
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical bonding within AIIIBVI materials under uniaxial compression†
Roman S.
Stepanov
 a,
Aleksandra D.
Radina
b,
Christian
Tantardini
*cde,
Alexander G.
Kvashnin
b and
Alexander V.
Kolobov
*af
a,
Aleksandra D.
Radina
b,
Christian
Tantardini
*cde,
Alexander G.
Kvashnin
b and
Alexander V.
Kolobov
*af
aResearch Institute of Physics, Institute of Physics, Herzen State Pedagogical University of Russia, 48 Moika emb., St Petersburg 191186, Russia. E-mail: akolobov@herzen.spb.ru
bProject Center for Energy Transition and ESG, Skolkovo Institute of Science and Technology, 30 Bolshoi Blv., bld. 1, Moscow 121205, Russia
cHylleraas center, Department of Chemistry, UiT The Arctic University of Norway, PO Box 6050 Langnes, N-9037 Tromsø, Norway. E-mail: christiantantardini@ymail.com
dDepartment of Materials Science and NanoEngineering, Rice University, Houston, Texas 77005, USA
eInstitute of Solid State Chemistry and Mechanochemistry SB RAS, 18 Kutateladze, Novosibirsk 630128, Russia
fDepartment of Electronics, Institute of Physics, Herzen University, 48 Moika emb., St. Petersburg 191186, Russia
First published on 24th July 2024
Abstract
The work provides a comprehensive explanation of the nature of chemical bonding through quantum chemical topology for multilayers of AIIIBVI compounds, such as GaSe, InSe, and GaTe, spanning pressures from 0 GPa to 30 GPa. These compounds are subjected to pressure orthogonal to the multilayers. Quantum chemical topological indices indicate that uniaxial pressure induces changes in hybridisation, leading to the disappearance of interlayer van der Waals forces. The distinct nature of the elements within the compounds results in different pressures at which van der Waals interactions disappear, as revealed by non-covalent interaction analysis. The presence or absence of chemical bonding is assessed by quantum topological indices as Espinosa indices, charge density distribution difference, and crystal orbital Hamilton populations. The varying changes in hybridisation, as indicated by topological indices, are corroborated by variations in the population of the electronic projected density of states. Ultimately, the type of chemical bonding is identified through the Espinosa indices in the field of Bader theory. This analysis confirms the existence of shared shell bonds between AIII and BVI atoms in vacuum that goes to an intermediate bond between shared and closed shells called the transition zone with increasing pressure. The implications and importance of this work extend beyond the presented results. It suggests that many other classes of two-dimensional materials may undergo phase transitions under uniaxial stress, leading to the formation of new phases with potentially interesting electronic properties.
1 Introduction
Two-dimensional (2D) layered (or quasi-2D) materials have attracted considerable attention in recent years due to their potential applications in next-generation electronics. The van der Waals (vdW) interaction is a crucial component that determines the properties and nature of the layered structure of such materials,1 allowing the creation of ultrathin films and heterostructures with atomically sharp interfaces, almost independently of the lattice constants.2 In addition, layered materials typically exhibit a strong monotonic dependence of the electronic structure on the vdW gap width,3 opening up the possibility of manipulating the properties and greatly expanding the design and research possibilities of transparent and flexible electronic devices.4–6 Examples include a thermally sensitive photodetector based on a mixed size vdW heterostructure GaSe/VO27 and an ultra-flexible photodetector based on a 2D-MoS2/Si heterojunction, successfully fabricated a few years ago,8 to name a few.In this context, we focus on GaSe, InSe, and GaTe, which are promising materials for various nanoelectronic applications.9 For example, interest in GaSe is mainly due to the strong anisotropy of its optical properties and its nonlinear optical response.10 GaSe has been investigated for potential applications in photoelectrochemistry and nanoscale photodetectors.11,12 InSe has been extensively used in many applications, particularly in optics.13,14 GaTe has been studied for its potential applications in optoelectronics, photochemistry, and thermoelectricity due to its low thermal conductivity.15,16
Significant structural changes, such as the collapse or reconfiguration of a vdW gap, can cause properties to switch, leading to interesting effects. It has been shown that reconfiguration of the vdW gap leads to significant changes in the electronic structure. This provides an exciting opportunity to manipulate material properties.17,18 For example, in a previous study,19 gallium selenide was uniaxially compressed to a pressure of 11 GPa, resulting in a decrease in the band gap with the formation of a gapless state. After the phase transition at 12 GPa, the band gap increased, and the fundamental band gap reappeared.
Several observations of the high-pressure cubic phases of these compounds have been made based on X-ray Bragg diffraction (XRD).20–22 Ghalouci et al.22 studied the structure of InSe under hydrostatic pressure and showed that β-InSe transforms to the rocksalt phase in the pressure range of 6 to 16 GPa and remains stable up to 30 GPa. Different transition pressures of β-GaSe to the cubic phase have been reported, ranging from 17 to 29.2 GPa.21,23 For GaTe, the transition to the rocksalt phase is expected at a pressure of about 7 GPa.24 However, it is important to note that low-angle XRD, which is commonly used to study two-dimensional materials, can be insensitive to local distortions and can sometimes lead to erroneous conclusions.25 Furthermore, XRD and X-ray absorption fine structure (XAFS) cannot provide information about the nature of chemical bonding. It is therefore necessary to obtain information about the chemical bonding by other methods.
External pressure can induce atomic reconfiguration and cause the collapse of the interlayer vdW interaction. Since the studied materials have strong structural anisotropy, one possible way to directly influence the vdW gap is to study the effect of uniaxial pressure. In addition, films of AIIIBVI structure with appropriate orientation can be grown on elastic substrates.26–28 In this case, the mechanical deformation produced in the film-substrate plane can generally be equivalent to uniaxial deformation. In a previous study,19 it was shown that under uniaxial pressure in GaSe the interlayer distance decreases and the bonding angles between the metal and chalcogen atoms also decrease. However, there were no significant changes until the pressure reached 11 GPa. Further increases in pressure led to the formation of bonds between chalcogen atoms across the vdW gap, and at 12 GPa a phase transition occurred. Based on the difference in charge density distribution (CDD) between the electron density of a system and that of its isolated atoms, which indicates the formation of chemical bonds, and a comparison of bond coordination according to the octet rule,29 the resulting phase has been interpreted as quasi-one-dimensional.30 However, this is not the only possible approach to take into account bonding in the studied structures. For example, the presence of hypervalent atoms that break the octet rule can be assumed, especially since in the cubic modification the structure can resemble a typical hypervalently bonded compound such as PF5.31,32 In this case, we actually get a rocksalt-like structure. On the other hand, the chemical bonding in the new phase may have a strong ionic component, and this result is not unprecedented; materials with inherently polar covalent bonding with a strong antibonding component have already been shown to have ionic bonding during phase transition in MgO and GaN.33 For the compounds under study, the transition to a new phase occurs at pressures of 6 GPa for InSe, 14 GPa for GaSe, and 10 GPa for GaTe. Independent methods such as Crystal Orbital Hamilton Population (COHP) and Bader analysis are used to provide a detailed and comprehensive explanation of the bonding nature in AIIIBVI materials under pressure.
2 Computational details
Ab initio calculations without pressure and under pressure were performed using a plane-wave basis set with the Perdew–Burke–Ernzerhoff (PBE) DFT functional34 and the ultrasoft pseudopotential35 implemented in the CASTEP simulation package.36 Two-point steepest descent (TPSD) algorithm was used for optimisation. This algorithm has previously37 shown the best trade-off between computational cost and agreement with experimental data. The first Brillouin zone was sampled with 15 × 15 × 3 k-points. The values of the kinetic cut-off energies were chosen to achieve convergence of the interatomic forces equal to 1 meV Å−1 at the above k-points grid: 600 eV for GaSe, 660 eV for GaTe and 700 eV for InSe. The vdW interactions were taken into account within ab initio calculations by the many-body dispersion (MBD) correction.38 Simulations under pressure were carried out by applying uniaxial pressure along the c-axis of conventional cells in the range 0 to 30 GPa with increments of 1 GPa.The study of the chemical bonding of atoms in the studied layered materials was carried out by crystal orbital Hamilton population (COHP)39 and Bader analysis40–42 on electronic density of states (DOS) and full-electron density from projected augmented waves (PAW)43 implemented in VASP.44–46 The DOS and electron density were calculated with VASP44–46 using MBD correction, and the kinetic energy cutoff was set to 500 eV, which was sufficient to achieve the same force accuracy obtained with CASTEP36 in the order of 1 meV Å−1. The first Brillouin zone was sampled using the Monkhorst–Pack k-point grid47 with a resolution of 2π × 0.05 Å−1.
The Bader analysis, which included the search for bond critical points, was carried out using the CRITIC2 code.48,49 Additionally, the analysis of the non-covalent interactions (NCI)50 through the calculations of the reduced density gradient of the electron density between the molecular fragments was conducted with the same code to estimate the strength of their non-covalent interactions.
Additionally, the charge density difference (CDD) was calculated on the electron density obtained from the VASP calculations.44–46
3 Results and discussion
The AIIIBVI compounds are layered crystals with non-covalent interactions between the layers (e.g., vdW).20 Many of these semiconductors have different polytypes, characterised by different stacking of the layers.51,52 In this study, we have carried out calculations for the β-phases of GaSe, InSe, and GaTe, all of which belong to the same space group P63/mmc.53 The choice of the β phase is based on the fact that calculations for it give qualitatively the same results as for the ε phase, with significantly reduced computational complexity due to the higher symmetry of the former with respect to the latter, as previously demonstrated by Ghalouci et al.21 and Rak et al.54 The higher symmetry of the β phase allows a reduction in computational cost due to the greater number of symmetry operations that can be used to introduce periodic boundary conditions compared to ε-phase. For clarity, we have added both β and ε phases in Fig. S1 (ESI†). The β-phase at 0 GPa is characterised by stacked layers along the c-axis, with 13th group metal atoms tetrahedrally coordinated with chalcogen atoms within each layer. Please refer to Fig. 2(a), (e), 3(a), (e), and 4(a), (e) for visual representations. The tetrahedral configuration of the metal atoms can be directly attributed to sp3 hybridisation. Conversely, the chalcogen atoms, characterised by two lone pairs (consisting of s- and p-electrons) and two singlets forming bonds to complete the octet, exhibit a distorted tetrahedral configuration, also due to sp3, similar to the well-known case of ammonia.55–57 While the interaction between the layers at 0 GPa is non-covalent and decreases with increasing uniaxial pressure along the c-axis, as shown by non-covalent interaction (NCI) analysis50 (see Fig. 1). The increase in pressure causes the interlayer spacings to approach the intralayer interatomic spacings, resulting in atomic hybridization and consequently a change in atomic coordination (see Fig. 2–4). If the interlayer non-covalent interactions could be easily studied, the chemical bonding within the intralayer atoms would require more in-depth analysis. | ||
| Fig. 1 Non-covalent interaction (NCI) analysis showing the isosurface of reduced density gradient equal to 0.3 of GaSe (a) and (b), GaTe (c) and (d), InSe (e) and (f). Legenda: Ga, blue; Se, red; Te, pink; In, green. | ||
 | ||
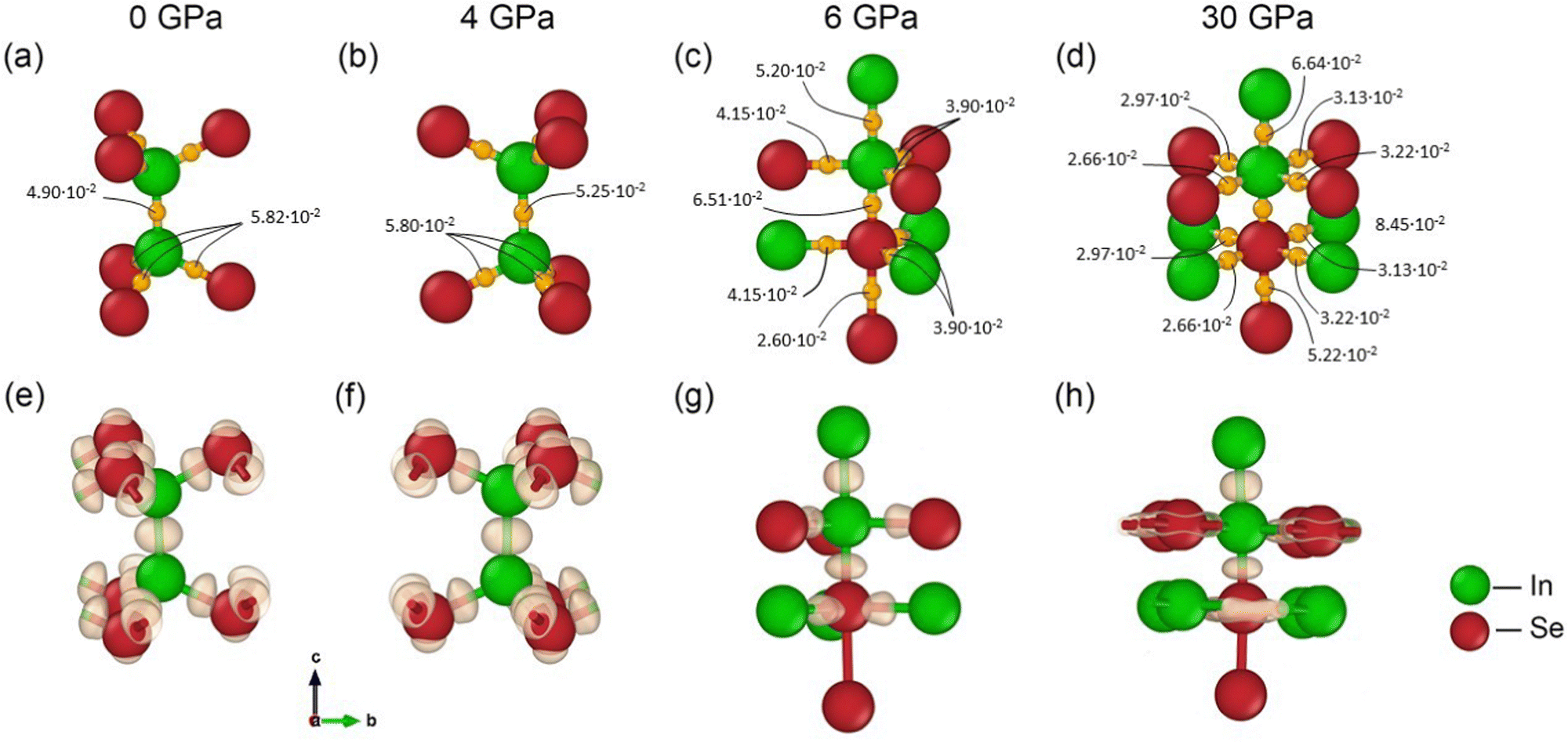
| Fig. 2 Bader analysis showing the bond critical points with the value of electron density in e·bohr−3 at each of them and the charge density difference (CDD) distribution for GaTe. | ||
 | ||
| Fig. 3 Bader analysis showing the bond critical points with the value of electron density in e·bohr−3 at each of them and charge density difference (CDD) distribution for InSe. | ||
 | ||
| Fig. 4 Bader analysis showing the bond critical points with the value of electron density in e·bohr−3 at each of them and charge density difference (CDD) distribution for GaSe. | ||
Initially, we have considered the Tantardini–Oganov thermochemical electronegativity scale (χTO), which overcomes the drawbacks of the Pauling scale.58 The Tantardini–Oganov scale, compared to the Pauling scale, predicts the degree of ionicity of chemical bonds, improves the separation of elements into metals and non-metals, and greatly improves the description of the thermochemistry of molecules and chemical reactions.58 We have therefore used this scale to analyse the nature of the chemical bonding within the intralayers of GaSe, GaTe, and InSe at 0 GPa.
In GaSe, the atoms have χTO values of 2.43 for Ga and 3.37 for Se, giving a difference between Ga and Se (ΔχTO) of 0.94. This difference characterises their chemical bond as polar covalent. For GaTe and InSe, the ΔχTO values are 0.71 and 1.08, respectively. While the electronegativity indicates that the chemical bonding between these atoms is covalent, a clear understanding also requires the application of various tools from the field of quantum chemical topology.59 We performed a quantum theory of atoms in molecules (QTAIM) analysis to identify bond critical points (BCPs) between intralayer atoms, and evaluated the Espinosa indices60 at BCPs to define the nature of the chemical bonding. Espinosa indices focus on the properties of the electron density at BCPs. In QTAIM, bond critical points are locations where the electron density reaches a saddle point between two atoms, indicating a bond path. The electron density at these points, denoted ρ(r), and its Laplacian, ∇2ρ(r), are key descriptors used to define the Espinosa indices.60 The value of ρ(r) at a BCP provides a measure of the electron density concentration, while the Laplacian of ρ(r) indicates whether the electron density is locally depleted or accumulated. Positive values of ∇2ρ(r) indicate depletion, typically associated with closed-shell interactions (i.e., ionic bonds), while negative values indicate accumulation, typically associated with shared shells (i.e., covalent bonds). Espinosa indices also include the potential energy density (V(r)) and the kinetic energy density, G(r), at the BCP. These quantities help to elucidate the energetic aspects of the interaction. The ratio |V(r)|/G(r) serves as an indicator of the type of bond: values around 1 indicate a purely covalent bond, values significantly less than 1 indicate a non-covalent interaction, and intermediate values indicate a partially covalent character, such as in hydrogen bonds. Espinosa's approach also extends to the study of the total energy density, H(r), which is defined as the sum of V(r) and G(r), obtained with the virial theorem, providing a comprehensive view of the stability and nature of the bond interaction. It is also worth noting that the BCP is always present in the case of covalent or ionic bonds, as in the case of metal bonds.61 However, it is not always present in the case of non-covalent interactions, as first proposed by Lane et al.62 and subsequently also by Tantardini.63 In particular, in the case of van der Waals (vdW) interactions, the concept of BCP is replaced by the vdW volume, which describes the volume of the vdW interaction.64 The Espinosa indices are given in Tables S1–S3 of the ESI,† and they are traceable by their electron density values at the BCPs shown in Fig. 2(a)–(d), 3(a)–(d), and 4(a)–(d). Further analysis of the nature of the chemical bonding was carried out by examining the charge density difference (CDD) distribution, which is the charge density minus the superposition of atomic densities. See Fig. 2(e)–(h), 3(e)–(h) and 4(e)–(h).
Let us first consider the effect of uniaxial pressure on GaTe, the material with the lowest ΔχTO. Increasing the pressure from 0 to 10 GPa leads to the disappearance of the non-covalent interaction between the layers, and the appearance of bonds between the metal and chalcogen atoms across the vdW gap (along the c-axis) see Fig. 2. According to the Espinosa indices (see Table S1 in ESI†), the BCP between Ga–Ga atoms at 0 and 8 GPa shows characteristics of shared electron shells, indicating a covalent bond. This is attributed to the negative values of the Hamiltonian (Hb) and the Laplacian of the electron density (∇2ρb) at the BCP, as well as to a ratio greater than 2.0 between the absolute value of |Vb| and the Gb at this point, see Table S1 (ESI†). At such pressures, the BCP between Ga–Te atoms exhibits a ratio between |Vb| and Gb in the range of 1.0 to 2.0, accompanied by positive ∇2ρb and negative Hb values, typical for a so-called transition zone between shared (i.e., covalent bond) and closed (i.e., ionic bond) shells.60 This zone is typical for H-bonds60 and metal–metal bonds.61 Increasing the pressure to 10 GPa keeps the characteristics of the Ga–Ga BCP unchanged, while the shift of the hybridisation from sp3 to sp3d in both Ga and Te, supported by the pDOS analysis indicating an increase of the d state population (see Fig. S1 in ESI†), shows Espinosa indices typical of a shared shell for the apical Ga–Te BCP, while all other BCPs show characteristics of a transition zone (see Table S1 in ESI†). At 30 GPa (see Fig. 2 and Fig. S1 in ESI†) the new hybridisation is retained but distorted, while the population of d states remains virtually unchanged. Here, we observe from the Espinosa indices (see Table S1 in ESI†) that the Ga–Te BCPs on the xy-plane (see Fig. 2(d) and (h)) are characteristic of a transition zone. However, two of them have a ratio between |Vb| and Gb lower than 1.00, which is usually expected for closed shell interactions. Nevertheless, the other two parameters, ∇2ρb and Hb, are negative, leaving no doubt about the nature of such a bond: a transition zone. If the Espinosa indices could provide such details, the CDD distribution shown in Fig. 2(e)–(h) would describe only two types of bonds: covalent and ionic. The covalent bonds are characterised by a concentration of CDD in the middle of the bond between two atoms.30 As observed in Fig. 2(e)–(h), GaTe compounds exhibit covalent bonds at all pressures with a lack of description between the axially bonded Te–Te with at pressure as seen in Fig. 2(g) and (h). The lack of CDD between Te–Te mentioned above can be justified by the unchanging distribution or charge density from the isolated to the bonded that by COHP39 (see Fig. S4c, ESI†) can be explained by the antibonding character between them.
The next compound is InSe, where the pressure increase at 6 GPa shows a transition from sp3 to sp3d with the disappearance of the interlayer non-covalent interactions (see Fig. 1 and 3). At high pressure (i.e. 30 GPa), In and Se change their hybridization from sp3d to sp3d2 (see Fig. 3), which is confirmed by pDOS with increasing d-state occupancy (see Fig. S2, ESI†). If the change in hybridization is clearly defined by the geometry, the nature of the chemical bonding, as indicated by the Espinosa indices, shows that from 0 to 4 GPa, there is only a transition zone at the In–In and In–Se BCPs, see Table S2 in the ESI.† The transition from sp3 to sp3d at 6 GPa indicates a closed-shell interaction (i.e., ionic bonding) between the apical Se–Se atoms, which is not supported by the CDD distribution in Fig. 3(g). While, the COHP (see Fig. S5, ESI†) show a bonding character of Se–Se at 6 GPa. Conversely, the transition in hybridization from sp3d to sp3d2 is accompanied by In–Se ionic bonds in the xy-plane, as indicated by the Espinosa indices (see Table S2 in ESI†), but not by the CDD distribution in Fig. 3(h), which again shows no CDD isosurface between the apical Se–Se. In InSe at high pressure we can observe from the COHP the same antibonding character between Se–Se as previously seen for Te–Te in GaTe (see Fig. S5, ESI†).
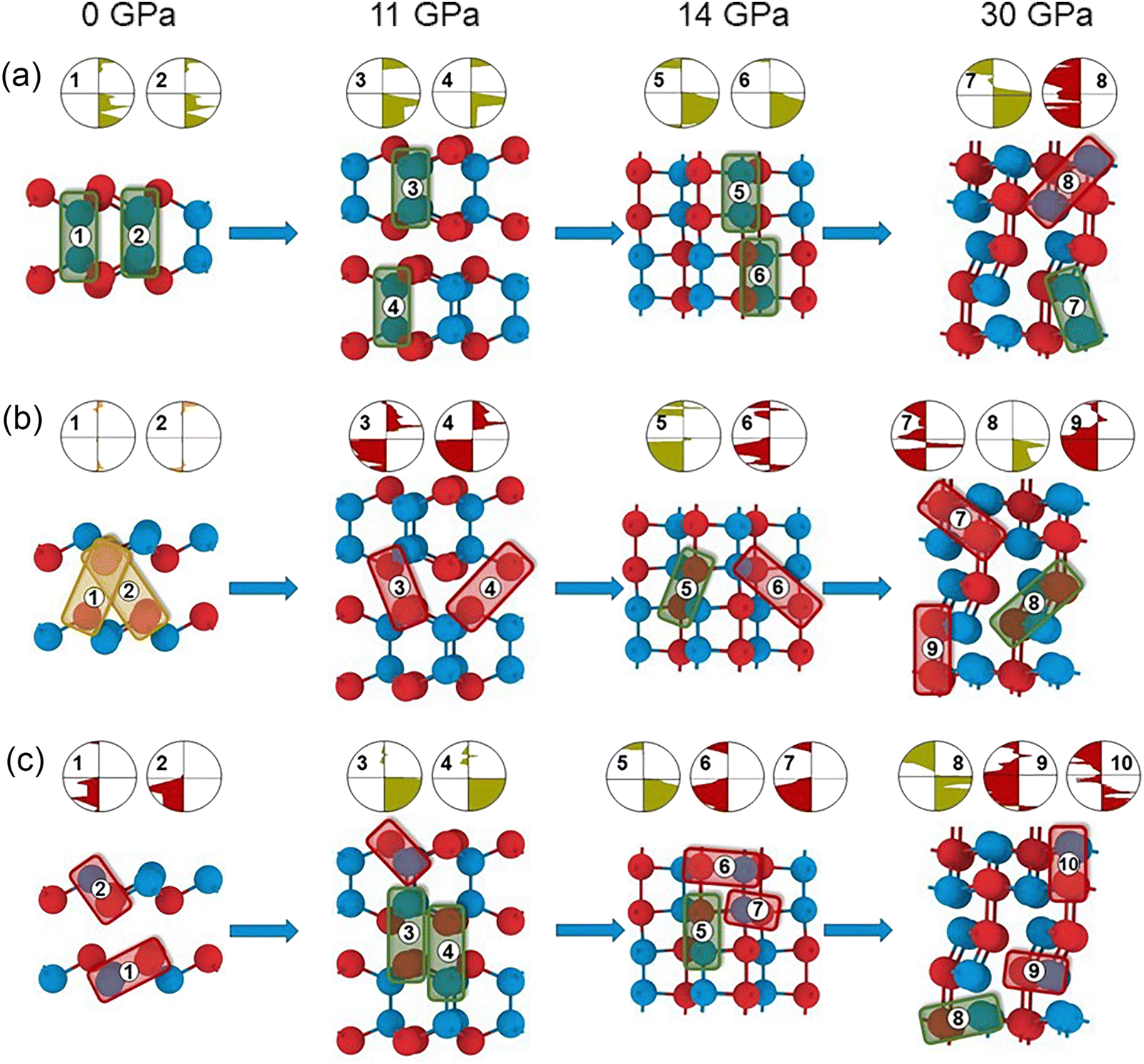
We have also studied GaSe, which at 12 GPa changes hybridization from sp3 to sp3d (see Fig. 4) showing that Se, like Ga, becomes fivefold coordinated. As noted above, this determines the end of non-covalent interactions and the disappearance of the layered structure, see Fig. 1. Apparently, changes in the bonding geometry are caused by changes in the hybridization, which can be analysed with pDOS (see Fig. 5). There is an increase in the number of d-states, suggesting that the hybridisation has changed from sp3 to sp3d, corresponding to a trigonal bipyramidal coordination (see Fig. 5). Increasing the pressure to 30 GPa shows an unexpected change in the hybridisation from sp3d to sp3 for both Ga and Se, the latter characterised by a strongly distorted tetrahedral geometry as shown by the BCPs in Fig. 4. This requires further investigation to confirm, even though the pDOS shows a decrease in the population of d-orbitals in the valence bands, see Fig. 5. In fact, as we know, the change in coordination predicted by the BCP analysis combined with the pDOS analysis leads to a violation of the octet rule due to the hypervalent atoms. The analysis of the pDOS, obtained here with a plane-wave basis set, which guarantees the completeness of this method, can give an indication of the change in the population of different orbitals. In particular, we can study the hybridisation due to the change of the d-orbital population without artefacts, such as an increase in the orbital population due to the large size of the Gaussian atomic orbital basis set, which is often a problem in all-electron calculations.65,66 The analysis of Espinosa indices shows that the Ga–Ga bond at 0 and 11 GPa is characterised by shared shell interaction, whereas the Ga–Se bond is characterised by transition zone interaction (see Table S3 in ESI†). The change in hybridisation from sp3 to sp3d at 14 GPa is accompanied by transition zone interactions between all atoms within the structure, except for the Ga-Ga bond, which remains characterised by shared shell interaction according to the Espinosa indices (see Table S3 in ESI†), but the CDD as seen in Fig. 4(g) does not show any isosurface between the apical Se–Se. Here, the COHP (see Fig. 6) shows a bonding character between the apical Se–Se, which was not previously seen in InSe at 6 GPa. The previously obtained sp3d hybridisation of both Ga and Se is maintained at 30 GPa and it is accompanied by a change in the bonding character between apical Se–Se from transition zone to a closed shell interaction (see Table S3 in ESI†), which is not confirmed by the CDD distribution in Fig. 4(h), due to the absence of an isosurface. Here the COHP (see Fig. 6) shows an antibonding character as seen before for the apical Se–Se in InSe at 30 GPa. Therefore, in order to deepen into our previous conclusions about the chemical bonding of GaSe, we have further investigated the COHP.39 Using the COHP calculations, we first focus on the covalent bonding between metal atoms. As expected from both CDD and BCP analyses, COHP clearly shows the remarkably robust nature of the Ga–Ga bond under pressure conditions (as shown in Fig. 6(a)). For the phase obtained at 11 GPa, the anti-bonding nature of the interaction is observed for all bonds in the XY plane (Fig. 6(c)). Taken together with the CDD data, this again indicates the possible non-covalent nature of the interaction between metal and chalcogen atoms in a plane perpendicular to the applied pressure. The result of the analysis of the BCP together with pCOHP confirms this reasoning and allows us to make an assumption about hypervalence.
 | ||
| Fig. 5 Density of electronic states projected to each atomic orbital of Ga and Se atoms GaSe under pressure: (a) and (b) 0 GPa; (c) and (d) 11 GPa; (e) and (f) 12 GPa; (g) and (h) 30 GPa. | ||
 | ||
| Fig. 6 The changes in (a) Ga–Ga; (b) Se–Se; (c) Ga–Se bonds under pressure (Se – red circle, Ga – blue circle). The bonding, antibonding, and neutral states between the atoms are marked by the green, red and yellow color of the background, respectively. The small circles above the structures represent the pCOHP near the Fermi level. The numbers correspond to the bonds marked on the structures. | ||
From the increasing occupancy values obtained from the pCOHP analysis, it is clear that a significant fraction of the electron density is concentrated around the Se atoms (Fig. 6(c)). Within the first structural layer, the metal–chalcogen bonds show a significant degree of brittleness, with a substantial fraction of partially filled antibonding orbitals remaining (Fig. 6(c)). This observation is consistent with the well-documented antibonding nature of chalcogen atoms.67
To quantitatively analyze the changes in bond strength, the integrated –COHP (ICOHP) was calculated as a function of pressure for different types of chemical bonds in GaSe, as shown in Fig. S6 (ESI†). In this case, such an analysis could be applied to show a tendency for bonds to strengthen or weaken under pressure. Thus, a decrease in the ICOHP value indicates an increase in bond strength, whereas an increase in the value indicates a decrease in bond strength. For example, the Ga–Ga bond becomes stronger (Fig. S6, ESI†) with increasing pressure until the phase transition. After the phase transition, the ICOHP value increases to that of the initial structure. A further increase in pressure leads to a strengthening of the bond.
In the case of Se–Se interactions there are no dramatic changes. The magnitude of the interlayer interaction does not change with increasing pressure until the phase transition occurs. In the new phase we observe two types of Se–Se bonding: along the Z-axis and orthogonal to the Z-axis (Fig. S6b, ESI†). It can be seen that the strength of the perpendicular bonds is significantly increased, indicating a decrease in the antibonding interaction, while the bond strength along the Z-axis changes slightly.
As shown in Fig. S6c (ESI†), the ICOHP for Ga–Se bonds along the Z axis illustrates a decrease in the bonding component with increasing pressure up to the phase transition, and the appearance of the non-covalent interaction between the layers at the 11 GPa pressure. Then, this interaction is relatively unchanged with increasing pressure up to the 30 GPa phase (for GaSe). It is noteworthy that in the new phases, all Ga–Se interactions along the XY axis show an antibonding component and an extremely weak one, as confirmed by the ICOHP analysis. This observation suggests a redistribution of electron density between vertical and horizontal Ga–Se bonding, which fundamentally affects the interaction and structure in the new phase.
In summary, COHP and ICOHP analyses coupled with CDD suggest that the altered distribution of chalcogen electron density plays a key role in the phase transition mechanism and the emergence of a new phase. The driving force behind this ongoing phase transition is the redistribution of electrons between the layers along the Z axis.
4 Conclusions
The nature of the chemical bonding in the β phases of AIIIBVI compounds has been investigated during the phase transformations induced by uniaxial compression along the c-axis. It has been shown that the vdW interaction between the layers disappears as the pressure brings the layers closer together forcing the change in the initial sp3 hybridization. Such a change in hybridisation was confirmed by pDOS with the involvement of d-orbitals forming covalent bonds as suggested by CDD, BCPs and COHP descriptors. The results obtained may have a significant impact on the understanding of the properties of this class of materials under pressure, opening up prospects for further research.Author contributions
Roman Stepanov – conceptualisation, data curation, formal analysis, investigation, methodology, project administration, validation, visualisation, writing – original draft, writing – review & editing. Aleksandra Radina – investigation, formal analysis, visualisation, writing – original draft. Christian Tantardini – conceptualisation, formal analysis, investigation, validation, visualisation, resources, writing – original draft, supervision. Alexander Kvashnin – data curation, formal analysis, validation, supervision, resources, writing – review & editing. Alexander Kolobov – conceptualisation, funding acquisition, project administration, resources, supervision, validation, writing – review & editing.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.† Further data and information can be asked to the corresponding authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. S. S. and A. V. K. are grateful to the Russian Science Foundation for support of this work under grant # 22-19-00766. Ch. T acknowledges support from the Research Council of Norway through its Centres of Excellence scheme (262695), through the FRIPRO grant ReMRChem (324590).Notes and references
- D. L. Duong, S. J. Yun and Y. H. Lee, ACS Nano, 2017, 11, 11803–11830 CrossRef CAS PubMed.
- T. Journot, H. Okuno, N. Mollard, A. Michon, R. Dagher, P. Gergaud, J. Dijon, A. V. Kolobov and B. Hyot, Nanotechnology, 2019, 30, 505603 CrossRef CAS PubMed.
- Y. He, Nano Lett., 2016, 16, 1530–6984 CrossRef PubMed.
- H. Qi, L. Wang, J. Sun, Y. Long, P. Hu, F. Liu and X. He, Crystals, 2018, 8, 35 CrossRef.
- J. Y. Lee, J.-H. Shin, G.-H. Lee and C.-H. Lee, Nanomaterials, 2016, 6, 193 CrossRef PubMed.
- S. Huang, Nat. Commun., 2019, 10, 2447 CrossRef PubMed.
- J. Zhou, M. Xie, H. Ji, A. Cui, Y. Ye, K. Jiang, L. Shang, J. Zhang, Z. Hu and J. Chu, ACS Appl. Mater. Interfaces, 2020, 12, 18674–18682 CrossRef CAS PubMed.
- J.-M. Choi, H. Y. Jang, A. R. Kim, J.-D. Kwon, B. Cho, M. H. Park and Y. Kim, Nanoscale, 2021, 13, 672–680 RSC.
- Z. Yang and J. Hao, Adv. Mater. Technol., 2019, 4, 1900108 CrossRef CAS.
- L. Karvonen, A. Säynätjoki, S. Mehravar, R. D. Rodriguez, S. Hartmann, D. R. T. Zahn, S. Honkanen, R. A. Norwood, N. Peyghambarian, K. Kieu, H. Lipsanen and J. Riikonen, Sci. Rep., 2015, 5, 10334 CrossRef CAS PubMed.
- C. Manfredotti, R. Murri, A. Quirini and L. Vasanelli, Nucl. Instrum. Methods, 1975, 131, 457–462 CrossRef CAS.
- S. Shigetomi and T. Ikari, J. Appl. Phys., 2000, 88, 1520–1524 CrossRef CAS.
- Z. Kudrynskyi, V. Khomyak, V. Katerynchuk, M. Kovalyuk, V. Netyaga and B. Kushnir, Thin Solid Films, 2015, 582, 253–257 CrossRef CAS.
- A. G. Kyazym-zade, A. A. Agaeva, V. M. Salmanov and A. G. Mokhtari, Tech. Phys., 2007, 52, 1611–1613 CrossRef CAS.
- E. Borisenko, D. Borisenko, A. Timonina and N. Kolesnikov, J. Cryst. Grow., 2020, 535, 125548 CrossRef CAS.
- U. Schwarz, K. Syassen and R. Kniep, J. Alloys Compd., 1995, 224, 212–216 CrossRef CAS.
- A. V. Kolobov, P. Fons, Y. Saito and J. Tominaga, ACS Omega, 2017, 2, 6223–6232 CrossRef CAS PubMed.
- N.-K. Chen, X.-B. Li, X.-P. Wang, S.-Y. Xie, W. Q. Tian, S. Zhang and H.-B. Sun, IEEE Trans. Nanotechnol., 2017, 17, 140–146 Search PubMed.
- R. S. Stepanov, V. A. Gerega, A. V. Suslov and A. A. V. Kolobov, Phys. Status Solidi RRL, 2013, 17, 2200430 CrossRef.
- A. Gouskov, J. Camassel and L. Gouskov, Prog. Cryst. Growth Charact., 1982, 5, 323–413 CrossRef CAS.
- L. Ghalouci, B. Benbahi, S. Hiadsi, B. Abidri, G. Vergoten and F. Ghalouci, Comput. Mater. Sci., 2013, 68, 73–82 CrossRef.
- L. Ghalouci, F. Taibi, F. Ghalouci and M. Bensaid, Comput. Mater. Sci., 2016, 124, 62–77 CrossRef CAS.
- V. K. Dien, N. T. Tien, N. D. Khanh, N. T. N. Han and M.-F. Lin, Phys. Rev. B, 2023, 108(20), 205150 CrossRef CAS.
- Y. Wang, Chin. Phys. B, 2019, 28, 056104 CrossRef.
- C. Kittel, Introduction to solid state physics, John Wiley & Sons, Inc., Hoboken, 8th edn, 2018 Search PubMed.
- R. Browning, N. Kuperman, B. Moon and R. Solanki, Electronics, 2017, 6, 27 CrossRef.
- S.-W. Hsiao, C.-S. Yang, H.-N. Yang, C.-H. Wu, S.-K. Wu, L.-Y. Chang, Y.-T. Ho, S.-J. Chang and W.-C. Chou, Front. Mater., 2022, 9, 871003 CrossRef.
- L. T. Vinh, M. Eddrief, J. E. Mahan, A. Vantomme, J. H. Song and M. A. Nicolet, J. Appl. Phys., 1997, 81, 7289–7294 CrossRef CAS.
- L. Pauling, J. Am. Chem. Soc., 1931, 53, 3225–3237 CrossRef CAS.
- D. Andrae, Phys. Rep., 2000, 336, 413–525 CrossRef CAS.
- R. H. Crabtree, Chem. Soc. Rev., 2017, 46, 1720–1729 RSC.
- C. Tantardini and E. Benassi, Phys. Chem. Chem. Phys., 2017, 19, 27779–27785 RSC.
- S. Limpijumnong and W. R. Lambrecht, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 104103 CrossRef.
- M. Ernzerhof and G. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892(R) CrossRef PubMed.
- S. J. Clark, M. D. Segall, C. P. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- J. Barzilai and J. M. Borwein, IMA J. Numer. Anal., 2012, 8, 141–148 CrossRef.
- A. Tkatchenko, R. A. DiStasio, J. R. Car and M. Scheffler, Phys. Rev. Lett., 2012, 108, 236402 CrossRef PubMed.
- R. Dronskowski and P. E. Bloechl, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, Oxford, 1990 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 16, 11169–11186 CrossRef PubMed.
- G. A. de Wijs, P. K. de Boer, R. A. de Groot and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 2684–2693 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- A. Otero-de-la-Roza, M. A. Blanco, A. M. Pendás and V. Luana, Comput. Phys. Commun., 2009, 180, 157–166 CrossRef CAS.
- A. Otero-de-la-Roza, E. R. Johnson and V. Luana, Comput. Phys. Commun., 2014, 185, 1007–10188 CrossRef CAS.
- E. R. Johnson, J. Am. Chem. Soc., 2010, 132, 0002–7863 CrossRef PubMed.
- J. Robertson, J. Phys. C-Solid State Phys., 1979, 12, 0022–3719 Search PubMed.
- R. S. Stepanov, Crystals, 2023, 13, 2073–4352 CrossRef.
- H. Arora and A. Erbe, InfoMat, 2021, 3, 662–693 CrossRef CAS.
- Z. Rak, S. D. Mahanti, K. C. Mandal and N. C. Fernelius, J. Phys. Chem. Solids, 2009, 70, 344–355 CrossRef CAS.
- A. J. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef.
- J. E. Carpenter and F. Weinhold, J. Am. Chem. Soc., 1988, 110, 368–372 CrossRef CAS.
- A. B. F. Duncan, J. Chem. Phys., 1957, 27, 423–429 CrossRef CAS.
- C. Tantardini and A. R. Oganov, Nat. Commun., 2021, 12, 2087 CrossRef CAS PubMed.
- P. L. Popelier, The chemical bond II: 100 years old and getting stronger, 2016, pp. 71–117 Search PubMed.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529–5542 CrossRef CAS.
- C. Gatti and D. Lasi, Faraday Discuss., 2007, 135, 55–78 RSC.
- J. R. Lane, J. Contreras-García, J.-P. Piquemal, B. J. Miller and H. G. Kjaergaard, J. Chem. Theory Comput., 2013, 9, 3263–3266 CrossRef CAS PubMed.
- C. Tantardini, J. Comput. Chem., 2019, 40, 937–943 CrossRef CAS PubMed.
- C. Tantardini, A. A. Michalchuk, A. Samtsevich, C. Rota and A. G. Kvashnin, Sci. Rep., 2020, 10, 7816 CrossRef CAS PubMed.
- E. Magnusson, J. Am. Chem. Soc., 1990, 112, 7940–7951 CrossRef CAS.
- C. Tantardini, E. V. Boldyreva and E. Benassi, J. Phys. Chem. A, 2016, 120, 10289–10296 CrossRef CAS PubMed.
- Y. Huang, J. Zhou, L. Peng, K. Li, S. R. Elliott and Z. Sun, J. Phys. Chem. C, 2021, 125, 19537–19543 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational information for all the materials, BCP analysis, and DOS calculations for GaTe and InSe. See DOI: https://doi.org/10.1039/d4cp00937a |
| This journal is © the Owner Societies 2024 |