
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photolytic splitting of homodimeric quinone-derived oxetanes studied by ultrafast transient absorption spectroscopy and quantum chemistry†
Alejandro
Blasco-Brusola
a,
Lorena
Tamarit
 a,
Miriam
Navarrete-Miguel
b,
Daniel
Roca-Sanjuán
b,
Miguel A.
Miranda
*a and
Ignacio
Vayá
*a
a,
Miriam
Navarrete-Miguel
b,
Daniel
Roca-Sanjuán
b,
Miguel A.
Miranda
*a and
Ignacio
Vayá
*a
aDepartamento de Química/Instituto de Tecnología Química UPV-CSIC, Universitat Politècnica de València, Camino de Vera s/n, València 46022, Spain. E-mail: mmiranda@qim.upv.es; igvapre@qim.upv.es
bInstituto de Ciencia Molecular, Universitat de València, P.O. Box 22085, València 46071, Spain
First published on 16th April 2024
Abstract
The photoinduced cycloreversion of oxetane derivatives is of considerable biological interest since these compounds are involved in the photochemical formation and repair of the highly mutagenic pyrimidine (6-4) pyrimidone DNA photoproducts ((6-4)PPs). Previous reports have dealt with the photoreactivity of heterodimeric oxetanes composed mainly of benzophenone (BP) and thymine (Thy) or uracil (Ura) derivatives. However, these models are far from the non-isolable Thy〈º〉Thy dimers, which are the real precursors of (6-4)PPs. Thus, we have synthesized two chemically stable homodimeric oxetanes through the Paternò–Büchi reaction between two identical enone units, i.e. 1,4-benzoquinone (BQ) and 1,4-naphthoquinone (NQ), that led to formation of BQ-Ox and NQ-Ox, respectively. Their photoreactivity has been studied by means of steady-state photolysis and transient absorption spectroscopy from the femtosecond to the microsecond time scale. Thus, photolysis of BQ-Ox and NQ-Ox led to formation of the monomeric BQ or NQ, respectively, through ring opening in a “non-adiabatic” process. Accordingly, the transient absorption spectra of the triplet excited quinones (3BQ* and 3NQ*) were not observed as a result of direct photolysis of the quinone-derived oxetanes. In the case of NQ-Ox, a minor signal corresponding to 3NQ* was detected; its formation was ascribed to minor photodegradation of the oxetane during acquisitions of the spectra during the laser experiments. These results are supported by computational analyses based on density functional theory and multiconfigurational quantum chemistry (CASSCF/CASPT2); here, an accessible conical intersection between the ground and excited singlet states has been characterized as the main structure leading to deactivation of excited BQ-Ox or NQ-Ox. This behavior contrasts with those previously observed for heterodimeric thymine-derived oxetanes, where a certain degree of ring opening into the excited triplet state is observed.
Introduction
The photoreactions of carbonyl compounds in solution constitute a key chapter in photochemistry textbooks.1 They are very broad and encompass a wide variety of reactions such as bond cleavage, hydrogen abstraction and cycloaddition. In this regard, α,β-unsaturated enones are known to undergo cycloaddition, including dimerization, upon irradiation.2 In the particular case of cyclic enones, the inter- and intramolecular [2+2]-photoreactions leading to the formation of cyclobutane adducts have been reported and applied to the synthesis of natural products and biologically active organic compounds.2–4In particular, the Paternò–Büchi reaction of α,β-unsaturated carbonyl compounds is of considerable biological interest since it is involved in the formation of the mutagenic pyrimidine (6-4) pyrimidone DNA photoproducts ((6-4)PPs). The precursor of (6-4)PPs is thought to be a thymine-derived oxetane (Thy〈º〉Thy) intermediate that subsequently undergoes ring opening with concomitant rearrangement.5–8 Formation of these (6-4)PPs photoproducts upon direct DNA exposure to UV light may result in genotoxic, mutagenic and carcinogenic effects.9–12
In view of their biological interest, oxetane derivatives have attracted considerable attention, focusing not only on their formation but also on their photolytic splitting, which has been related to the photoenzymatic repair of DNA (6-4)PPs.13–17 In this regard, since the primary Thy〈º〉Thy dimers are not isolable due to their chemical instability,18 a variety of model oxetane conjugates composed of benzophenone (BP) and thymine (Thy) or uracil (Ura) derivatives have been employed in order to investigate the mechanism of photoinduced ring opening by means of spectroscopic techniques.19–23 Although these models are far from the real Thy〈º〉Thy dimers, the BP moiety has been found to be convenient to follow the course of the photoreaction by transient absorption spectroscopy, which allows rapid and quantitative detection of the BP triplet excited state (3BP*),24 amongst other transient species. Interestingly, irradiation of BP-Thy and/or BP-Ura oxetanes results in some cases in a rare “adiabatic” ring splitting process involving diradical species and triplet exciplexes, which ultimately evolve leading to the formation of 3BP* and Thy (or Ura) in its ground state.19,20,22,23,25 Here, “adiabatic” must be understood as the process (in this case, ring splitting) in which the system exclusively remains in the excited state (either singlet or triplet), with no transitions to the ground state occurring during the course of the reaction; in contrast, “non-adiabatic” means decay to the ground state.
Based on this background, the aim of the present work is to investigate the photolytic mechanism of chemically stable oxetane derivatives sharing with Thy〈º〉Thy two structural features, namely (i) their homodimeric nature and (ii) their formation by the Paternò–Büchi reaction between two identical units of α,β-unsaturated carbonyl compounds. Accordingly, 1,4-benzoquinone (BQ) and 1,4-naphthoquinone (NQ) have been selected to synthesize the corresponding stable dimeric oxetanes (BQ-Ox and NQ-Ox, respectively; see Scheme 1). Their photobehavior has been monitored by means of transient absorption spectroscopy, from the femtosecond to the microsecond time scale. In the case of “adiabaticity”, the “repaired” photoproducts BQ and NQ should be conveniently detected in their triplet excited states (3BQ* and 3NQ*, respectively). Otherwise, the ground state would be obtained (BQ and NQ, respectively). In addition to the experimental characterization, computational analysis based on density functional theory and multiconfigurational quantum chemistry (complete-active-space self-consistent field/complete-active-space second-order perturbation theory, CASSCF/CASPT2) have been made to provide a rationale for the experimental observations.
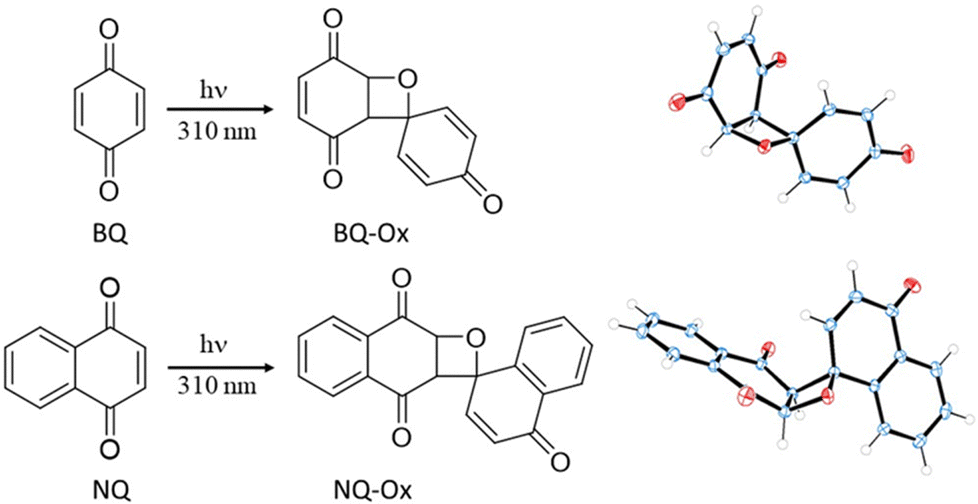 | ||
| Scheme 1 Schematic representation of the photoinduced formation of BQ-Ox and NQ-Ox in deaerated MeCN upon irradiation of the monomeric units BQ and NQ, respectively, through Pyrex tubes in a multilamp photoreactor emitting at λmax ∼ 310 nm. The structure of both oxetanes in the crystalline state is also shown: C blue, H white, O red. The thermal ellipsoids are drawn at the 50% probability level. | ||
Experimental section
Chemicals and reagents
1,4-Benzoquinone (BQ) and 1,4-naphthoquinone (NQ) were purchased from Sigma-Aldrich. Acetonitrile (HPLC grade), hexane, ethyl acetate, dichloromethane and methanol were purchased from Scharlab.Synthesis of the homodimeric oxetanes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 3:2). In the second step, the final product was purified by direct phase HPLC using dichloromethane/ethyl acetate (70:30) as eluent (flow rate 2 mL min−1).
:1 to 3:2). In the second step, the final product was purified by direct phase HPLC using dichloromethane/ethyl acetate (70:30) as eluent (flow rate 2 mL min−1).
1H-NMR (400 MHz, CDCl3): δ (ppm): 7.20 (d, J = 12 Hz, 1H), 6.97 (q, J = 16 Hz, 2H), 6.75 (d, J = 12 Hz, 1H), 6.18 (t, J = 16 Hz, 2H), 5.10 (d, J = 12 Hz, 1H), 4.01 (d, J = 12 Hz, 1H). 13C-NMR (100 MHz, CDCl3): δ (ppm): 190.9, 190.3, 182.8, 144.4, 141.6, 140.9, 140.2, 130.1, 127.9, 80.2, 71.6, 50.1. Yield: <2%. HRMS (ESI): m/z calcd for [M+H]+ C12H9O4:217.0501, found: 217.0498. CCDC 2026990.1
:5) as eluent (flow rate 2 mL min−1).
1H-NMR (400 MHz, CDCl3): δ (ppm): 8.31–7.58 (m, 8H), 6.81 (d, J = 8 Hz, 1H), 6.29 (d, J = 8 Hz, 1H), 5.55 (d, J = 8 Hz, 1H), 4.45 (d, J = 8 Hz, 1H). 13C-NMR (100 MHz, CDCl3): δ (ppm): 193.4, 191.7, 189.8, 183.0, 143.9, 135.6, 133.9, 130.8, 129.7, 128.5, 127.7, 126.9, 126.0, 83.3, 74.5, 56.8, 47.2, 31.6, 22.7, 14.1. Yield: <3%. HRMS (ESI): m/z calcd for [M+H]+ C20H13O4:317.0814, found: 317.0811. CCDC 2026991.†
Steady-state photolysis
Irradiations were performed in deaerated acetonitrile through Pyrex tubes in a multilamp Luzchem photoreactor (10 × 8 W lamps) emitting at λmax ∼ 310 nm.A semipreparative JASCO HPLC system (PU-2080 Plus pump, DG-2080-54-line degasser and LG-2080-04 gradient unit) connected to a JASCO (UV-1575) detector was used to separate and purify the different photoproducts, using an isocratic flux (2 mL min−1) of MeCN as an eluent, and a SEA18 Teknokroma column, 5 μm (25 × 1 cm2).
An analytical Waters HPLC system (Waters 1525 Binary HPLC Pump and a Waters 2-channel in-line degasser AF) connected to a Waters 2996 Photodiode Array Detector (wavelength fixed at 252 nm) was used to analyze the photoinduced break-down of the different products, using an isocratic flux (0.8 mL min−1) of MeCN as an eluent, and a reversed-phase Mediterranea Teknokroma column Sea18, 5 μm (25 × 0.46 cm2).
Spectroscopic techniques
The 1H- and 13C-NMR spectra were recorded at 400 and 100 MHz, respectively, using a Bruker AVANCE III instrument; chemical shifts are reported in ppm.High-resolution mass spectrometry (HRMS) was performed in an ultra performance liquid chromatography (UPLC) ACQUITY system (Waters Corp.) with a conditioned autosampler at 4 °C. The separation was accomplished on an ACQUITY UPLC BEH C18 column (50 mm × 2.1 mm i.d., 1.7 μm), which was maintained at 40 °C. The analysis was performed using acetonitrile and water (60:40 v/v containing 0.01% formic acid) as a mobile phase with a flow rate of 0.5 mL min−1, and the injection volume was 5 μL. The Waters ACQUITY™ XevoQToF Spectrometer (Waters Corp.) was connected to the UPLC system via an electrospray ionization (ESI) interface. This source was operated in positive ionization mode with the capillary voltage at 1.5 kV at 100 °C and the temperature of the desolvation was 300 °C. The cone and desolvation gas flows were 40 L h−1 and 800 L h−1, respectively. The collision gas flow and collision energy applied were 0.2 mL min−1 and 12 V, respectively. All data collected in centroid mode were acquired using Masslynx™ software (Waters Corp.). Leucine-enkephalin was used at a concentration of 500 pg μL−1 as the lock mass generating an [M+H]+ ion (m/z 556.2771) and fragment at m/z 120.0813 and a flow rate of 50 μL min−1 to ensure accuracy during the MS analysis.
Steady-state absorption spectra were recorded using a JASCO V-760 spectrophotometer.
Laser flash photolysis (LFP) measurements were performed using two different systems. One is a pulsed Nd:YAG L52137 V LOTIS TII at the excitation wavelength of 266 nm; this wavelength is achieved after a 1064 nm light beam has passed first through a second harmonic and then through a fourth harmonic crystal properly aligned to generate the 266 nm beam light. The single pulses were ca. 10 ns duration, and the energy was ∼10 mJ per pulse. All decays were measured upon accumulation of 5 consecutive pulses at the corresponding monitoring wavelength under continuous solution stirring in a deaerated atmosphere. The laser flash photolysis system consisted of the pulsed laser, a 77250 Oriel monochromator, and an oscilloscope DP04054 Tektronix. The output signal from the oscilloscope was transferred to a personal computer. Absorbances of all solutions were adjusted at ∼0.20 at 266 nm in acetonitrile (HPLC grade). The other is an LP980 laser system (Edinburgh instruments) consisting of an optical parametric oscillator (OPO, EKSPLA NT342) bombarded with the third harmonic of an Nd:YAG laser (EKSPLA PS5062), a pulsed Xenon lamp (150 W) as a detecting light source, a monochromator (TMS302-A, grating 150 lines per mm) and a PMT (Hamamatsu photonics). Experiments were carried out at an excitation wavelength of 310 nm (the single pulses were ca. 5 ns duration with an energy of ∼3 mJ). All components are controlled by the software L900 provided by Edinburgh. In this case, absorbances of all solutions were adjusted at ∼0.20 at 310 nm in acetonitrile (HPLC grade). All decays were measured upon accumulation of 20 consecutive pulses at the corresponding monitoring wavelength under continuous solution stirring in a deaerated atmosphere. All UV and LFP measurements were done using 10 × 10 mm2 quartz cuvettes at room temperature in deaerated acetonitrile (25 min N2 bubbling). The raw data files were exported to be treated with the OriginLab program.
Femtosecond transient absorption experiments were performed using a pump–probe system. The femtosecond pulses were generated using a mode-locked Ti-Sapphire laser of a compact Libra HE (4 W power at 4 kHz) regenerative amplifier delivering 100 fs pulses at 800 nm (1 mJ per pulse). The output of the laser was split into two parts to generate the pump and the probe beams. Thus, tunable femtosecond pump pulses were obtained by directing the 800 nm light into an optical parametric amplifier. In the present case, the pump was set at 265 nm and passed through a chopper prior to focusing onto a rotating cell (1 mm optical path) containing the samples in organic solution (acetonitrile HPLC grade). The white light used as a probe was produced after part of the 800 nm light from the amplifier travelled through a computer-controlled 8 ns variable optical delay line and impinged on a CaF2 rotating crystal. This white light was in turn split into two identical portions to generate reference and probe beams, which then were focused on the rotating cell containing the sample. The pump and the probe beams were made to coincide to interrogate the sample. The power of the pump beam was set to 200 μW. A computer-controlled imaging spectrometer was placed after this path to measure the probe and the reference pulses to obtain the transient absorption decays/spectra. The experimental data were treated and compensated by the chirp using the ExciPro program. The data files were exported as matrix format from ExciPro to be treated with the OriginLab program to obtain the time-resolved spectra and the ultrafast kinetic traces, which were fitted using a multi-exponential function following the Levenberg–Marquardt iteration algorithm:

Results and discussion
Although cyclic enones can in principle give the Paternò–Büchi reaction to form oxetanes, this is very rare, because other reactions such as cyclobutane formation or photoreductions are generally preferred.3,26–28 In the particular case of the parent BQ and NQ, homodimeric oxetanes BQ-Ox and NQ-Ox (Scheme 1) have not yet been reported; however, there is a brief report in the literature where formation of oxetanes upon irradiation of dimethyl-p-benzoquinone derivatives is claimed.29Irradiation of BQ and NQ in deaerated acetonitrile through Pyrex tubes in a multilamp photoreactor emitting at λmax ∼ 310 nm allowed us to isolate the oxetanes BQ-Ox and NQ-Ox, respectively (Scheme 1), albeit in very low yields (< 3%); they were fully characterized by 1H- and 13C-NMR, HRMS (see the Experimental section and Fig. S1 and S2 in the ESI†). More importantly, their structures (including 3D details) were resolved by X-ray crystallography. Remarkably, stereoselectivity to the exo dimer was noticed in the case of NQ-Ox.
Once the desired oxetanes BQ-Ox and NQ-Ox were available, their photobehavior was examined. Steady-state irradiation of both compounds resulted in ring splitting with formation of the corresponding quinones as the only photoproducts (Fig. S3 in the ESI†).
The photoinduced processes arising from excitation of BQ-Ox were also investigated by means of nanosecond laser flash photolysis (LFP) and compared with those of the reference compound BQ. Pulsed excitation of the latter in deaerated MeCN at different excitation wavelengths (310 and 266 nm) gave rise to its triplet excited state (3BQ*) with λmax ∼ 410 nm.30 However, no transient species were detected for BQ-Ox (Fig. 1 and Fig. S4 in the ESI†) under the same experimental conditions (the results obtained at λexc = 310 and 266 nm were the same); this revealed that the photocleavage of BQ-Ox decays to the ground state of the photoproducts, in contrast with previous results on BP-Thy or BP-Ura oxetanes, where certain degree of “adiabaticity” in the photoinduced cycloreversion process was observed.19,22,23,25
 | ||
| Fig. 1 (A) LFP transient absorption spectra for BQ at different times after the laser pulse (2, 10, 20 and 50 μs). (B) Decay traces for BQ (gray) and BQ-Ox (dark blue) at 410 nm. All measurements were performed in deaerated MeCN at λexc = 310 nm. | ||
To explain the lack of “adiabaticity” in the oxetane ring opening, theoretical calculations were performed. Thus, density functional theory and multiconfigurational quantum chemistry were efficiently combined and provided valuable insight into the involved mechanism (Fig. 2). The computational details, the active spaces, and the nature of the states at the stationary points can be found in the ESI,† (Fig. S5–S10 and Table S1). The photocycloreversion process is characterized by five main singular structures: the ground state (S0) oxetane (BQ-Ox) minimum, the triplet diradical (3BQ-Ox*) minimum, the triplet exciplex (3EXC*) minimum, the transition state (TS) between 3BQ-Ox* and 3EXC* and the crossing point (CP) involving S0, S1, and T1 states in between the TS and 3BQ-Ox*. From BQ-Ox to 3BQ-Ox*, the main deformation corresponds to the elongation of the oxetane C–C bond that is broken in the diradical structure. Then, 3BQ-Ox* and 3EXC* mainly differ in the C–O bond, which is broken in the latter. The CP region corresponds to an early elongation of the C–O bond from the diradical 3BQ-Ox*.
 | ||
| Fig. 2 Chemical mechanism of the photoinduced cycloreversion process for BQ-Ox. | ||
The mechanism proposed based on the computational determinations proceeds as follows: excitation of BQ-Ox induces the formation of its first singlet excited state S1 (1BQ-Ox*); this evolves towards a crossing point (CP), where S0, S1 and T1 are degenerated, corresponding, then, to a conical intersection (CI) and a singlet–triplet crossing (STC) point (Fig. S9 and Table S1 in the ESI† for the active space and the nature of the states at this structure, respectively). At this point, spin–orbit couplings (SOC) of SOCS0T1 = 35 cm−1 and SOCS1T1 = 22 cm−1 were obtained. If this CP region is reached, decay to the ground state S0 through internal conversion is expected to be highly efficient. Additionally, the obtained SOC values suggest that the population transfer S1 → T1 is less efficient as compared to T1 → S0. Furthermore, if T1 is populated in the CP region, there is an energy barrier of 0.40 eV that must be overcome to access 3EXC*. Consequently, the decay to S0 is the most favorable process in the CP region. From there, the energy profiles of Fig. 2 show two possible paths, either formation of the starting oxetane in its ground state or evolution towards the fully separated BQ units completing the ring opening through cleavage of the C–O bond in its ground state. Overall, these two routes are energetically more favorable than the “adiabatic” formation of 3BQ* through the triplet exciplex pathway. This clarifies the lack of “adiabaticity” observed experimentally in the photoinduced ring opening of BQ-Ox.
In view of the important differences between the photobehavior of the homodimeric oxetane derivative BQ-Ox and those of BP-Thy or BP-Ura, the photoinduced cycloreversion of NQ-Ox was also studied in detail. As in the previous case, the photobehavior was compared with that of its reference compound NQ. Thus, LFP measurements on NQ after excitation at 310 or 266 nm in deaerated MeCN gave rise to the formation of its triplet excited state (3NQ*) peaking at ca. 370 nm (Fig. 3 and Fig. S11, ESI† respectively).31 Interestingly, excitation of NQ-Ox resulted in the appearance of a much weaker signal with λmax ∼ 370 nm (the results obtained at λexc = 310 and 266 nm were the same).
 | ||
| Fig. 3 (A) LFP transient absorption spectra for NQ at different times after the laser pulse (0.5, 2, 5 and 15 μs). (B) Decay traces for NQ (black) and NQ-Ox (blue) at 370 nm. All measurements were performed in deaerated MeCN at λexc = 310 nm. | ||
The weak transient absorptions arising from excitation of NQ-Ox can be originated from either by inefficient “adiabatic” photocleavage of the oxetane or by excitation of a marginal amount of NQ arising from partial degradation of NQ-Ox during the LFP experiment. In order to better understand the origin of this species, femtosecond transient absorption spectroscopy was used with the aim of investigating the photoinduced processes arising from both NQ and NQ-Ox in the very early stages after excitation. In this regard, this technique has been demonstrated to be very useful to study in detail ultrafast photoinduced processes such as energy or electron transfer, charge separation or intersystem crossing.32 The photobehavior of NQ was first investigated since its photophysical properties at the ultrafast time-scale have not been studied previously.
As it is shown in Fig. 4A, excitation of NQ at 265 nm in MeCN resulted in the instantaneous formation of a species peaking at ca. 415 and 380 nm, which is assigned to the singlet–singlet absorption band (1NQ*). This species evolved towards the formation of a new transient peaking at ca. 370 nm, which matches with the formation of 3NQ*, also detected at longer time scales by means of LFP. This process can be explained from the kinetic traces shown in the inset of Fig. 4A; thus, the decay at 415 nm can be properly fitted with a two-component exponential law; the ultrafast one (∼400 fs) is associated with vibrational relaxation, while the longest component of ca. 13 ps, which coincides in lifetime with the growth at 370 nm, can be undoubtedly associated with the intersystem crossing process to form 3NQ*.
 | ||
| Fig. 4 Femtosecond transient absorption spectra for NQ (A) and for NQ-Ox (B) from 1 (black) to 50 ps (blue). The insets show the normalized kinetic traces at 415 (black) and 370 nm (blue). Measurements were performed in acetonitrile at λexc = 265 nm. | ||
The ultrafast photobehavior of NQ-Ox was found to be very similar to that observed for NQ (Fig. 4B), although the intensity of the absorbing species was much lower. Thus, absorption bands peaking at similar wavelengths and with coincident lifetimes, but exhibiting much less intensity than those of NQ, were detected for NQ-Ox. This is in line with the assumption that the small signal detected by LFP on NQ-Ox (see above) arises from 3NQ* generated upon photodegradation of the oxetane by the laser pulse rather than from the “adiabatic” photocleavage. Indeed, photodegradation of NQ-Ox to form NQ was found to be detectable at low irradiation times (Fig. S3 and S12 in the ESI†).
As in the case of BQ-Ox, theoretical calculations also provide an interpretation of the lack of “adiabaticity” in the photoinduced cycloreversion process in NQ-Ox (Fig. 5). See computational details, active spaces (Fig. S13–S20, ESI†) and the nature of the states in each stationary point (Table S2) in the ESI.† Five relevant points also characterize the photoinduced cycloreversion process of the naphthoquinone dimer: the ground state (S0) oxetane (NQ-Ox), the triplet diradical (3NQ-Ox*), the crossing point between S0, S1 and T1 (CP), the triplet exciplex (3EXC*) and the transition state (TS) between CP and 3EXC*, involving the same deformations corresponding to the elongation of the oxetane C–C and the C–O bonds.
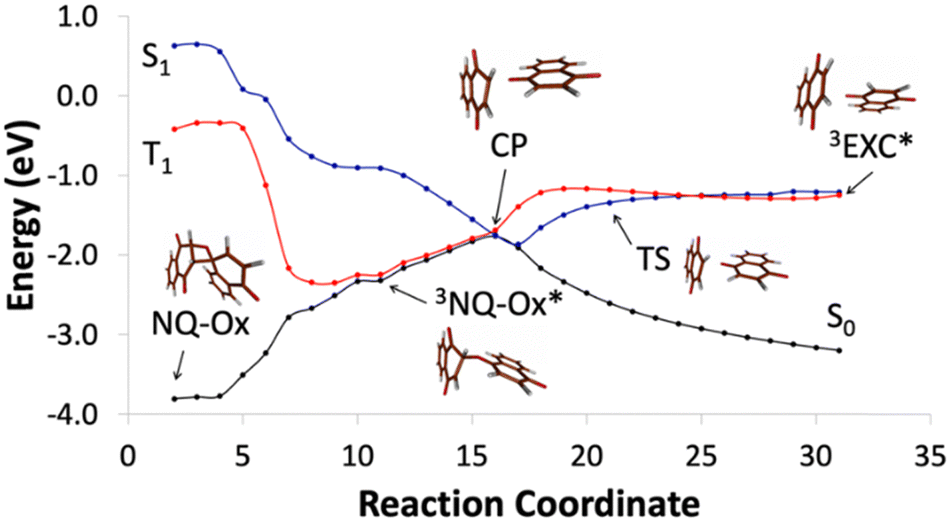 | ||
| Fig. 5 Chemical mechanism of the photoinduced cycloreversion process for NQ-Ox. | ||
The mechanism involved in the photocycloreversion of NQ-Ox shows also the same general qualitative trends as that of BQ-Ox, which agrees with the experimental observations regarding the comparison between the two systems. In this case, irradiation of the oxetane induces the population of 1NQ-Ox*, which evolves towards the CP between S0, S1 and T1 states. Here, SOC values of approximately 30 cm−1 and 5 cm−1 were found for the S1T1 and S0T1 STCs, respectively. This seems to point to a longer lifetime of the T1 in this region as compared with the case of BQ-Ox. However, C–O bond cleavage on the triplet state towards 3EXC* is hindered by an energy barrier of 0.52 eV, which also makes the “adiabatic” photocycloreversion process unfavorable.
Note finally that in both BQ-Ox and NQ-Ox systems, two distinct conformations of the so-called 3EXC* point were identified, one that exhibits a T-shaped arrangement of the monomers and another that shows a parallel orientation. The former is found directly from the minimum energy path on the T1 manifold from the TS. Further evolution on such a state shall bring the system to the parallel conformation which is the one energetically more stable and responsible for the excimer properties. Fig. S21 and S22 shown in the ESI† display the two different orientations for BQ-Ox and NQ-Ox excimers, respectively, and Table S3 (ESI†) provides a comparison of the energies.
A comparison of the current theoretical findings from BQ-Ox and NQ-Ox with those previously observed for head-to-head and heat-to-tail oxetane regioisomers composed of benzophenone and dimethylthymine (HH and HT, respectively)21 shows that the state crossing (CP) occurs prior to the TS region and after the 3BQ-Ox* diradical in BQ-Ox, NQ-Ox and HT, whereas it is located at the TS region for HH. This imposes an energetic barrier to reach the 3EXC* structure in BQ-Ox, NQ-Ox and HT, which is not present in HH. This result agrees with a highly efficient “adiabatic” photocycloreversion process only for HH. It is observed that the electronic structure of the excited singlet state in the diradical region of BQ-Ox and NQ-Ox resembles more the properties of HT, with a charge transfer nature between the two units of the dimer. In fact, there is a delocalization of the excitation in the BP unit in HT. The higher SOC values between S0 and T1 in the CP region for BQ-Ox and NQ-Ox compared with HT could result in a lack of “adiabaticity” in the quinone-derived oxetanes, whereas for HT, it is present to some extent.
Overall, taking the obtained results on homodimeric oxetanes BQ-Ox and NQ-Ox together with those previously obtained for the heterodimeric BP-Thy and BP-Ura oxetanes, the mechanism of photoinduced cycloreversion can be summarized as follows (Scheme 2): irradiation of the oxetane gives rise to the formation of its singlet excited state, which rapidly evolves towards cleavage of the C–C bond leading to the formation of singlet diradical species. From this point, heterodimeric oxetanes follow route III or IV, depending mainly on the regiochemistry and/or ring substitutions, inducing direct formation of triplet exciplexes or via triplet diradicals, finally resulting in an adiabatic oxetane photocleavage. Conversely, an alternative route (V) from triplet diradicals results in a “non-adiabatic” ring opening or recovery of the oxetane to its ground state. Indeed, route V seems to be the main path followed by the homodimeric oxetanes of the present work, since no “adiabaticity” has been observed in their photoinduced ring opening.
 | ||
| Scheme 2 Schematic representation of the photoinduced cycloreversion process for homodimeric (above) and heterodimeric (below) oxetane derivatives. The width of the colored arrows represents the efficiency of the process. | ||
Conclusions
The synthesis of chemically stable homodimeric oxetanes BQ-Ox and NQ-Ox has been performed for the first time upon irradiation of the monomeric precursors BQ and NQ, respectively. Steady-state photolysis of BQ-Ox or NQ-Ox leads to formation of the starting materials as single products. Irradiation of BQ-Ox by LFP does not result in the formation of detectable transient species, which agrees with a “non-adiabatic” photoinduced cycloreversion process. A similar result has been observed for NQ-Ox. In this case, the very tiny signal detected at 370 nm can be attributed to formation of 3NQ* from photodegradation of the oxetane during the laser experiments rather than from the “adiabatic” photocleavage. Indeed, this has been confirmed by means of ultrafast transient absorption measurements. The experimental results can be explained by theoretical calculations, which provide a clear interpretation of the lack of “adiabaticity” in the photoinduced cycloreversion process for BQ-Ox and NQ-Ox. In both cases, a conical intersection between the oxetane's first singlet excited state and the ground state is reached, instead of triplet exciplex formation, since an uphill energetic barrier has been determined; consequently, for both homodimeric oxetanes, the ring opening occurs in a “non-adiabatic” process or, alternatively, the oxetane is recovered in its ground state. These results contrast with those previously observed for heterodimeric oxetanes, where a certain degree of “adiabaticity” in the ring splitting is observed. These results may be relevant in the framework of the photolytic repair of (6-4)PPs and related DNA photoproducts.Author contributions
Research was conceived by all authors. Experiments were performed by A. B.-B., L. T. with the aid of I. V. The computational study was performed by M. N.-M. with the aid of D. R.-S. The research was supervised by I. V. and M. A. M. All authors contributed to the writing of the manuscript and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Grants PID2020-115010RB-I00 and PID2021-127199NB-I00 funded by MCIN/AEI/10.13039/501100011033 and grants from Conselleria d’Innovació, Universitats, Ciència i Societat Digital (CIAICO/2021/061, CIAICO/2022/121 and CIAPOS/2021/87) are gratefully acknowledged. M. N.-M. is thankful to the Universitat de València for her “Atracció de Talent 2020” predoctoral grant. The authors would like to thank the use of RIAIDT-USC analytical facilities for the X-ray crystallography analysis.References
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, California, 2010 Search PubMed.
- B. Dinda, Essentials of Pericyclic and Photochemical Reactions, Springer Cham, 2017, vol. 93, pp. 241–275 Search PubMed.
- A. C. Weedon, in Synthetic Organic Photochemistry, ed. W. M. Horspool, Springer US, Boston, MA, 1984, pp. 61–143 Search PubMed.
- K. Kokubo and T. Oshima, in CRC Handbook of Organic Photochemistry and Photobiology, ed W. M. Horspool and F. Lenci, CRC Press Taylor & Francis Group, Boca Ratón, 2003, vol. 1 & 2 Search PubMed.
- D. E. Brash and W. A. Haseltine, Nature, 1982, 298, 189–192 CrossRef CAS PubMed.
- J. Cadet, T. Douki, J.-P. Pouget and J.-L. Ravanat, in DNA Photolesions to Mutations, Skin Cancer and Cell Death, 2005 Search PubMed.
- J. Cadet, S. Mouret, J. L. Ravanat and T. Douki, Photochem. Photobiol., 2012, 88, 1048–1065 CrossRef CAS PubMed.
- N. Chatterjee and G. C. Walker, Environ. Mol. Mutagen., 2017, 58, 235–263 CrossRef CAS PubMed.
- M. G. Friedel, M. K. Cichon and T. Carell, DNA damage and repair: photochemistry, CRC Handbook of Organic Photochemistry and Photobiology CRC Press LLC, Boca Raton, 2004 Search PubMed.
- K. H. Kraemer, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 11–14 CrossRef CAS PubMed.
- J.-S. Taylor, Acc. Chem. Res., 1994, 27, 76–82 CrossRef CAS.
- S.-L. Yu and S.-K. Lee, Mol. Cell. Toxicol., 2017, 13, 21–28 CrossRef CAS.
- M. D'Auria and R. Racioppi, Molecules, 2013, 18, 11384–11428 CrossRef PubMed.
- J. Li, Z. Liu, C. Tan, X. Guo, L. Wang, A. Sancar and D. Zhong, Nature, 2010, 466, 887–890 CrossRef CAS PubMed.
- A. Sancar, Science, 1994, 266, 1954–1956 CrossRef CAS PubMed.
- S. A. Shrinivas, S. H. Shanta and B. B. Prajakta, Indo Global J. Pharm. Sci., 2017, 3, 555613 Search PubMed.
- X. Zhao and D. Mu, Histol. Histopathol., 1998, 13, 1179–1182 CAS.
- J. Cadet, A. Grand and T. Douki, Top. Curr. Chem., 2014, 356, 249–275 CrossRef PubMed.
- A. Joseph and D. E. Falvey, J. Am. Chem. Soc., 2001, 123, 3145–3146 CrossRef CAS PubMed.
- A. Joseph, G. Prakash and D. E. Falvey, J. Am. Chem. Soc., 2000, 122, 11219–11225 CrossRef CAS.
- G. Prakash and D. E. Falvey, J. Am. Chem. Soc., 1995, 117, 11375–11376 CrossRef CAS.
- A. Blasco-Brusola, M. Navarrete-Miguel, A. Giussani, D. Roca-Sanjuán, I. Vayá and M. A. Miranda, Phys. Chem. Chem. Phys., 2020, 22, 20037–20042 RSC.
- A. Blasco-Brusola, I. Vayá and M. A. Miranda, Org. Biomol. Chem., 2020, 18, 9117–9123 RSC.
- J. A. Bell and H. Linschitz, J. Am. Chem. Soc., 1963, 85, 528–532 CrossRef CAS.
- A. Blasco-Brusola, I. Vayá and M. A. Miranda, J. Org. Chem., 2020, 85, 14068–14076 CrossRef CAS PubMed.
- D. I. Schuster, in CRC Handbook of Organic Photochemistry and Photobiology, ed W. M. Horspool and F. Lenci, CRC Press Taylor & Francis Group, Boca Ratón, 2003, vol. 1 & 2 Search PubMed.
- Y. Ando and K. Suzuki, Chem.–Eur. J., 2018, 24, 15955–15964 CrossRef CAS PubMed.
- M. Fréneau and N. Hoffmann, J. Photochem. Photobiol., C, 2017, 33, 83–108 CrossRef.
- R. C. Cookson, J. J. Frankel and J. Hudec, Chem. Commun., 1965, 16 RSC.
- J.-C. Ronfard-Haret and R. V. Bensasson, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 2432–2436 RSC.
- I. Amada, M. Yamaji, M. Sase and H. Shizuka, J. Chem. Soc., Faraday Trans., 1995, 91, 2751–2759 RSC.
- C. Ruckebusch, M. Sliwa, P. Pernot, A. de Juan and R. Tauler, J. Photochem. Photobiol., C, 2012, 13, 1–27 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, chemical characterization of the investigated oxetanes by means of 1H- and 13C-NMR. CCDC 2026990 and 2026991. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cp00830h |
| This journal is © the Owner Societies 2024 |