
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural characterization of E22G Aβ1–42 fibrils via1H detected MAS NMR†
Natalie C.
Golota
 ab,
Brian
Michael‡
ab,
Edward P.
Saliba§
ab,
Sara
Linse
c and
Robert G.
Griffin
*ab
ab,
Brian
Michael‡
ab,
Edward P.
Saliba§
ab,
Sara
Linse
c and
Robert G.
Griffin
*ab
aDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: rgg@mit.edu
bFrancis Bitter Magnet Laboratory, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cBiochemistry and Structural Biology, Department of Chemistry, Lund University, Lund, SE 22100, Sweden
First published on 26th April 2024
Abstract
Amyloid fibrils have been implicated in the pathogenesis of several neurodegenerative diseases, the most prevalent example being Alzheimer's disease (AD). Despite the prevalence of AD, relatively little is known about the structure of the associated amyloid fibrils. This has motivated our studies of fibril structures, extended here to the familial Arctic mutant of Aβ1–42, E22G-Aβ1–42. We found E22G-AβM0,1–42 is toxic to Escherichia coli, thus we expressed E22G-Aβ1–42 fused to the self-cleavable tag NPro in the form of its EDDIE mutant. Since the high surface activity of E22G-Aβ1–42 makes it difficult to obtain more than sparse quantities of fibrils, we employed 1H detected magic angle spinning (MAS) nuclear magnetic resonance (NMR) experiments to characterize the protein. The 1H detected 13C–13C methods were first validated by application to fully protonated amyloidogenic nanocrystals of GNNQQNY, and then applied to fibrils of the Arctic mutant of Aβ, E22G-Aβ1–42. The MAS NMR spectra indicate that the biosynthetic samples of E22G-Aβ1–42 fibrils comprise a single conformation with 13C chemical shifts extracted from hCH, hNH, and hCCH spectra that are very similar to those of wild type Aβ1–42 fibrils. These results suggest that E22G-Aβ1–42 fibrils have a structure similar to that of wild type Aβ1–42.
1. Introduction
The formation of amyloid fibrils by peptide misfolding and aggregation is implicated in the development of over 40 diseases, including Alzheimer's disease (AD), Parkinson's disease, and dialysis related amyloidosis (DRA). As of 2022,1 it is estimated that 1 in 9 adults over 65 have AD, which rises to 1 in 3 adults over 85. This figure is expected to increase as the US population continues to age. The aggregation of amyloid-β (Aβ) in the brain has been correlated with pathogenesis of AD, and, although not proven as the cause of AD, it remains a critical biomarker of the disease.2–4 Aβ occurs most frequently as a small protein of 40 or 42 residues and is derived by proteolytic cleavage of amyloid precursor protein (APP).5–7 While Aβ1–42 is expressed at lower levels than Aβ1–40, it is associated with greater cellular toxicity and is a major component of plaque found in AD patients.8,9 The development of aggressive, early onset forms of AD is associated with alterations to the APP. In vitro, enhanced aggregation dynamics are observed for mutations A21G, E22Δ, E22K, E22G, or D23N.7,10–15 The Arctic mutant, E22G, is characterized by the most rapid aggregation and fibrillization, thought to be driven by reduced electrostatic repulsive forces and decreased sidechain size.16 Structural studies of E22G-Aβ1–40 have revealed extensive polymorphism,17,18 while the characterization of E22G-Aβ1–42 has been limited to cryogenic electron microscopy.19,20 The first study examined two human brain protofilament extracts consisting of residues V12-V40 and E11-G37 of E22G, in addition to filaments from brains of mice with the Arctic mutation E22G.19 More recently, cryo-electron microscopy and cryo-electron tomography were used to determine the structure of Aβ from APPNL-G-F mouse brains.20 In both cases, structural modifications were observed particularly in the case of the APPNL-G-F mouse brain samples. However, to date, there has not been a MAS NMR study of E22G-Aβ1–42.Since amyloid fibrils form a discontinuous solid phase of objects with high aspect ratio and are difficult to crystallize, they are not amenable to studies with either solution-state NMR or X-ray crystallography.21 However, because MAS NMR can address structural questions in such systems, it has become a valuable tool for probing dynamics, identifying polymorphism, and determining atomic resolution structures.15,22,23 In particular, development of dipole recoupling and fast magic angle spinning (MAS) methods24 have led to marked improvements in the resolution, sensitivity, and utility of biomolecular solid-state NMR.25–28 Applications to Aβ1–42 have revealed a monomorphic, dimeric structure of mirror image S-shaped monomers in which hydrophobic residues were isolated in the interior of the fibril core and ∼15 N-terminal residues form a dynamic tail.22,23,29,30 Only a few residues in the tail are observed in the MAS spectra, presumably because dynamics interferes with the 1H decoupling and broadens the signals.31,32 This structure represents one filament of two monomers per plane, while recent small angle scattering data reveal an elongated cross-section of four monomers per plane, representing two such filaments twisting together in a fibril.33 In addition, cryo-electron microscopy has been used to study amyloid fibrils and has proven to be particularly valuable in the study of brain derived samples.19,23,34,35
Only recently have applications of 1H-detected MAS methods to amyloid fibrils become more common. The advantages are an absolute sensitivity enhancement over conventional 13C detection36 and improved coherence lifetimes.37,38 The improved sensitivity has provided structural information about amyloid fibrils and membrane proteins beyond that available from directly detected 13C and 15N spectra.39–45 Conventional 13C detected methods have relied upon large sample volumes, moderate MAS frequencies (typically in the range ωr/2π ∼ 10–20 kHz), and radio frequency driven recoupling46,47 (RFDR) for homonuclear 13C–13C spin correlations and short-range resonance assignments. RFDR, a first order, zero quantum recoupling sequence that uses a single 180° pulse centered in the rotor period to recouple homonuclear dipolar couplings,46–48 has been frequently employed in probing amyloids, membrane proteins and viral particles.49–53 On the other hand, recent experiments combined homonuclear recoupling with MAS at ωr/2π ≥ 90 kHz to acquire 1H-detected multidimensional experiments on a few hundred micrograms of material. However, to date very few 1H detected studies were performed on fully protonated Aβ, demonstrating the need for continued development and application of dipolar recoupling at high spinning frequencies.38,54
In this study, we demonstrate the use of 1H detected NMR for high-resolution structural characterization of fully protonated GNNQQNY crystals and E22G-Aβ1–42 fibril samples. This work demonstrates the utility of the rigid crystal in optimizing the development of proton detected methods to the study of amyloidogenic proteins. A MAS frequency of ωr/2π = 90 kHz was sufficient to achieve 1H linewidths of ∼0.3 ppm in GNNQQNY using cross polarization single quantum correlation (CP-HSQC) 1H–13C and 1H–15N experiments. Furthermore, the hCCH-RFDR experiment was optimized on GNNQQNY, and we determined that the optimal mixing times for RFDR at ωr/2π = 90 kHz are comparable to those at ωr/2π = 20 kHz.
Biosynthetic expression of E22G-Aβ1–42 was difficult since the protein is toxic to Escherichia coli and leads to relatively poor yield of tag-free peptide.16 We therefore expressed E22G-Aβ1–42 as a fusion protein with the self-cleavable tag NPro in the form of its EDDIE mutant; that is, as EDDIE-E22G-Aβ1–42 (see the sequence provided below),55 which protects the cells by driving the expressed product to inclusion bodies. Nevertheless, the high surface activity of E22G-Aβ1–42 leads to high losses during purification, and the final yield of isotopically enriched E22G-Aβ1–42 were limited; thus, the improved sensitivity available from 1H-detected MAS NMR was essential for study of the E22G mutant.
The 1H–13C CP-HSQC spectra reveal a conformationally disordered A42, as previously observed in wild type Aβ1–42, and slight (≤1 ppm) chemical shift differences from wild type Aβ1–42. The 1H–15N CP-HSQC spectra have considerably broadened line shapes and reduced resolution, likely due to slight structural differences among fibrils due to the characteristic rapid fibril formation of Arctic mutant samples. 15N spectra are known to be sensitive to these effects since the 15N is directly involved in hydrogen bonding. Finally, we demonstrate the utility of homonuclear recoupling at ωr/2π = 90 kHz to determine that E22G-Aβ1–42 fibrils are largely monomorphic. The hCCH spectra, recorded using either RFDR or TOCSY mixing, confirm the small chemical shift perturbations observed in hCH spectra. Collectively, we use proton detected MAS NMR to conclude that E22G Aβ1–42 fibrils are monomorphic, with a fibril core structure similar to that of wild type Aβ1–42, in accordance with a recently published cryo-EM study.19
2. Materials and methods
2.1 GNNQQNY synthesis
Uniformly labeled 13C,15N-GNNQQNY was synthesized using solid phase peptide synthesis and purified via HPLC courtesy of the Swanson Biotechnology Center at the MIT Koch Institute. The sample purity was >98% as ascertained by mass spec analysis. Samples were crystallized at peptide concentrations < 10 mg mL−1 as previously described.56,57 The crystals were then packed into a 3.2 mm or 0.7 mm rotors using ultracentrifugation. The 3.2 mm rotors contained ∼20 mg of sample whereas the 0.7 mm rotor uses 0.5 mg.2.2 Expression and purification of EDDIE-Aβ1–42 E22G
As mentioned above E22G-AβM0,1–42 is toxic to Escherichia coli and leads to poor yield of tag-free peptide.16 Therefore to express E22G-Aβ1–42 we employed a self-cleavable tag NPro in the form of its EDDIE mutant.55 This approach, which protects the cells by driving the expressed product to inclusion bodies, enables the isolation of Aβ peptides starting with Asp1 at the N-terminus,58–60 as well as the expression of other toxic peptides such as IAPP.61 The amino acid sequence of the expressed construct, EDDIE-E22G-Aβ1–42, is as follows:MELNHFELLY KTSKQKPVGV EEPVYDTAGR PLFGNPSEVH PQSTLKLPHD RG![[E with combining low line]](https://www.rsc.org/images/entities/b_char_0045_0332.gif)
![[D with combining low line]](https://www.rsc.org/images/entities/b_char_0044_0332.gif) DITTL RDLPRKGDCR SGNHLGPVSG IYIKPGPVYY QDYTGPVYHR APLEFFDE
DITTL RDLPRKGDCR SGNHLGPVSG IYIKPGPVYY QDYTGPVYHR APLEFFDE![[T with combining low line]](https://www.rsc.org/images/entities/b_char_0054_0332.gif) Q FETKRIGR VTGSDGKLYH IYVVDGIL LK
Q FETKRIGR VTGSDGKLYH IYVVDGIL LK![[Q with combining low line]](https://www.rsc.org/images/entities/b_char_0051_0332.gif) AKRGTPR TLKWRNTN CPLWVTSC
AKRGTPR TLKWRNTN CPLWVTSC ![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif)
![[F with combining low line]](https://www.rsc.org/images/entities/b_char_0046_0332.gif)
![[R with combining low line]](https://www.rsc.org/images/entities/b_char_0052_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/b_char_0048_0332.gif)
![[S with combining low line]](https://www.rsc.org/images/entities/b_char_0053_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/b_char_0047_0332.gif)
![[Y with combining low line]](https://www.rsc.org/images/entities/b_char_0059_0332.gif)
![[V with combining low line]](https://www.rsc.org/images/entities/b_char_0056_0332.gif)
![[K with combining low line]](https://www.rsc.org/images/entities/b_char_004b_0332.gif)
![[L with combining low line]](https://www.rsc.org/images/entities/b_char_004c_0332.gif)
![[N with combining low line]](https://www.rsc.org/images/entities/b_char_004e_0332.gif)
![[I with combining low line]](https://www.rsc.org/images/entities/b_char_0049_0332.gif)
![[M with combining low line]](https://www.rsc.org/images/entities/b_char_004d_0332.gif) .
.
Here the EDDIE mutant is the first 158 residues62 with those that are bold and underlined being the mutated sites. The E22G-Aβ1–42 is entirely bold faced and underlined.
The gene construct was designed with E. coli-preferred codons in a pET3a plasmid (purchased from Genscript, Piscataway, New Jersey) and the fusion protein was expressed in E. coli BL21 DE3 PlysS star in M9 minimal medium with 13C6-glucose and 15NH4Cl as the sole carbon and nitrogen sources.
A cell pellet from 4 L of solution was sonicated 5 times in 80 mL 10 mM tris/HCl, 1 mM EDTA, pH 8.5 (buffer A) containing a trace of DNase, with centrifugation at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 7 min between sonication steps. The inclusion body pellet after the 5th sonication was dissolved in 150 mL buffer A with 10 M urea and 1 mM DTT, by sonication and stirring. The resulting solution (ca. 9.4–9.7 M urea) was diluted with 200 mL buffer A with 1 mM DTT (thus yielding ca. 4 M urea) and loaded onto 2 × 20 mL DEAE-sepharose FF columns (GE Healthcare (now Cytiva)) in tandem, equilibrated in buffer A with 4 M urea and 1 mM DTT. The columns were washed with 100 mL buffer A with 4 M urea and 1 mM DTT, and eluted with a 0–0.4 M NaCl gradient in the same buffer. Fractions containing EDDIE-E22G-Aβ1–42 were diluted 15 times with 1 M tris, 1 mM EDTA, 5 mM DTT, pH 7.9 in glass bottles and left at 4 °C for 48 h, total volume 1.0 L. During this time EDDIE slowly folded leading to auto-cleavage and release of E22G-Aβ1–42. The solution was then dialyzed in 3.5 kDa MW cutoff dialysis bags (boiled 4 times in Millipore water before use) against a total of 30 L of 5 mM tris/HCl, 0.5 mM EDTA, pH 8.5, in three shifts (10 L per shift). The dialyzed solution was supplemented with 50 g Q-sepharose big beads (GE Healthcare, equilibrated in buffer A) and incubated for 0.5 h in the cold room with occasional stirring using a glass rod. The beads were collected on a Büchner funnel and washed with 200 mL buffer A. E22G-Aβ1–42 was eluted in buffer A with 50 mM NaCl, 8 fractions of 50 mL each. The fractions were lyophilized, dissolved 2-by-2 in 10 mL 6 M GuHCl and isolated from any residual E. coli proteins, aggregates, and small molecule contaminants by size exclusion chromatography (SEC) in 20 mM sodium phosphate, 0.2 mM EDTA, 0.02% (w/v) NaN3, pH 8.5 using a Superdex 75 26/600 column. The eluted fractions were monitored by UV absorbance and SDS PAGE with Coomassie staining. Fractions corresponding to the center of the E22G-Aβ1–42 monomer peak were pooled in a glass bottle, pH adjusted to 8.0 by adding NaH2PO4 and the pool was incubated quiescently at 37 °C. Monomer from each of the four rounds of SEC were added to the same bottle to propagate the morph formed in the first of the four aliquots.
000g for 7 min between sonication steps. The inclusion body pellet after the 5th sonication was dissolved in 150 mL buffer A with 10 M urea and 1 mM DTT, by sonication and stirring. The resulting solution (ca. 9.4–9.7 M urea) was diluted with 200 mL buffer A with 1 mM DTT (thus yielding ca. 4 M urea) and loaded onto 2 × 20 mL DEAE-sepharose FF columns (GE Healthcare (now Cytiva)) in tandem, equilibrated in buffer A with 4 M urea and 1 mM DTT. The columns were washed with 100 mL buffer A with 4 M urea and 1 mM DTT, and eluted with a 0–0.4 M NaCl gradient in the same buffer. Fractions containing EDDIE-E22G-Aβ1–42 were diluted 15 times with 1 M tris, 1 mM EDTA, 5 mM DTT, pH 7.9 in glass bottles and left at 4 °C for 48 h, total volume 1.0 L. During this time EDDIE slowly folded leading to auto-cleavage and release of E22G-Aβ1–42. The solution was then dialyzed in 3.5 kDa MW cutoff dialysis bags (boiled 4 times in Millipore water before use) against a total of 30 L of 5 mM tris/HCl, 0.5 mM EDTA, pH 8.5, in three shifts (10 L per shift). The dialyzed solution was supplemented with 50 g Q-sepharose big beads (GE Healthcare, equilibrated in buffer A) and incubated for 0.5 h in the cold room with occasional stirring using a glass rod. The beads were collected on a Büchner funnel and washed with 200 mL buffer A. E22G-Aβ1–42 was eluted in buffer A with 50 mM NaCl, 8 fractions of 50 mL each. The fractions were lyophilized, dissolved 2-by-2 in 10 mL 6 M GuHCl and isolated from any residual E. coli proteins, aggregates, and small molecule contaminants by size exclusion chromatography (SEC) in 20 mM sodium phosphate, 0.2 mM EDTA, 0.02% (w/v) NaN3, pH 8.5 using a Superdex 75 26/600 column. The eluted fractions were monitored by UV absorbance and SDS PAGE with Coomassie staining. Fractions corresponding to the center of the E22G-Aβ1–42 monomer peak were pooled in a glass bottle, pH adjusted to 8.0 by adding NaH2PO4 and the pool was incubated quiescently at 37 °C. Monomer from each of the four rounds of SEC were added to the same bottle to propagate the morph formed in the first of the four aliquots.
The formation of fibrils was validated for a small sample using thioflavin T fluorescence in a PerkinElmer LS50B fluorescence spectrometer, as compared with the thioflavin T fluorescence in buffer. The morphology of the fibrils, as revealed by cryo-TEM, is indistinguishable from that of the wild type peptide, displaying a double-filament structure with a short twist distance between the apparent cross-over points.16
We also purified one aliquot of E22G-Aβ1–42 after expression of NT*-E22G-Aβ1–42 (synthetic gene with E. coli-preferred codons in pET3a plasmid purchased from Genscript) in E. coli BL21 DE3 PlysS star in 1 L M9 minimal medium with 13C-glucose and 15NH4Cl as the sole carbon and nitrogen sources using the published protocol.63 After the final SEC, we mixed this aliquot into the same solution as described above.
The samples were finally packed into 0.7 mm diameter rotors using ultracentrifugation. Tryptic digest and intact mass spectrometry, as described in the ESI† (Fig. S1, S2 and Table S1), were used to confirm the desired sequence with 97.3% isotopic enrichment and absence of Aβ1–42 wild type fibrils. The 0.7 mm rotors used to record the E22G spectra contain 0.5 mg peptide.
2.3 Magic angle spinning nuclear magnetic resonance
1H detected spectra were acquired at a static field of 18.8 T (800 MHz for 1H) with a three channel (HCN) 0.7 mm Bruker MAS probe. The spinning frequency of ωr/2π = 90 kHz was regulated using a MAS 3 controller and temperature was maintained at 273 K using a Bruker cooling unit. The Rabi fields (and 90° pulse durations) for 1H, 13C, and 15N were 133 kHz (1.875 μs), 83 kHz (3 μs), and 83 kHz (3 μs), respectively.Dipolar based CP-HSQC spectra were recorded using the pulse sequence previously described using the zero-quantum cross polarization condition at nutation frequencies near (5ωr/4) and (ωr/4), for 1H and 15N or 13C, respectively.44
For the hCCH experiments, the 1H–13C CP was performed using a tangential ramp on the proton RF power and a linear 10% ramp was used for the 13C–1H CP. For hCCH-TOCSY, WALTZ-16 was used with an RF field of ∼22.5 kHz corresponding to (ωr/4). For hCCH-RFDR experiments, illustrated in Fig. 1, the π-pulse width was 2.5 μs corresponding to an RF field of 200 kHz. At ωr/2π > 30 kHz, 1H decoupling is not required during the RFDR mixing period as the mixing pulses recouple and decouple simultaneously.53 Spectra were apodised using 60° shifted squared sine bells or Gaussian apodization and zero filled to at least twice the number of points in the indirect dimension. Details of the acquisition parameters are provided in Table S2 (ESI†).
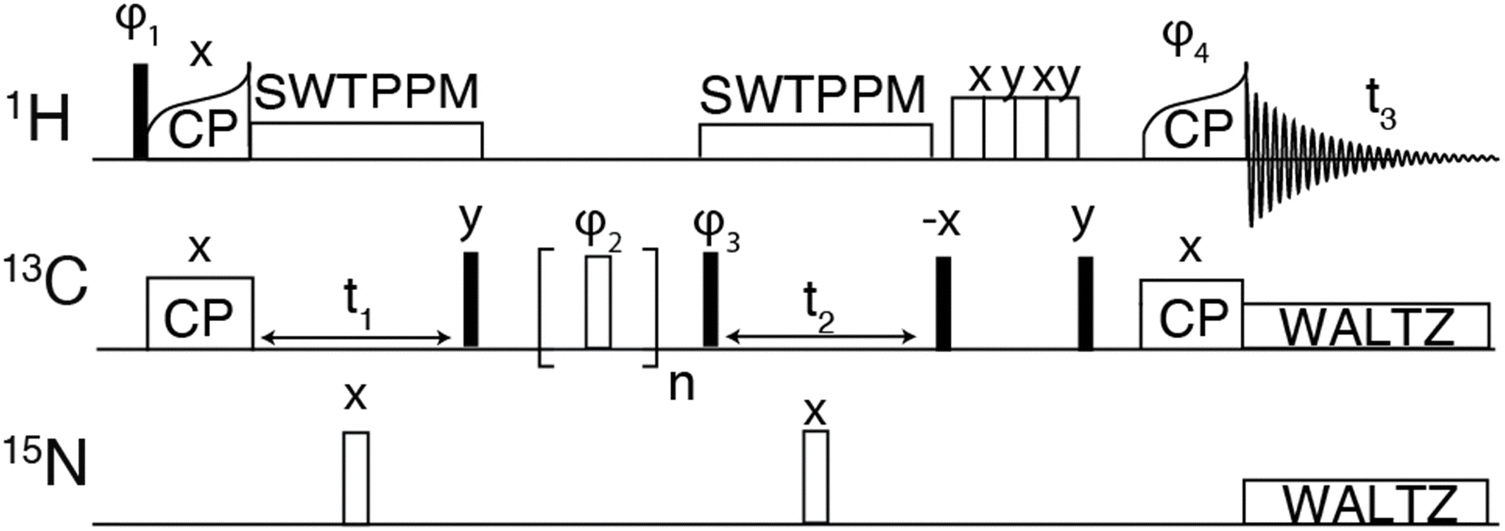 | ||
| Fig. 1 Pulse sequence for 3-dimensional hCCH experiment using RFDR for carbon–carbon mixing. The phase cycle φ1 was [y, −y], φ2 was XY-32[x, y, x, y, y, x, y, x, y, −x, y,−x, −x, y, −x, y, −x, −y, −x, −y, −y, −x, −y, −x, −y, x, −y, x, x, −y, x, −y], φ3 was [x, x, −x, −x], φ4 was [4*x, 4*−x]. The receiver phase cycle was [x, −x, −x, x, −x, x, x, −x]. | ||
In all 1H detected experiments, swept-low power TPPM (SW-TPPM) at an RF field amplitude of ω1H/2π ∼ 24 kHz was used for proton decoupling during t1 and t2, and WALTZ-16 was applied to 13C and 15N at ω1H/1C/2π = 10 kHz, respectively.64,65 Water suppression was achieved through the MISSISSIPPI pulse sequence without homospoil gradients,66 using a 24 kHz RF field and 200 ms pulse duration. Conventional hCC-RFDR spectra were acquired at a field of 18.8 T (800 MHz for 1H) with a three channel (HCN) 3.2 mm Bruker MAS probe at a MAS rate of 20 kHz and a temperature of 273 K. For the 3.2 mm probe, Rabi fields were 83 kHz for 1H and 13C. RFDR mixing was performed using a 180° pulse width of ∼6 μs and a CW proton decoupling field of 83 kHz.
3. Results and discussion
In order to validate the pulse experiment shown in Fig. 1, we use the amyloidogenic peptide GNNQQNY which is part of the terminal domain of Sup35p, an amyloid forming yeast prion protein.56 Early studies found the monoclinic microcrystals of GNNQQNY, as used herein, have amyloidogenic properties including binding of Congo red and formation of β-sheet and steric zipper structures. High resolution X-ray crystal structures have been obtained, in addition to 13C and 15N assignments via MAS NMR.57,67–70 The monoclinic crystals are rigid and offer an amyloidogenic system with which to optimize high resolution protein spectroscopic methods.The 1H–13C and 1H–15N heteronuclear correlation spectra of monoclinic crystals of GNNQQNY and the associated proton assignments are shown in Fig. 2. These are the first 1H detected experiments of GNNQQNY and linewidths of 0.33 ppm and 0.27 ppm were obtained for the Cα proton of N12 and the NH proton of N9, respectively. The degeneracy of amino acids in this sample results in spectral overlap, reducing the effective resolution of many resonances. Typically, proton linewidths were less than 0.3 ppm for individual resonances, where all linewidths are reported without apodization. The narrow line shapes in spectra of the fully protonated sample are indicative of the highly ordered crystal structure of GNNQQNY, while further reduction in linewidth is expected with application of higher MAS rates.
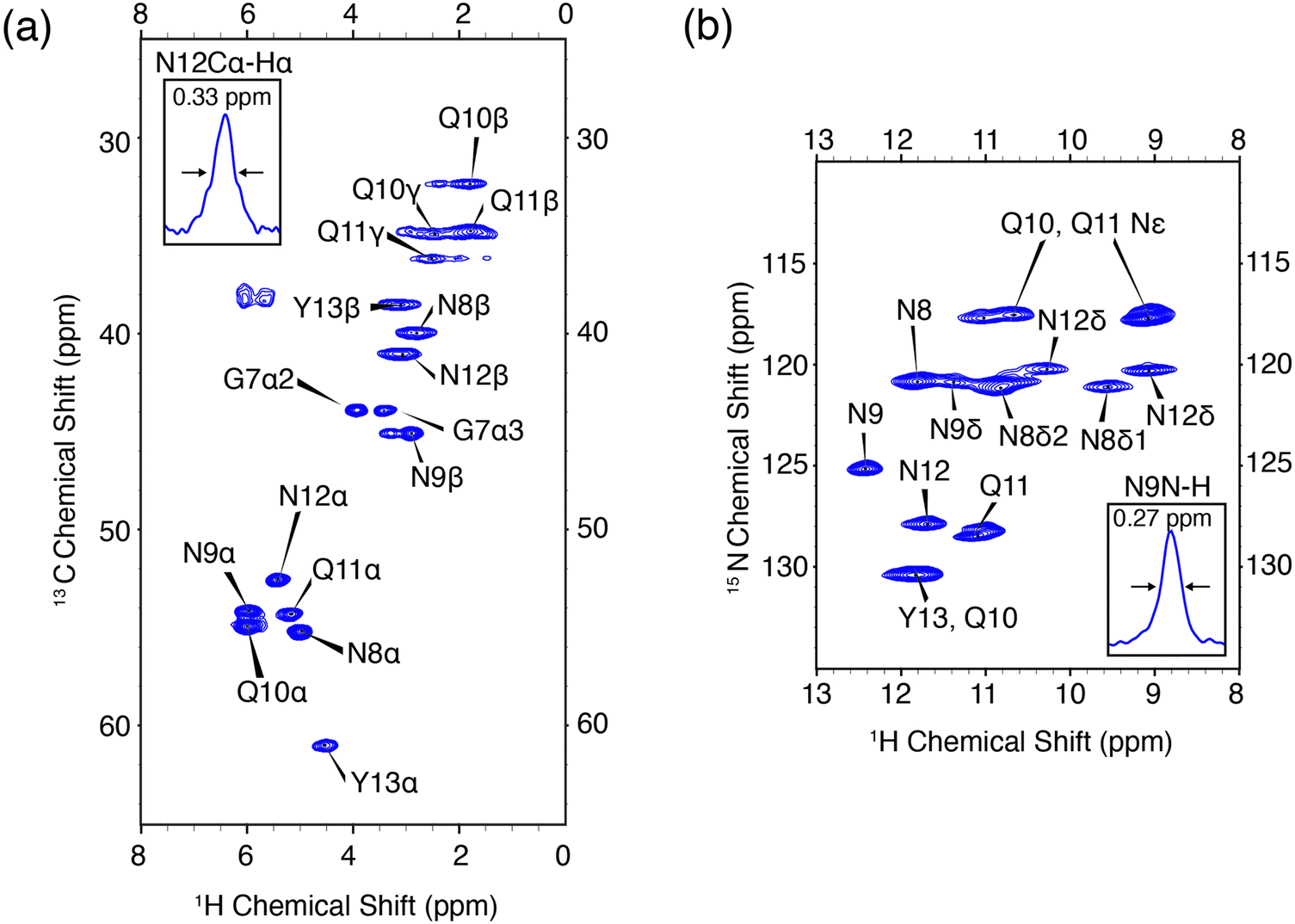 | ||
| Fig. 2 High resolution 1H detected CP-HSQC spectra of nanocrystalline U-13C,15N-GNNQQNY at ωr/2π = 90 kHz, ω0H/2π = 800 MHz. (a) 1H–13C correlations. (b) 1H–15N correlations. The mixing times and other spectral acquisition parameters are provided in Table S2 (ESI†). | ||
The hCH experiment was then used as a building block for the hCCH-RFDR experiment initially implemented on GNNQQNY, using the pulse sequence shown in Fig. 1. A MAS frequency of ωr/2π = 90 kHz and ω1C/2π = 200 kHz RFDR pulses were sufficient to recouple 13C–13C and decouple 1H–13C. Thus, conventional 1H decoupling during the RFDR mixing period was not required. At a MAS frequency of ωr/2π = 90 kHz, the theoretical description describing RFDR requires accounting for the finite pulse effects, given that the 2.5 μs 180° pulse used occupies a considerable fraction of the 11.11 μs rotor period. For a further description of finite pulse RFDR, we refer the reader to the original paper by Bennett, et al. where finite pulses were initially discussed.47
As shown in Fig. 3(A), the hCCH-RFDR at ωr/2π = 90 kHz reproduces all intra-residue correlations that are expected in a one-bond RFDR. While numerous other heteronuclear and homonuclear correlation methods have been developed for 1H detected MAS NMR, the direct comparison of hCCH to hCC spectra acquired at 20 kHz MAS is a valuable spectroscopic tool for spectral fingerprinting of limited quantity samples. However, we note that in the hCCH-RFDR spectrum, the very weakly detected inter-residue correlations of N12Cα-Y13Cα and G7Cα-Q10Cα were not visible. These longer-range correlations may be more effectively probed at high spinning frequencies by other homonuclear correlation schemes or with additional transients per point. In the present spectrum, 8 scans per point were acquired for the hCCH-RFDR leading to a total experiment time of 2 days and 15 hours.
 | ||
| Fig. 3 hCCH-RFDR spectra of nanocrystalline GNNQQNY. (a) The 13C–13C correlations of hCCH-RFDR (red) and hCC RFDR (blue) of U[13C,15N]-GNNQQNY crystals. The hCCH-RFDR spectrum was acquired at ωr/2π = 90 kHz using a mixing time of 1.7 ms. The conventional 2-dimensional hCC-RFDR spectrum was acquired at ωr/2π = 20 kHz MAS using a mixing time of 1.6 ms. The N9Cβ-Cγ* and G7Cα-C0* peaks are folded. (b) Signal intensity as a function of RFDR mixing time for hCCH-RFDR. For each contact in I–IV, the normalized signal intensity is plotted. | ||
As shown in Fig. 3(B), most residues have achieved maximum signal intensity between 1.7 and 2.5 ms, which is comparable to optimal mixing times at lower MAS frequencies, such as ωr/2π = 20 kHz. The longest optimal mixing times are observed for the G7 and Y13 residues, which are excluded from the steric zipper core interface between peptide dimers. Thus, the hCCH-RFDR detected spectra of GNNQQNY at high resolution permitted us to optimize spectral fingerprinting and determine optimal mixing times, which are comparable to those canonically used at lower MAS frequencies.
We then employed the 1H detected experiments to characterize E22G-Aβ1–42 by MAS NMR. As detailed above, the E22G-Aβ1–42 mutant was expressed using a fusion construct with the EDDIE mutant of Npro expressed in E. Coli for uniform 13C and 15N isotopic enrichment. However, due to the very high surface activity of the free peptide, only limited sample quantities could be purified after cleveage, and as such, proton detected MAS NMR were essential for reasons of sensitivity. The expression protocol was previously validated with for example wild type Aβ1–4260,71 and cryo-TEM revealed the morphology of the E22G-AβM1–42 fibrils to be indistinguishable from the wild type AβM1–42 fibrils, with both fibrils displaying a double filament structure with short twist distance between apparent cross over points.16
CP-HSQC spectra were acquired for E22G-Aβ1–42 and illustrated in Fig. 4. The hCH spectra showed only small chemical shift perturbations from the previously published wild type AβM1–42 fibril spectra.38 We note an overall reduction in the spectral resolution of the E22G sample acquired at ωr/2π = 90 kHz and ω1H/2π = 800 MHz, relative to the wild type sample at ωr/2π = 111 kHz and ω0/2π = 1 GHz.38 Two resonances were assigned to the A42 13Cα in both the E22G spectrum (Fig. 4) and the previously studied wild type sample.23,72 This peak doubling, which is attributed to two conformations of the A42 residue, was observed in wild type AβM1–42 only at ωr/2π = 111 kHz and was not observed at ωr/2π = 20 kHz. E22G-Aβ1–42 cross peak assignments (Fig. 4) were rapidly obtained due to small chemical shift perturbations (Fig. 6) in the hCH spectra of E22G-Aβ1–42 relative to previously obtained assignments of wild type samples. The shared presence of two conformations of the C-terminal segment in both wild type and E22G-Aβ1–42 initially suggested a conserved C-terminal structural fold. There are several unassigned, narrow resonances that could also be attributable to dynamic residues in the N-terminal segment. These observations underscore the importance of developing high spinning frequency MAS spectroscopy alone, or in combination with low temperature MAS, to study the N- and C-terminal segment structures and dynamics.
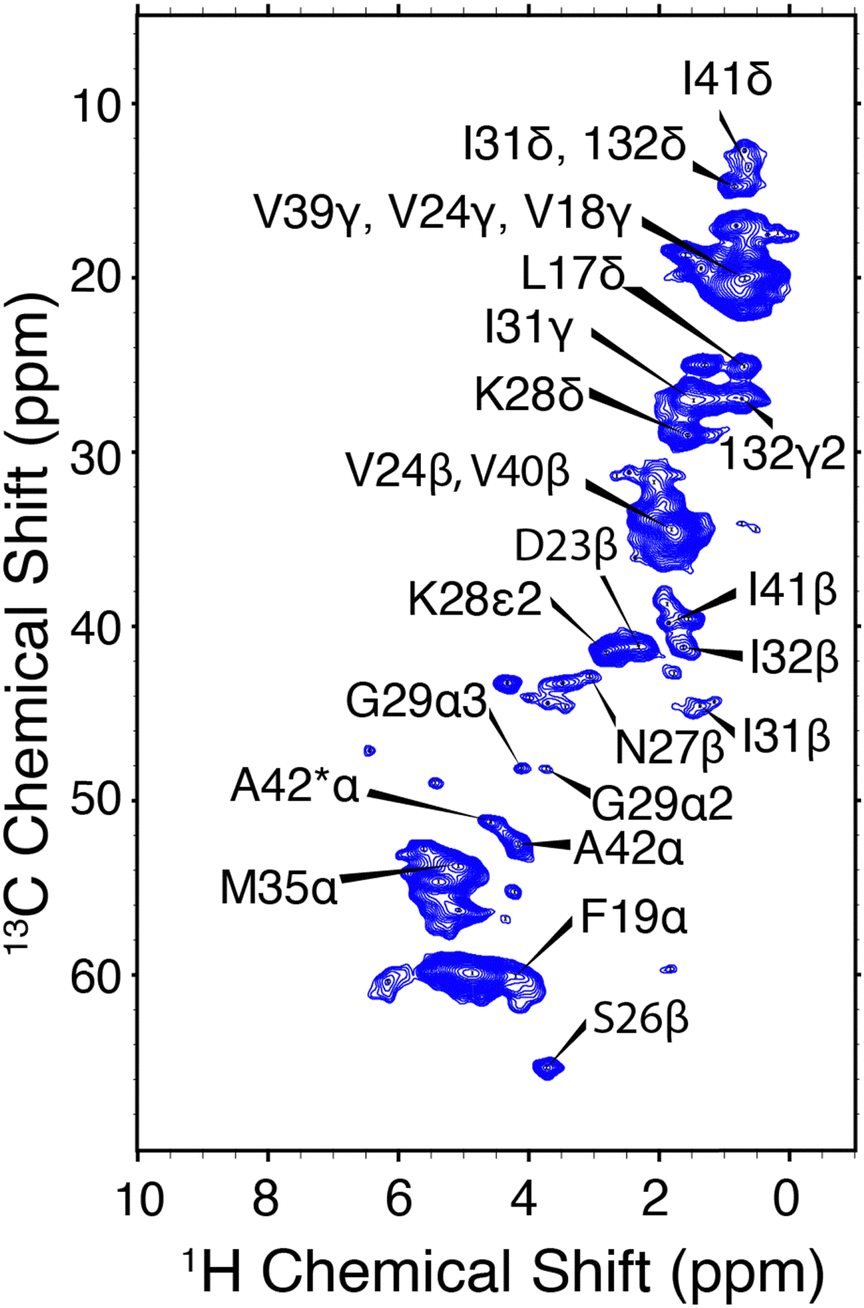 | ||
| Fig. 4 hCH correlations of E22G-Aβ1–42 amyloid fibrils. The assigned chemical shifts are consistent with a structure similar to Aβ1–42. ωr/2π = 90 kHz and ω0H/2π = 800 MHz. Mixing times and other spectral acquisition parameters are tabulated in Table S2 (ESI†). | ||
The hNH spectrum of E22G-Aβ1–42 (Fig. S3, ESI†) shows a reduction in resolution, the only cross peak that can presently be unambiguously assigned being due to A42. The lower resolution of the 15N spectral dimension has been observed previously in Aβ fibrils,54 and is likely due to structural heterogeneity, which results from the rapid fibril formation; thus, many studies of Aβ amyloid do not report 15N spectra. It is possible that polymorphism, sample impurities, and dynamics also contribute to the broadening.
To probe the role of potential polymorphism beyond the likely structural doubling of A42, we employed the hCCH-RFDR and hCCH-TOCSY experiments. hCCH-RFDR offers a direct spectral comparison to the wealth of previously acquired hCC spectra of WT Aβ1–42 in the literature.22,23,72 Despite the common occurrence of hCC-RFDR, the hCCH-RFDR has to date been sparsely applied in fully protonated samples54,73 or in selectively deuterated samples.74 Instead, RFDR is most often incorporated into hCHH or hNHH pulse sequences which offer increased sensitivity and the potential for longer range quantitative 1H–1H distance measurements.38,39,45,75 However, in fully protonated samples the continued development of high spinning frequency MAS rotors and pulse sequence methodology is required.
A comparison between the hCCH-RFDR and hCCH-TOCSY spectra is illustrated in Fig. 5. The TOCSY based hCCH was described previously, with mixing largely mediated by 13C–13C scalar couplings43 and is based on total through bond correlation pulse sequences.76 As such, MAS rates ωr/2π > 90 kHz are expected to improve the performance of the pulse sequence on a dynamic sample such as the E22G-Aβ1–42 fibrils. Because it relies on dipolar rather than J-couplings the performance of RFDR is expected to be superior to that of TOCSY when a large chemical shift difference is present. Thus, in Fig. 5 we observe more cross peaks with the hCCH-RFDR experiment. For both pulse sequences, the C–C projection of the hCCH experiment displays improved resolution over the hCH detected experiments, although site specific resolution is still not achieved with 2D spectra in this sample. Thus, higher MAS frequencies, access to higher magnetic fields and higher dimensional spectroscopy remain critical steps for complete structural studies of pathologically relevant Aβ fibrils. Despite these limitations, several important preliminary biological conclusions can be drawn using the spectra presented here.
 | ||
| Fig. 5 13C–13C hCCH spectra of E22G-Aβ1–42 amyloid fibrils using TOCSY (red) or RFDR (blue) for carbon homonuclear mixing. Mixing times and other spectral acquisition parameters are tabulated in Table S2 (ESI†). | ||
In Aβ1–42 fibrils serine residues, S8 and S26, were previously shown to be sensitive to polymorphism.22,72 Specifically, when polymorphs are present, then the Cα–Cβ cross region (∼55/57 ppm in Fig. 5) shows multiple peaks. In the present E22G-Aβ1–42 sample, the S26 Cα–Cβ correlation was observed as a single resonance, with ∼1 ppm in chemical shift difference relative to wild type Aβ1–42. Aside from the structural doubling of A42Cα, no further resonance doubling was observed. Compared to wild type Aβ1–42 RFDR fingerprint spectra, E22G-Aβ1–42 is similarly monomorphic. This observation is further supported by the single S26β hCH cross peak present in Fig. 4.
In a previous study17 of E22G-Aβ1–40, which employed a sample prepared synthetically, rather than biosynthetically, six specific residues (F19,A21, I32, L34, V36, and G38) in the core of the fibril structure were 13C/15N labeled. The spectra were well resolved and clearly showed four different cross peaks for the labeled residues. Although it is difficult to discern the reasons for the polymorphism in samples used in other studies, two factors, the purity of synthetic samples and the possibility of racemization of the three His residues in Aβ, are discussed in the literature.77–79 And in a separate investigation,18 it was shown that folding of E22G-Aβ1-40 fibrils could be enhanced with seeds of Aβ1–42, whereas WT Aβ1–40 was not affected. The spectra in these experiments obtained from synthetic samples (labeled as G22, V24, A30 and I31) displayed two-fold polymorphism in two of the four selectively labeled sites. Thus, polymorphism is observed in synthetic Aβ1–40 samples, e.g. E22G-Aβ1–40, whereas we did not detect it in E22G-Aβ1–42 prepared using biosynthetic methods. Nevertheless, these experiments suggested the dominant form of the Arctic mutant may adopt β-strand conformations similar to the wild type Aβ1–42, and this hypothesis is further supported by the MAS NMR data presented here.
Another notable feature in the present E22G-Aβ1–42 is the minimal chemical shift perturbations associated with the K28 residue (∼0.2 ppm), suggesting that the K28-A42 salt bridge observed previously in wild type Aβ1–42 is likely conserved in E22G-Aβ1–42. A detailed compilation of the shifts in E22G-Aβ1–42 and Aβ1–42 is presented in Tables S3 and S4 (ESI†) In addition, the 15N–1H correlation spectra (Fig. S3, ESI†) also show a doublet at (135.5 ppm,9.5 ppm) in excellent agreement with the shifts observed in Aβ1–42.23,72 The figure allows a comparison of the NH spectra of E22G-Aβ1–42 with those of WT Aβ1–42. Similar salt bridges are present in the E22Δ-Aβ1–39 mutant structure linking E3 and K28.15 We do note that the largest observed chemical shift perturbations occur in adjacent residues S26 and N27, but they remained limited to ∼1 ppm. This supports the hypothesis that the mutation conserves fibril core features while inducing electrostatic perturbations that would make the wild-type fibril fold even more stable for the E22G mutant due to reduced electrostatic repulsion.16 As for the wild-type, the main driving force for fibril formation of E22G-Aβ1–42 are thus hydrophobic interactions due to burial of a large number of non-polar residues in the fibril core and the associated increase in entropy of the released water molecules.16 Overall, the core residues of the E22G-Aβ1–42 fibril display minimal chemical shift perturbations, on the order of ≤1 ppm, as illustrated in Fig. 6.
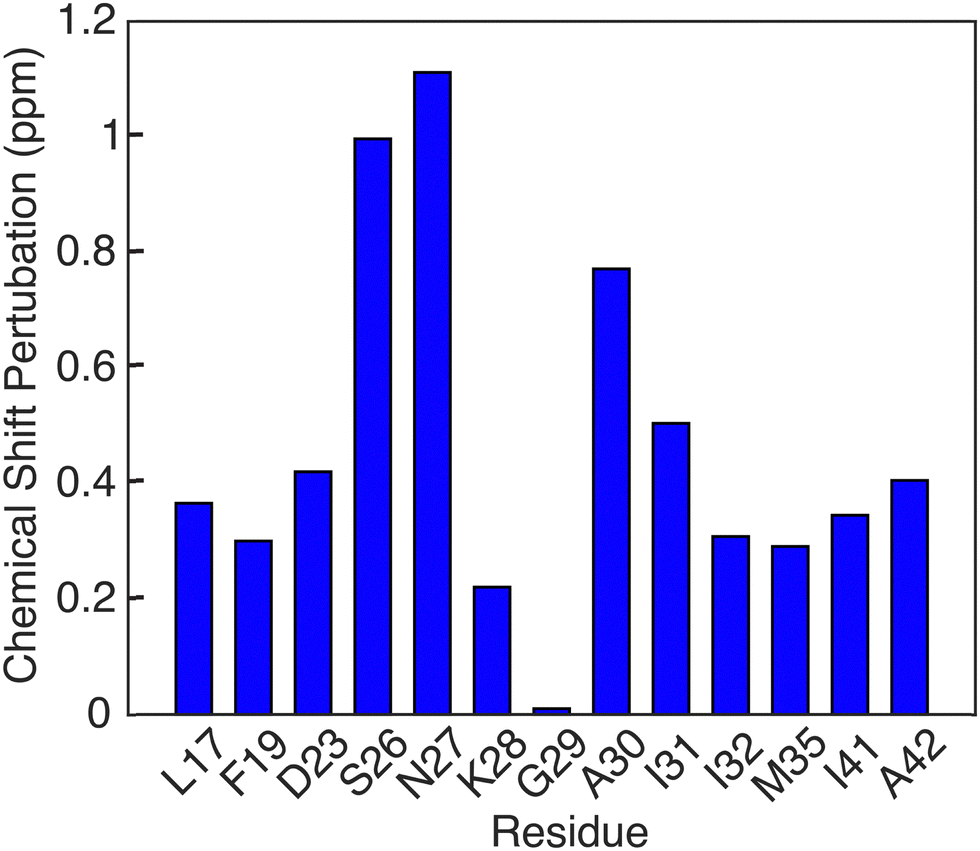 | ||
| Fig. 6 Chemical shift pertubations of E22G Aβ1–42 fibrils relative to wild type Aβ1–42 fibrils found in [ref. 23, 38, 50]. | ||
4. Conclusion
We present the initial 1H-detected MAS NMR results on two amyloidogenic systems, GNNQQNY monoclinic crystals and E22G-Aβ1–42 fibrils. Linewidths typically <0.3 ppm were observed in CP-HSQC spectra of the rigid peptide GNNQQNY. This high-resolution model system was employed to optimize hCCH-RFDR experiments, and an optimal mixing period in this system was observed to be ∼2 ms, on par with mixing periods used at lower MAS frequencies. These methods were then extended to the study of E22G-Aβ1–42 fibrils. The spectral resolution was reduced relative to wild type Aβ1–42, indicating an increase in sample heterogeneity perhaps due to the rate at which the fibrils are formed. The use of MAS at ωr/2π = 90 kHz confirmed the observation of dynamics in the A42 residue as observed also in wild type samples, indicating the C-terminus is not strongly modulated by the E22G mutation. hCCH-RFDR demonstrated the sample to be largely monomorphic, in contrast to studies of synthetic E22G-Aβ1–40 in which multiple polymorphs were observed. Overall, the chemical shift perturbations observed across numerous residues are <1 ppm between the previously reported wild type Aβ1–42 and the present E22G-Aβ1–42. This result suggests a conserved core fibril structure, in accordance with recent cryoEM results. Taken as whole, this study presents further evidence that the Arctic mutant has a propensity for adopting Aβ1–42 like fibril features as previously observed in Aβ1–40 fibrils of E22G mutant. In the future, the use of dynamic nuclear polarization and access to increased MAS rates and higher fields could aid in the determination of the complete three-dimensional structure of the sample quantity limited E22G mutant of Aβ1–42.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Salima Bahri and Ravi Shankar Palani for helpful conversations regarding this project. This project is funded by the National Institutes of Health under grants AG058504 and GM132997 to RGG, and by the Swedish Research Council, VR 2015-00143 to SL. NCG is supported by the National Science Foundation Graduate Research Fellowship under Grant No. 1744302. EPS is supported by an NIH F32 fellowship under award number 1 F32 GM139304-01. We thank members of the MIT Koch institute for assistance with GNNQQNY synthesis and characterization of the E22G-Aβ sample by mass spectrometry.References
- Alzheimer’s Association, 2022 Alzheimer’s Disease Facts and Figures, Alzheimer’s Dementia, 2022, 18, 700–789 CrossRef PubMed.
- D. Eisenberg and M. Jucker, The Amyloid State of Proteins in Human Diseases, Cell, 2012, 148, 1188–1203 CrossRef CAS PubMed.
- H. Hampel, J. Hardy, K. Blennow, C. Chen, G. Perry, S. H. Kim, V. L. Villemagne, P. Aisen, M. Vendruscolo, T. Iwatsubo, C. L. Masters, M. Cho, L. Lannfelt, J. L. Cummings and A. Vergallo, The Amyloid-B Pathway in Alzheimer's Disease, Mol. Psychiatry, 2021, 26, 5481–5503 CrossRef CAS PubMed.
- M. P. Murphy and H. LeVine Iii, Alzheimer's Disease and the Amyloid-B Peptide, J. Alzheimer's Dis., 2010, 19, 311–323 Search PubMed.
- T. Oltersdorf, P. Ward, T. Henriksson, E. Beattie, R. Neve, I. Lieberburg and L. Fritz, The Alzheimer Amyloid Precursor Protein. Identification of a Stable Intermediate in the Biosynthetic/Degradative Pathway, J. Biol. Chem., 1990, 265, 4492–4497 CrossRef CAS.
- D. J. Selkoe, Alzheimer's Disease: Genes, Proteins, and Therapy, Physiol. Rev., 2001, 81, 741–766 CrossRef CAS PubMed.
- T. Tomiyama and H. Shimada, App Osaka Mutation in Familial Alzheimer’s Disease-Its Discovery, Phenotypes, and Mechanism of Recessive Inheritance, Int. J. Mol. Sci., 2020, 21, 1413 CrossRef CAS PubMed.
- A. E. Roher, J. D. Lowenson, S. Clarke, A. S. Woods, R. J. Cotter, E. Gowing and M. J. Ball, Beta-Amyloid-(1–42) Is a Major Component of Cerebrovascular Amyloid Deposits: Implications for the Pathology of Alzheimer Disease, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 10836–10840 CrossRef CAS PubMed.
- G. Brinkmalm, W. Hong, Z. M. Wang, W. Liu, T. T. O’Malley, X. Sun, M. P. Frosch, D. J. Selkoe, E. Portelius, H. Zetterberg, K. Blennow and D. M. Walsh, Identification of Neurotoxic Cross-Linked Amyloid-B Dimers in the Alzheimer's Brain, Brain, 2019, 142, 1441–1457 CrossRef PubMed.
- C. Nilsberth, A. Westlind-Danielsson, C. B. Eckman, M. M. Condron, K. Axelman, C. Forsell, C. Stenh, J. Luthman, D. B. Teplow, S. G. Younkin, J. Naslund and L. Lannfelt, The ‘Arctic’ App Mutation (E693g) Causes Alzheimer's Disease by Enhanced a Beta Protofibril Formation, Nat. Neurosci., 2001, 4, 887–893 CrossRef CAS PubMed.
- K. Kamino, H. T. Orr, H. Payami, E. M. Wijsman, M. E. Alonso, S. M. Pulst, L. Anderson, S. Odahl, E. Nemens, J. A. White, A. D. Sadovnick, M. J. Ball, J. Kaye, A. Warren, M. McInnis, S. E. Antonarakis, J. R. Korenberg, V. Sharma, W. Kukull, E. Larson, L. L. Heston, G. M. Martin, T. D. Bird and G. D. Schellenberg, Linkage and Mutational Analysis of Familial Alzheimer-Disease Kindreds for the App Gene Region, Am. J. Hum. Genet., 1992, 51, 998–1014 CAS.
- O. Bugiani, G. Giaccone, G. Rossi, M. Mangieri, R. Capobianco, M. Morbin, G. Mazzoleni, C. Cupidi, G. Marcon and A. Giovagnoli, Hereditary Cerebral Hemorrhage with Amyloidosis Associated with the E693k Mutation of App, Arch. Neurol., 2010, 67, 987–995 CrossRef PubMed.
- T. J. Grabowski, H. S. Cho, J. P. G. Vonsattel, G. W. Rebeck and S. M. Greenberg, Novel Amyloid Precursor Protein Mutation in an Iowa Family with Dementia and Severe Cerebral Amyloid Angiopathy, Ann. Neurol., 2001, 49, 697–705 CrossRef CAS PubMed.
- C. Van Broeckhoven, J. Haan, E. Bakker, J. Hardy, W. Van Hul, A. Wehnert, M. Vegter-Van der Vlis and R. Roos, Amyloid B Protein Precursor Gene and Hereditary Cerebral Hemorrhage with Amyloidosis (Dutch), Science, 1990, 248, 1120–1122 CrossRef CAS PubMed.
- A. K. Schütz, T. Vagt, M. Huber, O. Y. Ovchinnikova, R. Cadalbert, J. Wall, P. Güntert, A. Böckmann, R. Glockshuber and B. H. Meier, Atomic-Resolution Three-Dimensional Structure of Amyloid B Fibrils Bearing the Osaka Mutation, Angew. Chem., Int. Ed., 2015, 54, 331–335 CrossRef PubMed.
- X. Yang, G. Meisl, B. Frohm, E. Thulin, T. P. J. Knowles and S. Linse, On the Role of Sidechain Size and Charge in the Aggregation of Aβ42 with Familial Mutations, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5849–E5858 CrossRef CAS PubMed.
- M. R. Elkins, T. Wang, M. Nick, H. Jo, T. Lemmin, S. B. Prusiner, W. F. DeGrado, J. Stohr and M. Hong, Structural Polymorphism of Alzheimer's Beta-Amyloid Fibrils as Controlled by an E22 Switch: A Solid-State Nmr Study, J. Am. Chem. Soc., 2016, 138, 9840–9852 CrossRef CAS PubMed.
- B. K. Yoo, Y. Xiao, D. McElheny and Y. Ishii, E22g Pathogenic Mutation of B-Amyloid (Aβ) Enhances Misfolding of Aβ40 by Unexpected Prion-Like Cross Talk between Aβ42 and Aβ40, J. Am. Chem. Soc., 2018, 140, 2781–2784 CrossRef PubMed.
- Y. Yang, W. J. Zhang, A. G. Murzin, M. Schweighauser, M. Huang, S. Lovestam, S. Y. Peak-Chew, T. Saito, T. C. Saido, J. Macdonald, I. Lavenir, B. Ghetti, C. Graff, A. Kumar, A. Nordberg, M. Goedert and S. H. W. Scheres, Cryo-Em Structures of Amyloid-Beta Filaments with the Arctic Mutation (E22g) from Human and Mouse Brains, Acta Neuropathol., 2023, 145, 325–333 CrossRef CAS PubMed.
- C. Leistner, M. Wilkinson, A. Burgess, M. Lovatt, S. Goodbody, Y. Xu, S. Deuchars, S. E. Radford, N. A. Ranson and R. A. W. Frank, The in-Tissue Molecular Architecture of B-Amyloid Pathology in the Mammalian Brain, Nat. Commun., 2023, 14, 2833 CrossRef CAS PubMed.
- P. H. Nguyen, A. Ramamoorthy, B. R. Sahoo, J. Zheng, P. Faller, J. E. Straub, L. Dominguez, J. E. Shea, N. V. Dokholyan, A. De Simone, B. Y. Ma, R. Nussinov, S. Najafi, S. T. Ngo, A. Loquet, M. Chiricotto, P. Ganguly, J. McCarty, M. S. Li, C. Hall, Y. M. Wang, Y. Miller, S. Melchionna, B. Habenstein, S. Timr, J. X. Chen, B. Hnath, B. Strodel, R. Kayed, S. Lesné, G. H. Wei, F. Sterpone, A. J. Doig and P. Derreumaux, Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer's Disease, Parkinson's Disease, Type Ii Diabetes, and Amyotrophic Lateral Sclerosis, Chem. Rev., 2021, 121, 2545–2647 CrossRef CAS PubMed.
- M. A. Wälti, F. Ravotti, H. Arai, C. G. Glabe, J. S. Wall, A. Böckmann, P. Güntert, B. H. Meier and R. Riek, Atomic-Resolution Structure of a Disease-Relevant Aβ(1–42) Amyloid Fibril, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4976–E4984 CrossRef PubMed.
- M. T. Colvin, R. Silvers, Q. Z. Ni, T. V. Can, I. Sergeyev, M. Rosay, K. J. Donovan, B. Michael, J. Wall, S. Linse and R. G. Griffin, Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils, J. Am. Chem. Soc., 2016, 138, 9663–9674 CrossRef CAS PubMed.
- Y. Nishiyama, G. J. Hou, V. Agarwal, Y. C. Su and A. Ramamoorthy, Ultrafast Magic Angle Spinning Solid-State Nmr Spectroscopy: Advances in Methodology and Applications, Chem. Rev., 2023, 123, 918–988 CrossRef CAS PubMed.
- A. E. Bennett; R. G. Griffin and S. Vega, Recoupling of Homo- and Heteronuclear Dipolar Interactions in Rotating Solids, inSolid-State Nmr Iv Methods and Applications of Solid-State Nmr, ed. B. Blümich, Springer Berlin Heidelberg, Berlin, Heidelberg, 1994, pp.1–77 Search PubMed.
- R. Griffin, Dipolar Recoupling in Mas Spectra of Biological Solids, Nat. Struct. Biol., 1998, 5, 508–512 CrossRef CAS PubMed.
- Y. Ji, L. Liang, X. Bao and G. Hou, Recent Progress in Dipolar Recoupling Techniques under Fast Mas in Solid-State Nmr Spectroscopy, Solid State Nucl. Magn. Reson., 2021, 112, 101711 CrossRef CAS PubMed.
- Y. Su, L. Andreas and R. G. Griffin, Magic Angle Spinning Nmr of Proteins: High-Frequency Dynamic Nuclear Polarization and 1h Detection, Annu. Rev. Biochem., 2015, 84, 465–497 CrossRef CAS PubMed.
- T. Lührs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Döbeli, D. Schubert and R. Riek, 3d Structure of Alzheimer's Amyloid-B(1–42) Fibrils, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef PubMed.
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Aβ(1–42) Fibril Structure Illuminates Self-Recognition and Replication of Amyloid in Alzheimer's Disease, Nat. Struct. Mol. Biol., 2015, 22, 499–505 CrossRef CAS PubMed.
- J. R. Long, B. Q. Sun, A. Bowen and R. G. Griffin, Molecular-Dynamics and Magic-Angle-Spinning Nmr, J. Am. Chem. Soc., 1994, 116, 11950–11956 CrossRef CAS.
- Q. Z. Ni, E. Markhasin, T. V. Can, B. Corzilius, K. O. Tan, A. B. Barnes, E. Daviso, Y. C. Su, J. Herzfeld and R. G. Griffin, Peptide and Protein Dynamics and Low-Temperature/Dnp Magic Angle Spinning Nmr, J. Phys. Chem. B, 2017, 121, 4997–5006 CrossRef CAS PubMed.
- V. Lattanzi, I. Andre, U. Gasser, M. Dubackic, U. Olsson and S. Linse, Amyloid Beta 42 Fibril Structure Based on Small-Angle Scattering, Proc. Natl. Acad. Sci. U. S. A., 2021, 118 Search PubMed.
- L. Gremer, D. Scholzel, C. Schenk, E. Reinartz, J. Labahn, R. B. G. Ravelli, M. Tusche, C. Lopez-Iglesias, W. Hoyer, H. Heise, D. Willbold and G. F. Schroder, Fibril Structure of Amyloid-Beta(1–42) by Cryo-Electron Microscopy, Science, 2017, 358, 116–119 CrossRef CAS PubMed.
- M. Kollmer, W. Close, L. Funk, J. Rasmussen, A. Bsoul, A. Schierhorn, M. Schmidt, C. J. Sigurdson, M. Jucker and M. Fändrich, Cryo-Em Structure and Polymorphism of Aβ Amyloid Fibrils Purified from Alzheimer's Brain Tissue, Nat. Commun., 2019, 10, 4760 CrossRef PubMed.
- A. Bax, S. W. Sparks and D. A. Torchia, Detection of Insensitive Nuclei, Methods Enzymol., 1989, 176, 134–150 CAS.
- T. Le Marchand, T. Schubeis, M. Bonaccorsi, P. Paluch, D. Lalli, A. J. Pell, L. B. Andreas, K. Jaudzems, J. Stanek and G. Pintacuda, 1h-Detected Biomolecular Nmr under Fast Magic-Angle Spinning, Chem. Rev., 2022, 122, 9943–10018 CrossRef CAS PubMed.
- S. Bahri, R. Silvers, B. Michael, K. Jaudzems, D. Lalli, G. Casano, O. Ouari, A. Lesage, G. Pintacuda, S. Linse and R. G. Griffin, 1h Detection and Dynamic Nuclear Polarization–Enhanced Nmr of Aβ1–42 Fibrils, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2114413119 CrossRef CAS PubMed.
- L. B. Andreas, K. Jaudzems, J. Stanek, D. Lalli, A. Bertarello, T. Le Marchand, D. Cala-De Paepe, S. Kotelovica, I. Akopjana, B. Knott, S. Wegner, F. Engelke, A. Lesage, L. Emsley, K. Tars, T. Herrmann and G. Pintacuda, Structure of Fully Protonated Proteins by Proton-Detected Magic-Angle Spinning Nmr, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 9187–9192 CrossRef CAS PubMed.
- A. Loquet, N. El Mammeri, J. Stanek, M. Berbon, B. Bardiaux, G. Pintacuda and B. Habenstein, 3d Structure Determination of Amyloid Fibrils Using Solid-State Nmr Spectroscopy, Methods, 2018, 138, 26–38 CrossRef PubMed.
- J. Struppe, C. M. Quinn, M. M. Lu, M. Z. Wang, G. J. Hou, X. Y. Lu, J. Kraus, L. B. Andreas, J. Stanek, D. Lalli, A. Lesage, G. Pintacuda, W. Maas, A. M. Gronenborn and T. Polenova, Expanding the Horizons for Structural Analysis of Fully Protonated Protein Assemblies by Nmr Spectroscopy at Mas Frequencies above 100 Khz, Solid State Nucl. Magn. Reson., 2017, 87, 117–125 CrossRef CAS PubMed.
- R. Linser, M. Dasari, M. Hiller, V. Higman, U. Fink, J.-M. Lopez del Amo, S. Markovic, L. Handel, B. Kessler, P. Schmieder, D. Oesterhelt, H. Oschkinat and B. Reif, Proton-Detected Solid-State Nmr Spectroscopy of Fibrillar and Membrane Proteins, Angew. Chem., Int. Ed., 2011, 50, 4508–4512 CrossRef CAS PubMed.
- J. Stanek, L. B. Andreas, K. Jaudzems, D. Cala, D. Lalli, A. Bertarello, T. Schubeis, I. Akopjana, S. Kotelovica, K. Tars, A. Pica, S. Leone, D. Picone, Z.-Q. Xu, N. E. Dixon, D. Martinez, M. Berbon, N. El Mammeri, A. Noubhani, S. Saupe, B. Habenstein, A. Loquet and G. Pintacuda, Nmr Spectroscopic Assignment of Backbone and Side-Chain Protons in Fully Protonated Proteins: Microcrystals, Sedimented Assemblies, and Amyloid Fibrils, Angew. Chem., Int. Ed., 2016, 55, 15504–15509 CrossRef CAS PubMed.
- E. Barbet-Massin, A. J. Pell, J. S. Retel, L. B. Andreas, K. Jaudzems, W. T. Franks, A. J. Nieuwkoop, M. Hiller, V. Higman, P. Guerry, A. Bertarello, M. J. Knight, M. Felletti, T. Le Marchand, S. Kotelovica, I. Akopjana, K. Tars, M. Stoppini, V. Bellotti, M. Bolognesi, S. Ricagno, J. J. Chou, R. G. Griffin, H. Oschkinat, A. Lesage, L. Emsley, T. Herrmann and G. Pintacuda, Rapid Proton-Detected Nmr Assignment for Proteins with Fast Magic Angle Spinning, J. Am. Chem. Soc., 2014, 136, 12489–12497 CrossRef CAS PubMed.
- V. Agarwal, S. Penzel, K. Szekely, R. Cadalbert, E. Testori, A. Oss, J. Past, A. Samoson, M. Ernst, A. Böckmann and B. H. Meier, De Novo 3d Structure Determination from Sub-Milligram Protein Samples by Solid-State 100 Khz Mas Nmr Spectroscopy, Angew. Chem., Int. Ed., 2014, 53, 12253–12256 CrossRef CAS PubMed.
- A. E. Bennett, R. G. Griffin, J. H. Ok and S. Vega, Chemical Shift Correlation Spectroscopy in Rotating Solids: Radio Frequency-Driven Dipolar Recoupling and Longitudinal Exchange, J. Chem. Phys., 1992, 96, 8624–8627 CrossRef CAS.
- A. E. Bennett, C. M. Rienstra, J. M. Griffiths, W. Zhen, P. T. Lansbury and R. G. Griffin, Homonuclear Radio Frequency-Driven Recoupling in Rotating Solids, J. Chem. Phys., 1998, 108, 9463–9479 CrossRef CAS.
- T. Gullion and S. Vega, A Simple Magic Angle Spinning Nmr Experiment for the Dephasing of Rotational Echoes of Dipolar Coupled Homonuclear Spin Pairs, Chem. Phys. Lett., 1992, 194, 423–428 CrossRef CAS.
- M. T. Eddy, T.-Y. Yu, G. Wagner and R. G. Griffin, Structural Characterization of the Human Membrane Protein Vdac2 in Lipid Bilayers by Mas Nmr, J. Biomol. NMR, 2019, 73, 451–460 CrossRef CAS PubMed.
- R. Silvers, M. T. Colvin, K. K. Frederick, A. C. Jacavone, S. Lindquist, S. Linse and R. G. Griffin, Aggregation and Fibril Structure of Aβm01–42 and Aβ1–42, Biochemistry, 2017, 56, 4850–4859 CrossRef CAS PubMed.
- L. B. Andreas, M. Reese, M. T. Eddy, V. Gelev, Q. Z. Ni, E. A. Miller, L. Emsley, G. Pintacuda, J. J. Chou and R. G. Griffin, Structure and Mechanism of the Influenza a M218–60 Dimer of Dimers, J. Am. Chem. Soc., 2015, 137, 14877–14886 CrossRef CAS PubMed.
- M. Roos, V. S. Mandala and M. Hong, Determination of Long-Range Distances by Fast Magic-Angle-Spinning Radiofrequency-Driven 19f–19f Dipolar Recoupling Nmr, J. Phys. Chem. B, 2018, 122, 9302–9313 CrossRef CAS PubMed.
- M. J. Bayro, R. Ramachandran, M. A. Caporini, M. T. Eddy and R. G. Griffin, Radio Frequency-Driven Recoupling at High Magic-Angle Spinning Frequencies: Homonuclear Recoupling Sans Heteronuclear Decoupling, J. Chem. Phys., 2008, 128, 052321 CrossRef PubMed.
- A. Wickramasinghe, Y. Xiao, N. Kobayashi, S. Wang, K. P. Scherpelz, T. Yamazaki, S. C. Meredith and Y. Ishii, Sensitivity-Enhanced Solid-State Nmr Detection of Structural Differences and Unique Polymorphs in Pico- to Nanomolar Amounts of Brain-Derived and Synthetic 42-Residue Amyloid-B Fibrils, J. Am. Chem. Soc., 2021, 143, 11462–11472 CrossRef CAS PubMed.
- W. Kaar, K. Ahrer, A. Dürauer, S. Greinstetter, W. Sprinzl, P. Wechner, F. Clementschitsch, K. Bayer, C. Achmüller, B. Auer, R. Hahn and A. Jungbauer, Refolding of Npro Fusion Proteins, Biotechnol. Bioeng., 2009, 104, 774–784 CrossRef CAS PubMed.
- M. Balbirnie, R. Grothe and D. S. Eisenberg, An Amyloid-Forming Peptide from the Yeast Prion Sup35 Reveals a Dehydrated B-Sheet Structure for Amyloid, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2375–2380 CrossRef CAS PubMed.
- P. C. A. van der Wel, J. R. Lewandowski and R. G. Griffin, Solid-State Nmr Study of Amyloid Nanocrystals and Fibrils Formed by the Peptide Gnnqqny from Yeast Prion Protein Sup35p, J. Am. Chem. Soc., 2007, 129, 5117–5130 CrossRef CAS PubMed.
- D. Thacker, K. Sanagavarapu, B. Frohm, G. Meisl, T. P. J. Knowles and S. Linse, The Role of Fibril Structure and Surface Hydrophobicity in Secondary Nucleation of Amyloid Fibrils, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 25272–25283 CrossRef CAS PubMed.
- S. Linse, T. Scheidt, K. Bernfur, M. Vendruscolo, C. M. Dobson, S. I. A. Cohen, E. Sileikis, M. Lundqvist, F. Qian, T. O’Malley, T. Bussiere, P. H. Weinreb, C. K. Xu, G. Meisl, S. R. A. Devenish, T. P. J. Knowles and O. Hansson, Kinetic Fingerprints Differentiate the Mechanisms of Action of Anti-Aβ Antibodies, Nat. Struct. Mol. Biol., 2020, 27, 1125–1133 CrossRef CAS PubMed.
- M. Lindberg, E. Axell, E. Sparr, S. Linse and A. Label-Free High-Throughput, Protein Solubility Assay and Its Application to Abeta40, Biophys. Chem., 2024, 307, 107165 CrossRef CAS PubMed.
- M. Lundqvist, D. C. Rodriguez Camargo, K. Bernfur, S. Chia and S. Linse, Expression, Purification and Characterisation of Large Quantities of Recombinant Human Iapp for Mechanistic Studies, Biophys. Chem., 2021, 269, 106511 CrossRef CAS PubMed.
- R. Ueberbacher, A. Dürauer, K. Ahrer, S. Mayer, W. Sprinzl, A. Jungbauer and R. Hahn, Eddie Fusion Proteins: Triggering Autoproteolytic Cleavage, Process Biochem., 2009, 44, 1217–1224 CrossRef CAS.
- X. Zhong, R. Kumar, Y. Wang, H. Biverstål, C. Ingeborg Jegerschöld, P. J B Koeck, J. Johansson, A. Abelein and G. Chen, Amyloid Fibril Formation of Arctic Amyloid-B 1–42 Peptide Is Efficiently Inhibited by the Brichos Domain, ACS Chem. Biol., 2022, 17, 2201–2211 CrossRef CAS PubMed.
- P. C. van der Wel, J. R. Lewandowski and R. G. Griffin, Structural Characterization of Gnnqqny Amyloid Fibrils by Magic Angle Spinning Nmr, Biochemistry, 2010, 49, 9457–9469 CrossRef CAS PubMed.
- A. J. Shaka, J. Keeler and R. Freeman, Evaluation of a New Broad-Band Decoupling Sequence – Waltz-16, J. Magn. Reson., 1983, 53, 313–340 CAS.
- D. H. Zhou and C. M. Rienstra, High-Performance Solvent Suppression for Proton Detected Solid-State Nmr, J. Magn. Reson., 2008, 192, 167–172 CrossRef CAS PubMed.
- P. C. A. van der Wel, K.-N. Hu, J. Lewandowski and R. G. Griffin, Dynamic Nuclear Polarization of Amyloidogenic Peptide Nanocrystals: Gnnqqny, a Core Segment of the Yeast Prion Protein Sup35p, J. Am. Chem. Soc., 2006, 128, 10840–10846 CrossRef CAS PubMed.
- P. C. A. van der Wel, J. R. Lewandowski and R. G. Griffin, Structural Characterization of Gnnqqny Amyloid Fibrils by Magic Angle Spinning Nmr, Biochemistry, 2010, 49, 9457–9469 CrossRef CAS PubMed.
- R. Nelson, M. R. Sawaya, M. Balbirnie, A. Ø. Madsen, C. Riekel, R. Grothe and D. Eisenberg, Structure of the Cross-Beta Spine of Amyloid-Like Fibrils, Nature, 2005, 435, 773–778 CrossRef CAS PubMed.
- M. R. Sawaya, S. Sambashivan, R. Nelson, M. I. Ivanova, S. A. Sievers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. W. Wiltzius, H. T. McFarlane, A. O. Madsen, C. Riekel and D. Eisenberg, Atomic Structures of Amyloid Cross-Beta Spines Reveal Varied Steric Zippers, Nature, 2007, 447, 453–457 CrossRef CAS PubMed.
- D. Thacker, K. Sanagavarapu, B. Frohm, G. Meisl, T. P. J. Knowles and S. Linse, The Role of Fibril Structure and Surface Hydrophobicity in Secondary Nucleation of Amyloid Fibrils, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 25272–25283 CrossRef CAS PubMed.
- M. T. Colvin, R. Silvers, B. Frohm, Y. Su, S. Linse and R. G. Griffin, High Resolution Structural Characterization of Abeta42 Amyloid Fibrils by Magic Angle Spinning Nmr, J. Am. Chem. Soc., 2015, 137, 7509–7518 CrossRef CAS PubMed.
- S. Wang, S. Parthasarathy, Y. Nishiyama, Y. Endo, T. Nemoto, K. Yamauchi, T. Asakura, M. Takeda, T. Terauchi, M. Kainosho and Y. Ishii, Nano-Mole Scale Side-Chain Signal Assignment by 1h-Detected Protein Solid-State Nmr by Ultra-Fast Magic-Angle Spinning and Stereo-Array Isotope Labeling, PLoS One, 2015, 10, e0122714 CrossRef PubMed.
- P. Paluch, R. Augustyniak, M. L. Org, K. Vanatalu, A. Kaldma, A. Samoson and J. Stanek, Nmr Assignment of Methyl Groups in Immobilized Proteins Using Multiple-Bond C-13 Homonuclear Transfers, Proton Detection, and Very Fast Mas, Front. Mol. Biosci., 2022, 9, 828785 CrossRef CAS PubMed.
- Y. Nishiyama, M. Malon, Y. Ishii and A. Ramamoorthy, 3d N-15/N-15/H-1 Chemical Shift Correlation Experiment Utilizing an Rfdr-Based H-1/H-1 Mixing Period at 100 Khz Mas, J. Magn. Reson., 2014, 244, 1–5 CrossRef CAS PubMed.
- M. Baldus and B. H. Meier, Total Correlation Spectroscopy in the Solid State. The Use of Scalar Couplings to Determine the through-Bond Connectivity, J. Magn. Reson., Ser. A, 1996, 121, 65–69 CrossRef CAS.
- T. Vagt and R. Glockshuber, Where Purity Matters: Recombinant Versus Synthetic Peptides in Beta Amyloid Formation, Methods Protein Biochem., 2012, 187–196 Search PubMed.
- V. H. Finder, I. Vodopivec, R. M. Nitsch and R. Glockshuber, The Recombinant Amyloid-Beta Peptide a Beta 1–42 Aggregates Faster and Is More Neurotoxic Than Synthetic a Beta 1–42, J. Mol. Biol., 2010, 396, 9–18 CrossRef CAS PubMed.
- D. J. Adams, T. G. Nemkov, J. P. Mayer, W. M. Old and M. H. B. Stowell, Identification of the primary peptide contaminant that inhibits fibrillation and toxicity in synthetic amyloid-β42, PLoS One, 2017, 12(8), e0182804 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00553h |
| ‡ Department of Chemistry, Brandeis University, Waltham, MA 02453 USA. |
| § Department of Chemistry and Applied Biosciences, ETH Zürich, 8093 Zürich, CH, Sweden. |
| This journal is © the Owner Societies 2024 |