (ESI†). For these measurements, the chemicals were introduced in a flame sealed 3 mm capillary inside a 5 mm conventional NMR tube which contained D2O for field lock and accurate referencing of chemical shifts of the samples. This setup was also used to avoid the interaction of Co2+ with the D2O, which would have affected the field locking or referencing, thereby maintaining the accuracy of the experimental analysis. In the case of ChCl, due to its solid state, the sample was dissolved in D2O.
A detailed analysis of the 1H NMR spectra was performed to identify the protons in the solvents and possible interactions in the DES and are provided in the ESI.†
1H NMR spectroscopy was then performed for EG:ChCl (4.5:1) in the presence of the two Co salts under study to investigate the effect of the salt chemistry on the Co coordination (shown in Fig. 4 and Fig. S7, ESI†) using the same experimental setup as described above.
 | ||
| Fig. 4
1H NMR spectra of EG:ChCl (4.5:1) (bottom), 0.1 mol L−1 CoCl2·6H2O in EG:ChCl (4.5:1) (middle) and 0.1 mol L−1 CoSO4·7H2O in EG:ChCl (4.5:1) (top) analysed at room temperature. | ||
In general, a broadening and shift to high ppm values of all the peaks were observed, consistent with the presence of a paramagnetic complex in the two Co containing systems compared to the neat EG:ChCl (4.5:1).69–71 All the chemical shift values of protons in the neat solvent system and the two Co containing systems are given in Table 1.
:ChCl (4.5:1), 0.1 mol L−1 CoCl2·6H2O and 0.1 mol L−1 CoSO4·7H2O in EG:ChCl (4.5:1) and the extent of chemical shift from the EG:ChCl (4.5:1) for the chloride and sulfate system
|
|
Chemical shift of the protons “d” (ppm) | Chemical shift of the protons “c” (ppm) | Chemical shift of the protons “b” (ppm) | Chemical shift of the protons “a” (ppm) | Chemical shift of the protons “x” (ppm) | Chemical shift of the protons “z” (ppm) |
|---|---|---|---|---|---|---|
| EG:ChCl (4.5:1) |
3.30 | 3.64 | 4.05 | 5.31 | 3.65 | 5.01 |
| CoCl2·6H2O in EG:ChCl (4.5:1) |
7.24 | 7.55 | 8.07 | 9.19 | 7.86 | 9.07 |
| CoSO4·7H2O in EG:ChCl (4.5:1) |
7.30 | 7.62 | 8.14 (merged with x) | 9.34 (merged with z) | 8.14 (merged with b) | 9.34 (merged with a) |
| Extent of the shift from EG:ChCl (4.5:1) for chloride system |
3.94 | 3.91 | 4.02 | 3.88 | 4.21 | 4.06 |
| Extent of the shift from EG:ChCl (4.5:1) for sulfate system |
4.00 | 3.98 | 4.09 | 4.03 | 4.49 | 4.33 |

The aliphatic protons (x) of EG in both systems showed a significant increase in the shift to high ppm values from the neat solvent system (by 4.21 ppm and 4.49 ppm for chloride and sulfate systems, respectively), compared to the protons of ChCl, suggesting a probable coordination of EG with Co2+. However, the extent of the peak shift to high ppm values for all the peaks, and the broader peaks from EG in the sulfate system compared to the chloride system, indicate a shorter average distance to the paramagnetic center (Co2+) in the sulfate system.70 For instance, in the sulfate system, the aliphatic protons (x) and hydroxyl protons (z) of EG show broader peaks overlapping the protons (b) and (a) of ChCl compared to the chloride system. Therefore, proton shifts in the sulfate system have been affected more by the paramagnetic properties of Co2+ than in the chloride system, indicating a difference in the chemical environment between the two systems based on the type of Co salt. This suggests a higher probability of EG coordination to Co2+ in the sulfate system compared to the chloride system. However, this EG coordination to Co2+ in the sulfate system was not observed from FTIR analysis, likely reflecting differences in the sensitivity of the two techniques.
Molecular dynamics simulations
Both force field molecular dynamics (FFMD) and ab initio molecular dynamics (AIMD) simulations were conducted to gain more insight into the speciation of Co2+ in the two systems under study. Two scenarios were studied for the EG:ChCl (4.5:1): one with CoSO4 salt and one with CoCl2 salt, with the number of different molecule types summarized in the ESI† (Table S1). The FFMD simulations are more approximate and more computationally economical, whereas the AIMD simulations are potentially more accurate and relatively computationally expensive. The FFMD simulations allow consideration of larger system sizes over longer timescales, whereas AIMD is confined to smaller system sizes and short timescales. The AIMD simulations have been used here to explore the stability of structures identified using the longer, larger, but more approximate FFMD simulations. Both sets of simulations were designed to predict the equilibrium structures of the Co/liquid complex.
Force field molecular dynamics simulations
Theoretically, there are 120 EG molecules for every Co2+ in the EG:ChCl (4.5:1) system corresponding to a concentration of 0.1 mol L−1 of Co2+. However, to allow for reasonable sampling of possible structures, the systems were initially studied using FFMD simulations at a higher concentration of 1 Co2+ to 45 EG.
First, the liquid density was compared with experiment to validate the force fields used in the FFMD simulations.45,46,72 The experimental density (1.1157 g cm−3) tallied favourably with the simulated bulk density (1.115 g cm−3) at 300 K. Following this, the average coordination environment of the Co2+ was quantified as shown in Fig. 5.
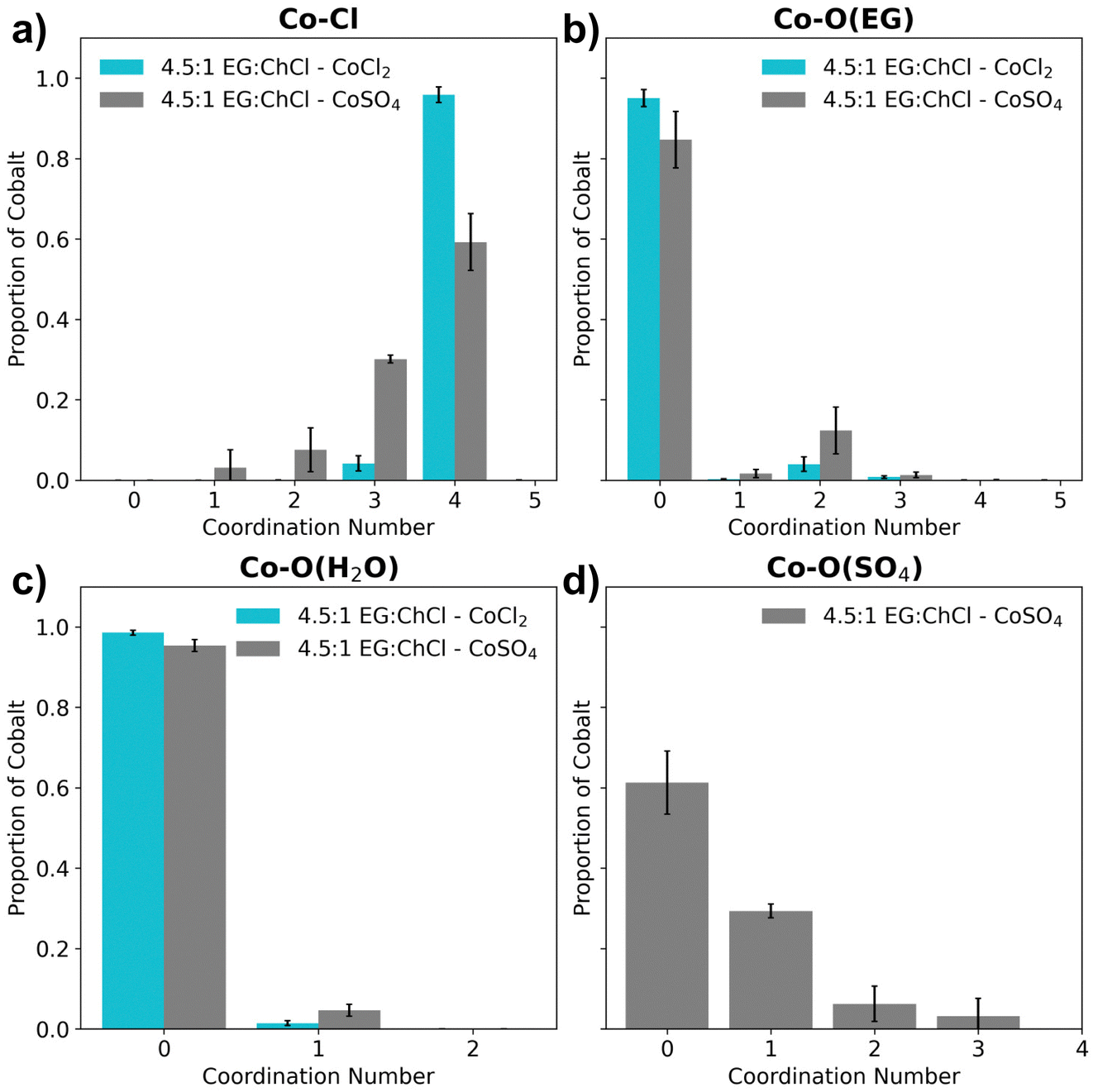 | ||
| Fig. 5 Coordination number distributions of the first shell of the Co2+ for the 4.5:1 EG:ChCl system for (a) Co–Cl, (b) Co–O(EG), (c) Co–O(H2O) and (d) Co–O(SO4). | ||
For this analysis, coordination of the Co2+ site to each relevant component of the solvent was defined by a cut-off distance in each case, established using radial distribution data (Fig. S8, ESI†). Coordination analysis of the Co2+–Cl first shell (Fig. 5a) revealed an almost exclusive preference for a 4-coordinated arrangement in the chloride system that was confirmed to be tetrahedral and is consistent with the experimental UV-vis and EPR data. In contrast, the sulfate system supports a relatively marked increase in the 3-coordinated chloride population counterbalanced with a decrease in the proportion of 4-coordinated complexes, suggesting another species has replaced a chloride ion in the first shell. Since the coordination of EG and water is not dominant (Fig. 5b and c), the replacement of a chloride in the sulfate system is due to coordination by a single sulfate (Fig. 5d).
This [CoCl3SO4]3− complex is predicted to comprise ∼30% of the sample in the sulfate system, supporting the hypothesis of mixed ligand complexes playing a role and consistent with the FTIR data. The coordination data also indicates a minor role for EG coordination to Co2+ in the sulfate-containing system with a ∼10% population of bidentate coordinated EG (coordination number is 2 as shown in Fig. 5b), which is in agreement with the observations from 1H NMR spectroscopic analysis. In contrast, the chloride system showed a relatively reduced amount of EG bidentate coordination to Co2+ (Fig. 5b).
The sulfate-containing system was subsequently studied using a ratio of 1 Co2+ to 120 EG, corresponding to the experimental concentration (0.1 mol L−1). All three replicates of the production run yielded Co–sulfate coordination. The RDF for this system further confirmed a higher likelihood of a monodentate sulfate–Co coordination and furthermore, suggested a mixed ligand environment in the first coordination shell (Fig. S9, ESI†).
Some of the mixed ligand complexes noted above, including H2O and EG in the first coordination shell, were observed in an octahedral geometry (Fig. S10, ESI†). Nonetheless, this proportion is a minority with only ∼14% of the Co2+ featuring this geometry. However, this is still a significant increase compared with its chloride salt counterpart where ∼3% of the cobalt were in the octahedral geometry. In this 14% octahedral population, EG was almost always coordinated to the Co2+ (Fig. S11a, ESI†). Further probing this octahedral coordination in the sulfate system, it was noted that water coordination was almost always accompanied by the EG and sulfate being coordinated to the Co2+ (Fig. S11b, ESI†). Water coordination was also observed in FFMD studies reported by Bezerra-Neto et al. and Bezerra et al. of Cu2+ and Co2+ respectively in the 2:1 EG:ChCl composition where both studies contained approximately similar amounts of water molecules as our study.40,73
The sulfate oxygen (O(SO4)–O(H2O)) RDFs (obtained for any sulfate in the liquid, coordinated or not), shown in Fig. 6a, confirm the dominance of sulfate–water interactions compared to the other species in the liquid. H-bonding analysis of the water interacting with the sulfate that is coordinated to Co2+ showed that over the 10000 frames, there were 145511 H-bonds between a water and a SO42− anion coordinated to a Co2+, indicating an average number of H-bonds at any point in time in the sample of ∼15. The probabilities of water being in the second sphere supporting sulfate coordination to the Co2+ are also higher than 50% as shown in Fig. 6b.
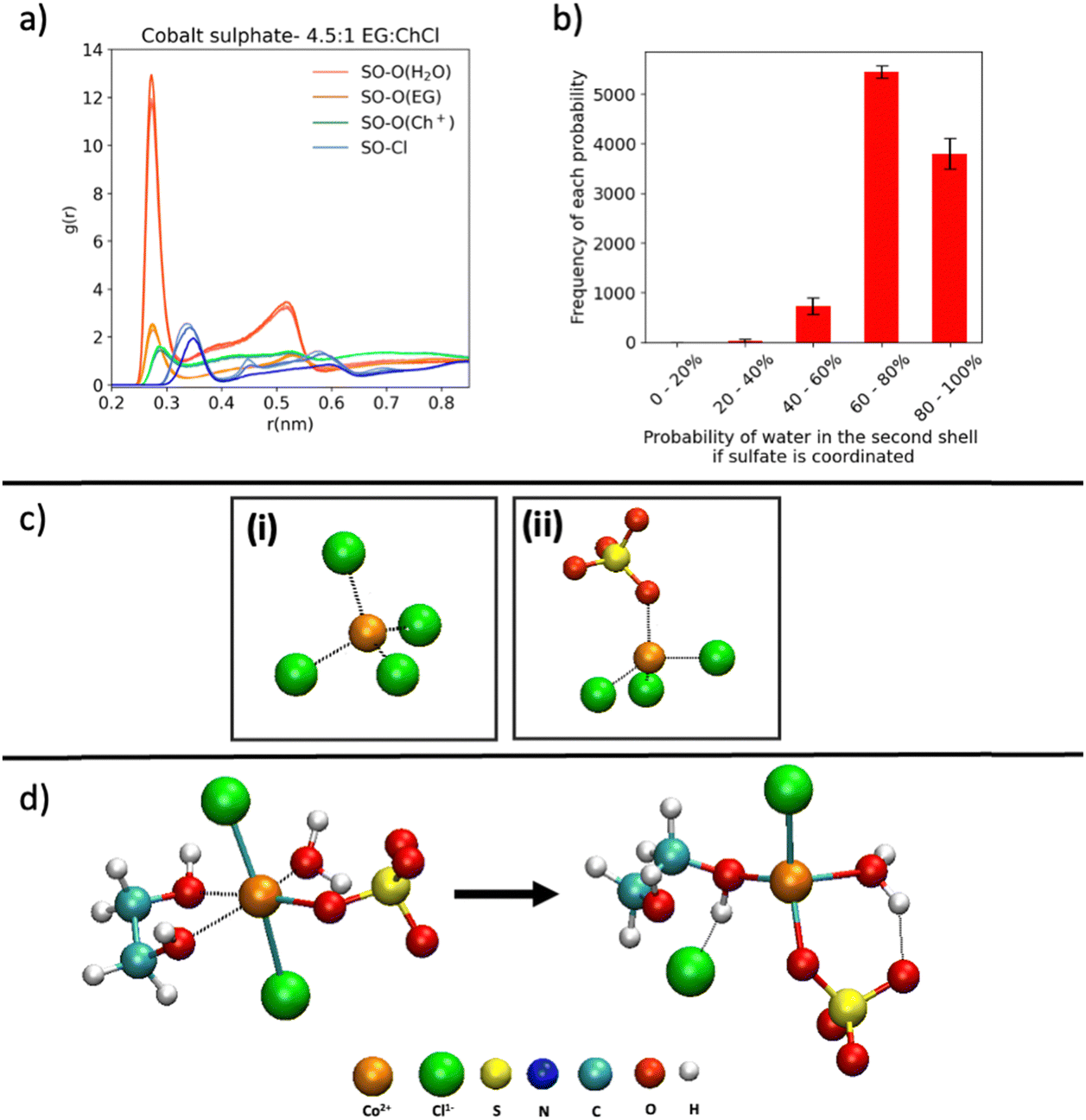 | ||
| Fig. 6 (a) RDFs of SO-X (where X is the other species of interest) in the sulfate system obtained from FFMD simulations. (b) Probabilities of finding water in the second shell of the Co2+ when sulfate is already coordinated to the cobalt obtained from FFMD simulations. (c) (i) [CoCl4]2− complex used in AIMD trajectories for the chloride system, and (ii) [CoCl3(SO4)]3− complex used in the AIMD trajectory for the sulfate system. (d) Initial (octahedral) and final (tetrahedral) state of the complex [CoCl2(EG)(SO4)(H2O)]2− studied for the sulfate system using AIMD. | ||
Ab initio molecular dynamics (AIMD) simulations
To further investigate the stability of the structures predicted using the FFMD simulations, AIMD simulations based on 1 Co2+ were explored. Therefore, AIMD was carried out based on initial geometries in 2 different tetrahedral complexes; [CoCl4]2− (Fig. 6c(i)), [CoCl3(SO4)]3− (Fig. 6c(ii)) and one octahedral complex composed of possible mixed ligands from the electrolyte, [CoCl2(EG)(SO4)(H2O)]2− (Fig. 6d).The tetrahedral complexes [CoCl4]2− and [CoCl3(SO4)]3− showed no significant structural variation over the duration of the AIMD simulations (∼20 ps) indicating that these complexes are stable. The calculated bond lengths of Co–Cl were found to be 2.25 Å in both systems, and the bond length of Co–OSO3 in the sulfate system was 1.95 Å.
The octahedral complex [CoCl2(EG)(SO4)(H2O)]2− (initial structure in Fig. 6d) begins to convert into a tetrahedral structure after 6 ps; the initial and final structures for this trajectory are shown in Fig. 6d. The main changes were the removal of a Cl− from the first coordination shell of the cobalt. Additionally, the EG, which was previously bidentate, changed to a monodentate coordination. One of the oxygens of the EG continued to undergo coordination and decoordination to the Co2+ at a high rate. However, the coordination of the H2O and SO42− to the Co2+ remained stable throughout the trajectory of length 10 ps. This suggests that a tetrahedral mixed ligand Co complex with EG, H2O, SO42− and Cl− coordination can exist. The sulfate coordination in the resultant tetrahedral complex described above can be stabilised through the hydrogen bonding conferred by the presence of water (Fig. 6d).
Additionally, this mixed ligand complex is anticipated to be electrochemically active as opposed to the homoleptic [CoCl4]2− complex which has been reported as electrochemically inactive in the literature.20,23 Even though the chloride system forms [CoCl4]2− as the dominant complex, the presence of high amounts of EG in excess compared to Cl− (Co2+:EG = 1:120 vs. Co2+:Cl− = 1:28) in the electrolyte could have an impact on Co electrochemistry. For instance, 1H NMR spectroscopic study shows the probability of EG coordination to the Co2+ in the chloride system.
As a control study, AIMD simulations of a cobalt complex consisting of two EGs, a monodentate sulfate, a Cl− and no H2O in the first coordination shell were conducted (Fig. S12, ESI†). Over 10 ps of AIMD trajectory, this geometry featured the removal of a sulfate from the first coordination shell, and the replacement of an EG with another EG molecule. Therefore, in this scenario the sulfate coordination is unlikely and provides further evidence that the presence of water may stabilise sulfate coordination.
In summary, Co2+ in both systems supported tetrahedral coordination, in agreement with the results obtained by UV-vis and EPR spectroscopy. The sulfate anion exclusively supported monodentate coordination to Co2+ in the sulfate system, which also agreed with the obtained FTIR spectra. However, using FFMD and AIMD simulations, the possibility of other coordination structures was indicated, which may be present in small quantities. These include structures supporting a bidentate EG, monodentate sulfate, a Cl− and a H2O. AIMD simulations further suggest that these structures can exist as tetrahedral structures.
Conclusions
The correlation of electrochemistry with Co2+ speciation has been studied for an all chloride electrolyte (CoCl2·6H2O) and a sulfate system (CoSO4·7H2O) both in EG:ChCl (4.5:1). Our previous study showed that, among both systems, the sulfate system is more favourable for Co2+ reduction and also leads to an electrodeposit containing higher Co metal content, probably due to the presence of sulfate anion as an additive in the electrolyte. The results of this study obtained from UV-vis spectroscopy, EPR spectroscopy, FFMD simulations and AIMD simulations showed that Co2+ forms a tetrahedral complex as the dominant structure in both sulfate and chloride systems. A difference in the chemical shift values of the protons was observed in 1H NMR spectra between the two systems, demonstrating a different chemical environment of Co2+ in the two systems. For instance, a higher probability of EG coordination to Co2+ in the sulfate system was indicated compared to the chloride system.
FTIR spectroscopy and MD simulations suggest the monodentate coordination of SO42− to Co2+ in the sulfate system, indicating the existence of [CoCl3(SO4)]3− as the dominant structure (80%), whereas in the chloride system, CoCl42− was the dominant structure (95%), corroborating the difference in the coordinated ligands to Co2+ in the two systems. Interestingly, high H2O content in the sulfate system could promote the Co2+ coordination with the SO42− and EG revealing the importance of the presence of H2O in an electrolyte for forming a mixed ligand coordinated Co2+ and easing the electrochemical reduction to Co metal. Additionally, simulations suggest the presence of sulfate can support mixed ligand cobalt complexes involving monodentate sulfate, EG, water and chloride.
Data availability
The data supporting this article are available on request from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Australian Research Council Training Centre for Future Energy Storage Technologies (IC180100049) and funded by the Australian Government. The theoretical work for this project was undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI), which is supported by the Australian Government. The authors also acknowledge Prof. Gosia Swadzba- Kwasny from Queen's University Belfast for technical discussions on cobalt speciation.References
- L. Gaines, Q. Dai, J. T. Vaughey and S. Gillard, Direct Recycling R&D at the ReCell Center, Recycling, 2021, 6(2), 31 CrossRef.
- C. Padwal, H. D. Pham, S. Jadhav, T. T. Do, J. Nerkar, L. T. M. Hoang, A. Kumar Nanjundan, S. G. Mundree and D. P. Dubal, Deep Eutectic Solvents: Green Approach for Cathode Recycling of Li-Ion Batteries, Adv. Energy Sustainability Res., 2021, 3(1), 2100133 CrossRef.
- X. Zheng, Z. Zhu, X. Lin, Y. Zhang, Y. He, H. Cao and Z. Sun, A Mini-Review on Metal Recycling from Spent Lithium Ion Batteries, Engineering, 2018, 4(3), 361–370 CrossRef CAS.
- S. Suriyanarayanan, M. P. Babu, R. Murugan, D. Muthuraj, K. Ramanujam and I. A. Nicholls, Highly Efficient Recovery and Recycling of Cobalt from Spent Lithium-Ion Batteries Using an N-Methylurea-Acetamide Nonionic Deep Eutectic Solvent, ACS Omega, 2023, 8(7), 6959–6967 CrossRef CAS PubMed.
- A. B. Botelho Junior, S. Stopic, B. Friedrich, J. A. S. Tenório and D. C. R. Espinosa, Cobalt Recovery from Li-Ion Battery Recycling: A Critical Review, Metals, 2021, 11(12), 1999 CrossRef CAS.
- Y. Chen, C. Liu, Y. Wang, Y. Tian, Y. Li, M. Feng, Y. Guo, J. Han and T. Mu, Efficient Recovery of Valuable Metals from Lithium-Ion Battery Cathodes Using Phytic Acid-Based Deep Eutectic Solvents at a Mild Temperature, Energy Fuels, 2023, 37(7), 5361–5369 CrossRef CAS.
- G. R. T. Jenkin, A. Z. M. Al-Bassam, R. C. Harris, A. P. Abbott, D. J. Smith, D. A. Holwell, R. J. Chapman and C. J. Stanley, The application of deep eutectic solvent ionic liquids for environmentally-friendly dissolution and recovery of precious metals, Miner. Eng., 2016, 87, 18–24 CrossRef CAS.
- B. Li, Q. Li, Q. Wang, X. Yan, M. Shi and C. Wu, Deep eutectic solvent for spent lithium-ion battery recycling: comparison with inorganic acid leaching, Phys. Chem. Chem. Phys., 2022, 24(32), 19029–19051 RSC.
- G. Zante and M. Boltoeva, Review on Hydrometallurgical Recovery of Metals with Deep Eutectic Solvents, Sustainable Chem., 2020, 1(3), 238–255 CrossRef.
- A. Zhu, X. Bian, W. Han, D. Cao, Y. Wen, K. Zhu and S. Wang, The application of deep eutectic solvents in lithium-ion battery recycling: A comprehensive review, Resour., Conserv. Recycl., 2023, 188, 106690 CrossRef CAS.
- X. Cao, L. Xu, Y. Shi, Y. Wang and X. Xue, Electrochemical behavior and electrodeposition of cobalt from choline chloride-urea deep eutectic solvent, Electrochim. Acta, 2019, 295, 550–557 CrossRef CAS.
- A. Cojocaru, M. L. Mares, P. Prioteasa, L. Anicai and T. Visan, Study of electrode processes and deposition of cobalt thin films from ionic liquid analogues based on choline chloride, J. Solid State Electrochem., 2014, 19(4), 1001–1014 CrossRef.
- M. Landa-Castro, P. Sebastián, M. I. Giannotti, A. Serrà and E. Gómez, Electrodeposition of nanostructured cobalt films from a deep eutectic solvent: Influence of the substrate and deposition potential range, Electrochim. Acta, 2020, 359, 136928 CrossRef CAS.
- M. Li, Z. Wang and R. G. Reddy, Cobalt electrodeposition using urea and choline chloride, Electrochim. Acta, 2014, 123, 325–331 CrossRef CAS.
- A. Serrà, P. Sebastián-Pascual, M. Landa-Castro and E. Gómez, Electrochemical assessment of high active area of cobalt deposited in deep eutectic solvent, J. Electroanal. Chem., 2021, 896, 115177 CrossRef.
- A. M. Sakita, R. Della Noce, C. S. Fugivara and A. V. Benedetti, On the cobalt and cobalt oxide electrodeposition from a glyceline deep eutectic solvent, Phys. Chem. Chem. Phys., 2016, 18(36), 25048–25057 RSC.
- I. N. Perera, J. M. Pringle, K. Periyapperuma, A. Somers, A. Siriwardana, G. Pozo and C. Pozo-Gonzalo, Enhanced Electrochemical Properties of Cobalt by Varying the Ethylene Glycol - Choline Chloride Composition in a Deep Eutectic Solvent Mixture, J. Electrochem. Soc., 2023, 170(5), 052503 CrossRef CAS.
- L. Sanchez-Cupido, J. M. Pringle, A. I. Siriwardana, M. Hilder, M. Forsyth and C. Pozo-Gonzalo, Correlating Electrochemical Behavior and Speciation in Neodymium Ionic Liquid Electrolyte Mixtures in the Presence of Water, ACS Sustainable Chem. Eng., 2020, 8(37), 14047–14057 CrossRef CAS.
- R. Fukui, Y. Katayama and T. Miura, The effect of organic additives in electrodeposition of Co from an amide-type ionic liquid, Electrochim. Acta, 2011, 56(3), 1190–1196 CrossRef CAS.
- S. Schaltin, P. Nockemann, B. Thijs, K. Binnemans and J. Fransaer, Influence of the Anion on the Electrodeposition of Cobalt, from Imidazolium Ionic Liquids, Electrochem. Solid-State Lett., 2007, 10(10), D104–D107 CrossRef CAS.
- M. Forsyth, M. Hilder, Y. Zhang, F. Chen, L. Carre, D. A. Rakov, M. Armand, D. R. Macfarlane, C. Pozo-Gonzalo and P. C. Howlett, Tuning Sodium Interfacial Chemistry with Mixed-Anion Ionic Liquid Electrolytes, ACS Appl. Mater. Interfaces, 2019, 11(46), 43093–43106 CrossRef CAS PubMed.
- L.-G. Lin, J.-W. Yan, Y. Wang, Y.-C. Fu and B.-W. Mao, Anin situSTM study of cobalt electrodeposition on Au(111) in BMIBF4ionic liquid, J. Exp. Nanosci., 2006, 1(3), 269–278 CrossRef CAS.
- Y.-T. Hsieh, M.-C. Lai, H.-L. Huang and I. W. Sun, Speciation of cobalt-chloride-based ionic liquids and electrodeposition of Co wires, Electrochim. Acta, 2014, 117, 217–223 CrossRef CAS.
- A. P. Abbott, J. C. Barron, G. Frisch, K. S. Ryder and A. F. Silva, The effect of additives on zinc electrodeposition from deep eutectic solvents, Electrochim. Acta, 2011, 56(14), 5272–5279 CrossRef CAS.
- A. Sorgho, M. Bougouma, G. De Leener, J. Vander Steen and T. Doneux, Impact of speciation on the tellurium electrochemistry in choline chloride-based deep eutectic solvents, Electrochem. Commun., 2022, 140, 107327 CrossRef CAS.
- J. M. Hartley, S. Scott, Z. Dilruba, A. J. Lucio, P. J. Bird, R. C. Harris, G. R. T. Jenkin and A. P. Abbott, Iodine speciation in deep eutectic solvents, Phys. Chem. Chem. Phys., 2022, 24(39), 24105–24115 RSC.
- A. P. Abbott, A. Ballantyne, R. C. Harris, J. A. Juma, K. S. Ryder and G. Forrest, A Comparative Study of Nickel Electrodeposition Using Deep Eutectic Solvents and Aqueous Solutions, Electrochim. Acta, 2015, 176, 718–726 CrossRef CAS.
- R. Gupta, B. Vats, A. K. Pandey, M. K. Sharma, P. Sahu, A. K. Yadav, S. M. Ali and S. Kannan, Insight into Speciation and Electrochemistry of Uranyl Ions in Deep Eutectic Solvents, J. Phys. Chem. B, 2020, 124(1), 181–189 CrossRef CAS PubMed.
- G. Qadr, M. I. Awad, K. Haji, J. A. Jumaa and H. H. Abdallah, Nickel electrodeposition from deep eutectic solvents containing copper ions at a high temperature, J. Mol. Liq., 2023, 378, 121584 CrossRef CAS.
- S. Cihangir, K. S. Ryder and A. Unal, Detailed Investigation of Zinc Coating Behaviours in Various Deep Eutectic Solvents, Electrochim. Acta, 2023, 142708 CrossRef CAS.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Deep eutectic solvents (DESs) and their applications, Chem. Rev., 2014, 114(21), 11060 CrossRef CAS PubMed.
- J. T. M. Amphlett, M. D. Ogden, W. Yang and S. Choi, Probing Ni2+ and Co2+ speciation in carboxylic acid based deep eutectic solvents using UV/Vis and FT-IR spectroscopy, J. Mol. Liq., 2020, 318, 114217 CrossRef CAS.
- M. K. Tran, M.-T. F. Rodrigues, K. Kato, G. Babu and P. M. Ajayan, Deep eutectic solvents for cathode recycling of Li-ion batteries, Nat. Energy, 2019, 4(4), 339–345 CrossRef CAS.
- M. J. Roldán-Ruiz, M. L. Ferrer, M. C. Gutiérrez and F. Monte, d., Highly Efficient p-Toluenesulfonic Acid-Based Deep-Eutectic Solvents for Cathode Recycling of Li-Ion Batteries, ACS Sustainable Chem. Eng., 2020, 8(14), 5437–5445 CrossRef.
- S. Wang, Z. Zhang, Z. Lu and Z. Xu, A novel method for screening deep eutectic solvent to recycle the cathode of Li-ion batteries, Green Chem., 2020, 22(14), 4473–4482 RSC.
- J. T. M. Amphlett and S. Choi, The effect of increasing water content on transition metal speciation in deep eutectic solvents, J. Mol. Liq., 2021, 332, 115845 CrossRef CAS.
- V. Alizadeh, F. Malberg, A. A. H. Padua and B. Kirchner, Are There Magic Compositions in Deep Eutectic Solvents? Effects of Composition and Water Content in Choline Chloride/Ethylene Glycol from ab initio molecular dynamics, J. Phys. Chem. B, 2020, 124(34), 7433–7443 CrossRef CAS PubMed.
- Y. Zhang, D. Poe, L. Heroux, H. Squire, B. W. Doherty, Z. Long, M. Dadmun, B. Gurkan, M. E. Tuckerman and E. J. Maginn, Liquid Structure and Transport Properties of the Deep Eutectic Solvent Ethaline, J. Phys. Chem. B, 2020, 124(25), 5251–5264 CrossRef CAS PubMed.
- A. T. Celebi, T. J. H. Vlugt and O. A. Moultos, Structural, Thermodynamic, and Transport Properties of Aqueous Reline and Ethaline Solutions from Molecular Dynamics Simulations, J. Phys. Chem. B, 2019, 123(51), 11014–11025 CrossRef CAS PubMed.
- L. L. Bezerra, F. G. S. Oliveira, L. P. M. Dos Santos, H. B. de Sant’Ana, F. X. Feitosa, A. N. Correia, W. Schwarzacher, E. S. Marinho, P. de Lima-Neto and N. K. V. Monteiro, Electrochemical and theoretical investigation on the behavior of the Co(2+) ion in three eutectic solvents, J. Mol. Graphics Modell., 2022, 112, 108137 CrossRef CAS PubMed.
- H. Bastos, N. Schaeffer, J. M. Pringle, J. A. P. Coutinho and C. Pozo-Gonzalo, Enhanced Dissolution of Metal Oxides in Hydroxylated Solvents - Towards Application in Lithium-Ion Battery Leaching, ChemSusChem, 2023, e202300455 CrossRef CAS PubMed.
- A. M. Wilson, P. J. Bailey, P. A. Tasker, J. R. Turkington, R. A. Grant and J. B. Love, Solvent extraction: the coordination chemistry behind extractive metallurgy, Chem. Soc. Rev., 2014, 43(1), 123 RSC.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids, J. Am. Chem. Soc., 1996, 118(45), 11225–11236 CrossRef CAS.
- B. Doherty and O. Acevedo, OPLS Force Field for Choline Chloride-Based Deep Eutectic Solvents, J. Phys. Chem. B, 2018, 122(43), 9982–9993 CrossRef CAS PubMed.
- W. R. Cannon, B. M. Pettitt and J. A. McCammon, Sulfate Anion in Water: Model Structural, Thermodynamic, and Dynamic Properties, J. Phys. Chem., 1994, 98(24), 6225–6230 CrossRef CAS.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, MDAnalysis: a toolkit for the analysis of molecular dynamics simulations, J. Comput. Chem., 2011, 32(10), 2319 CrossRef CAS PubMed.
- R. J. Gowers, M. Linke, J. Barnoud, T. J. E. Reddy, M. N. Melo, S. L. Seyler, J. Domanski, D. L. Dotson, S. Buchoux, I. M. Kenney and O. Beckstein, MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Conference: PROC. OF THE 15th PYTHON IN SCIENCE CONF. (SCIPY 2016); 2016-07-11 – 2016-07-11, United States, 2019; p Medium: ED; Size: 98.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47(1), 558–561 CrossRef CAS PubMed.
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33(12), 8822–8824 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 CrossRef CAS PubMed.
- J. Hafner, Ab-initio simulations of materials using VASP: Density-functional theory and beyond, J. Comput. Chem., 2008, 29(13), 2044–2078 CrossRef CAS PubMed.
- W. G. Hoover and B. L. Holian, Kinetic moments method for the canonical ensemble distribution, Phys. Lett. A, 1996, 211(5), 253–257 CrossRef CAS.
- S. Nosé, A unified formulation of the constant temperature molecular dynamics methods, J. Chem. Phys., 1984, 81(1), 511–519 CrossRef.
- A. Y. M. Al-Murshedi, A. Al-Yasari, H. F. Alesary and H. K. Ismail, Electrochemical fabrication of cobalt films in a choline chloride–ethylene glycol deep eutectic solvent containing water, Chem. Pap., 2019, 74(2), 699–709 CrossRef.
- Z. Wang, Y. Chen, F. Zhou, R. Qin, Y. Tian, Z. Xue and T. Mu, Upcycling spent lithium-ion battery cathodes into cobalt-polyphenol networks by DES dissolution and solvent-induced crystallization, Green Chemistry, 2024, 26(10), 5988–5996 RSC.
- K. E. Prosser and C. J. Walsby, Electron Paramagnetic Resonance as a Tool for Studying the Mechanisms of Paramagnetic Anticancer Metallodrugs, Eur. J. Inorg. Chem., 2016,(12), 1573–1585 CrossRef.
- H. Drulis, K. Dyrek, K. P. Hoffmann, S. K. Hoffmann and A. Weselucha-Birczynska, EPR spectra of low-symmetry tetrahedral high-spin cobalt(II) in a cinchoninium tetrachlorocobaltate(II) dihydrate single crystal, Inorg. Chem., 1985, 24(24), 4009–4012 CrossRef CAS.
- S. K. Hoffmann, J. Goslar and S. Lijewski, Electron Paramagnetic Resonance and Electron Spin Echo Studies of Co(2+) Coordination by Nicotinamide Adenine Dinucleotide (NAD(+)) in Water Solution, Appl. Magn. Reson., 2013, 44(7), 817–826 CrossRef CAS PubMed.
- J. Degenhardt and A. E. McQuillan, In Situ ATR-FTIR Spectroscopic Study of Adsorption of Perchlorate, Sulfate, and Thiosulfate Ions onto Chromium(III) Oxide Hydroxide Thin Films, Langmuir, 1999, 15, 4595–4602 CrossRef CAS.
- F. P. Franguelli, B. Barta-Holló, V. M. Petruševski, I. E. Sajó, S. Klébert, A. Farkas, E. Bódis, I. M. Szilágyi, R. P. Pawar and L. Kótai, Thermal decomposition and spectral characterization of di[carbonatotetraamminecobalt(III)] sulfate trihydrate and the nature of its thermal decomposition products, J. Therm. Anal. Calorim., 2020, 145(6), 2907–2923 CrossRef.
- A. M. Fry, O. T. Sweeney, W. Adam Phelan, N. Drichko, M. A. Siegler and T. M. McQueen, Unique edge-sharing sulfate-transition metal coordination in Na2M(SO4)2 (M = Ni and Co), J. Solid State Chem., 2015, 222, 129–135 CrossRef CAS.
- P. Roonasi and A. Holmgren, An ATR-FTIR study of sulphate sorption on magnetite; rate of adsorption, surface speciation, and effect of calcium ions, J. Colloid Interface Sci., 2009, 333(1), 27–32 CrossRef CAS PubMed.
- C. M. Eggleston, S. Hug, W. Stumm, B. Sulzberger and M. Dos Santos Afonso, Surface Complexation of Sulfate by Hematite Surfaces: FTIR and STM Observations, Geochim. Cosmochim. Acta, 1998, 62(4), 585–593 CrossRef CAS.
- A. M. Banerjee, M. R. Pai, S. S. Meena, A. K. Tripathi and S. R. Bharadwaj, Catalytic activities of cobalt, nickel and copper ferrospinels for sulfuric acid decomposition: The high temperature step in the sulfur based thermochemical water splitting cycles, Int. J. Hydrogen Energy, 2011, 36(8), 4768–4780 CrossRef CAS.
- A. V. Radha, L. Lander, G. Rousse, J. M. Tarascon and A. Navrotsky, Thermodynamic stability and correlation with synthesis conditions, structure and phase transformations in orthorhombic and monoclinic Li2M(SO4)2 (M = Mn, Fe, Co, Ni) polymorphs, J. Mater. Chem. A, 2015, 3(6), 2601–2608 RSC.
- H. H. Adler and P. F. Kerr, Variations in infrared spectra, melecular symmetry and the site symmetry of sulfate minerals, Mineral, 1965, 50, 132–147 CAS.
- R. J. Abraham, I. Marsden and L. Xiujing, NMR spectra of the porphyrins. Part 39. Paramagnetic shifts in cobalt(II) porphyrins, Magn. Reson. Chem., 1990, 28(12), 1051–1057 CrossRef CAS.
- E. N. Zapolotsky, S. P. Babailov and G. A. Kostin, Paramagnetic Properties and Moderately RapidConformational Dynamics in the Cobalt(II) Calix[4]arene Complex by NMR, Molecules, 2022, 27(14), 4668 CrossRef CAS PubMed.
- V. K. Voronov, I. A. Ushakov and E. A. Funtikova, The heterospin cobalt complexes: peculiarities of high-resolution NMR spectra, Heliyon, 2022, 8(4), e09202 CrossRef CAS PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations, J. Am. Chem. Soc., 1992, 114(25), 10024–10035 CrossRef CAS.
- J. R. Bezerra-Neto, N. G. Sousa, L. P. M. Dos Santos, A. N. Correia and P. de Lima-Neto, The effect of water on the physicochemical properties of an ethylene glycol and choline chloride mixture containing Cu(2+) ions: electrochemical results and dynamic molecular simulation approach, Phys. Chem. Chem. Phys., 2018, 20(14), 9321–9327 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp01471e |
| This journal is © the Owner Societies 2024 |