
Theoretical insight into H2O impact on V2O5/TiO2 catalysts for selective catalytic reduction of NOx†
Boyu
Wu
 ,
Shengen
Zhang
*,
Mingtian
Huang
,
Shengyang
Zhang
,
Bo
Liu
and
Bolin
Zhang
*
,
Shengen
Zhang
*,
Mingtian
Huang
,
Shengyang
Zhang
,
Bo
Liu
and
Bolin
Zhang
*
Institute for Advanced Materials and Technology, University of Science and Technology Beijing, Beijing 100083, China. E-mail: zhangshengen@mater.ustb.edu.cn; zhangbolin@ustb.edu.cn
First published on 22nd April 2024
Abstract
H2O in flue gas causes the deactivation of V2O5/TiO2 catalysts for selective catalytic reduction (SCR) of NOx with NH3 at low temperatures. Developing water resistance requires understanding the theoretical mechanism of H2O impact on the catalysts. The aim of this work was to clarify the adsorption process of H2O and the deactivation mechanism induced by H2O through density functional theory (DFT). The process of H2O adsorption was studied based on a modeled V2O5/TiO2 catalyst surface. It was found that H2O had a strong interaction with exposed titanium atoms. Water adsorption on the catalyst surface significantly alters the electronic structure of VOx sites, transforming Lewis acid sites into Brønsted acid sites. Exposed titanium sites contribute to the decrease of Lewis acidity via adsorbed water. Ab initio thermodynamic calculations show that H2O adsorption on V2O5/TiO2 is stable at low coverage but less favorable at high coverage. Adsorption of NH3 is the most critical step for the SCR of NOx, and the adsorption of H2O can hinder this process. The H2O coverage below 15% of adsorption sites could enhance the NH3 adsorption rate and have a limited effect on the acidity, while higher coverage impeded the adsorption ability of VOx sites. This work provided electron-scale insight into the adsorption impact of H2O on the surface of V2O5/TiO2 catalysts, presented thermodynamic analysis of the adsorption of H2O and NH3, paving the way for the exploration of V2O5/TiO2 catalysts with improved water resistance.
1. Introduction
Nitrogen oxides (NOx) are major atmospheric pollutants that lead to significant environmental issues including acid rain, smog, global warming and depletion of the ozone layer.1–4 Over the last four decades, substantial research has been invested in addressing the NOx dilemma. Among the various NOx reduction technologies developed, the V2O5/TiO2 selective catalytic reduction (SCR) catalyst has become particularly notable.5,6 It has been widely implemented to control NOx emissions from diverse sources, such as coal-fired power plants, sintering plants, vehicles, and multiple industrial operations. The commercial vanadium-based catalysts can achieve excellent NOx removal across a broad temperature spectrum (150–450 °C), and provide better resistance towards poisoning factors than other catalysts.7–9The impact of water vapor on the performance of low-temperature SCR systems presents a complex picture. Numerous studies have consistently shown that water vapor significantly reduces the NOx removal rate of SCR catalysts at low temperatures, 5% of water vapor in the gas stream can cause 10–20% percent of decrease of NOx conversion rate at low temperatures (150–200 °C).9,10 There has been an active effort to develop catalysts more resistant towards water, however these studies primarily focus on the reactivity and active species involved.11 An understanding of the effect of water molecules on the surface is lacking, despite water being commonly present in industrial exhausts.
The consensus attributes this decrease to water molecules interfering with the adsorption of reactants or their subsequent reactions on the catalyst surface, thereby decreasing NOx removal rate.12–14 Qing et al.14 found that the presence of H2O inhibited NH3 adsorption, resulting in a significant decrease in NO conversion. However, Inomata et al.15 suggests water vapor may also facilitate the formation of additional Brønsted acid sites on certain vanadium-based catalysts, thus improving the low temperature activity. Additionally, water has a lower adsorption energy than NH3 on vanadium oxide surfaces. This discrepancy highlights critical gaps in understanding the intricate interactions between water vapor and catalytic surfaces at low-temperature, and how it can affect the catalytic performance.
Numerous studies have explored the configuration of vanadium species on surfaces. The calcination temperature for an SCR catalyst typically falls within the range of 300–600 °C.16 Within this range, V2O5 tends to develop a crystalline film on the support structure. However, this temperature is not sufficiently high to facilitate the formation of V2O5 “towers” – vertically oriented crystal structures.17,18 This observation has been confirmed through X-ray adsorption spectroscopy.19–21 Catalayud et al.22 suggest that on the TiO2 anatase (001) surface, the most stable arrangement for V2O5 is linear, lying in the [010] direction. Wilcox et al.23 proposed a sub monolayer configuration to represent a low loading (<2 wt%) vanadium surface with free surface titanium atoms exposed. This model, by intentionally exposing free surface titanium atoms, enabled the investigation of water's molecular interactions with the catalyst surface and the subsequent effects on mercury oxidation. Importantly, this configuration explicitly considered the significant interaction between TiO2 and water molecules, a critical factor at low vanadium loadings. Lian et al.24 suggest that a V2O5 loading of 4 to 8 wt% achieves satisfactory low-temperature NOx reduction rates. Yet, at higher loadings, experimental results indicate that the TiO2 surface becomes less exposed,18,25 leading to fewer free titanium atoms available to interact with water molecules. Saleh et al.18 observed a high V/Ti ratio on the surface of a low-temperature SCR catalyst, as suggested by XPS and XANES results. They found that at high vanadium loadings (7 wt%) on anatase surfaces, polymeric vanadium stripes almost completely cover the anatase (001) surface.
The (001) surface of anatase TiO2 is widely employed in commercial SCR catalysts as a support for active species, and the (001) surface has been chosen for most of the theoretical works. The anatase surface has a high surface area and strong metal–support interaction,16,26,27 attracting active species to enhance catalytic activity and stability.26,28–30 Furthermore, it has a well-defined structure and rich surface chemistry, facilitating theoretical modeling and analysis of reaction mechanisms.
The leading theories for the SCR reaction pathway include the Eley–Rideal mechanism, characterized by the reaction of adsorbed ammonia with gaseous NO molecules, and the Langmuir–Hinshelwood mechanism, which involves interaction between adsorbed ammonia and adsorbed NO species.31–33 Regardless of the reaction pathway NH3 adsorption is an initial and crucial step in the SCR reaction, and in particular its adsorption rate on the Lewis site is the most significant to the catalytic reaction cycle.14,33,34 In this study, we evaluate the adsorption process and the thermodynamic stability of models with varying levels of water coverage. The influence of the TiO2 (001) surface and active species during the water adsorption process is explored through DFT calculations and ab initio thermodynamic analyses. Models featuring different degrees of water coverage on the V2O5/TiO2 surface were developed to assess their impact on catalytic reactivity and stability. The adsorption behavior of NH3 derived from these computational results were compared with results from existing experimental and theoretical works. Several paths to enhance the resistance toward water induced deactivation are proposed.
2. Computational methodology
2.1. DFT calculations and modeling
The calculations were performed using the CP2K package (2023.2).35 We used the mixed Gaussian and plane wave approach to DFT, together with the revised Perdew–Burke–Ernzerhof (revPBE) functional parameters.36,37 Geometry optimization and vibrational analysis was performed at the triple-ζ basis level (pob-TZVP-rev2);38 for adsorption position finding, double-ζ basis sets (pob-DZVP-rev2)38 have been used. An energy cutoff of 400 Ry has been used; the convergence criterion for geometry optimization was set to 1.0 × 10−7. The semi-empirical dispersion corrections for the van der Waals interactions were applied by employing the Grimme's approach (so called DFT-D3) with Becke and Johnson (BJ) damping.39,40 To eliminate convergence problems, Fermi–Dirac smearing was employed.For the stability of the supported systems, V2O5 and TiO2 in bulk form were fully optimized. A 3-layer anatase TiO2 (001) surface was constructed as the support. To mitigate any potential dipole moments, a vacuum space of 20 Å was introduced along the Z-axis. It is widely accepted that three layers of TiO2 support can adequately represent the support structure.23,41–44 The K-points were selected based on a Monkhorst–Pack grid of 2 × 7 × 1 in real space. For modeling the supported catalyst surface, vanadium species were placed atop the supporting TiO2, and the model of the supported system was subsequently optimized. The adsorption was studied with extended supercell (15.13 × 15.13 Å2). Multiwfn (Version 3.8, release date: 2023-Dec-1) was used to aid input generation and postprocessing of the results.45
Water and NH3 molecules were relaxed separately as isolated molecules in a 30 × 30 × 30 Å3 periodic cell. The calculated bond lengths for the O–H bond in the H2O molecule and the N–H bond in the NH3 molecule are 0.97 Å and 1.02 Å, respectively. These values demonstrate excellent agreement with the corresponding experimental measurements, which are 0.96 Å for H2O46 and 1.01 Å for NH3.47 The slight difference in bond length might be attributed to the revPBE functional and tends to overestimate the bond length.48
2.2. Ab initio thermodynamic calculations
DFT calculations have limitations, particularly in simulating real-world conditions. A key limitation is that these calculations do not account for temperature and pressure effects. Consequently, the physical quantities evaluated through DFT are strictly valid only at absolute zero temperature and no pressure. To actually describe the effect of water adsorption at finite temperatures and pressures, DFT calculations should be combined with thermodynamic considerations, and we took a similar path described in previous literature to calculate the Gibbs free energy of adsorption.23,49–53 The stability of various configurations can be described by the Gibbs free energy of adsorption, at a temperature T and pressure p. The Gibbs free energy terms are defined as G(T,p) = Etot + Fvib − TSconf + pV. The first term is the total energy Etot, which is directly obtained from the DFT calculations; Fvib accounts for the vibrational contributions, and TSconf includes configurational entropy. As discussed by Cheng et al.52 and Rogal et al.,50 when calculating corrected Gibbs free energy of adsorption, the vibrational contribution of the substrate surface can be neglected given the substrate surface is not being reconstructed in a significant way. Furthermore, the vibrational contribution of the metal oxide surface remains fairly constant at application temperatures for low temperature NOx removal.54 However, the vibrational contribution of adsorbates is not neglectable.23,52,55 According to the harmonic approximation, the vibrational contribution to Gibbs free energy is defined as a frequency and temperature dependent function in eqn (1), where ωi is the frequency of adsorbate, ℏ is the reduced Planck constant, and kB is the Boltzmann constant.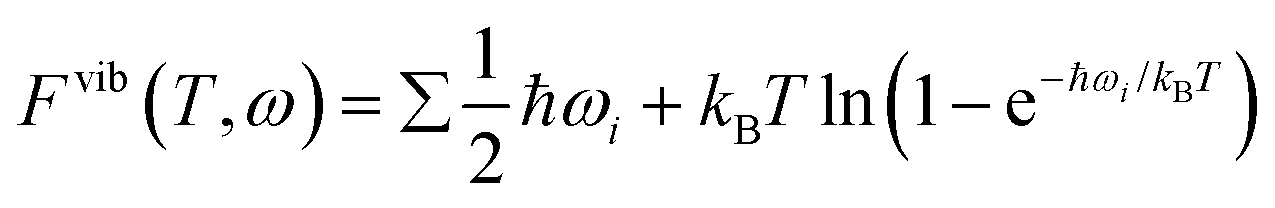 | (1) |
Gibbs free energy of adsorption ΔG(T,p) is defined as in eqn (2), where A is the surface area, Esystem is the Gibbs free energy of the surface with adsorbates, Eclean is the free energy of the surface, Ni is the quantity of corresponding adsorbate, and μi(T,pi) is the chemical potential of the corresponding adsorbate at a given T and pi.
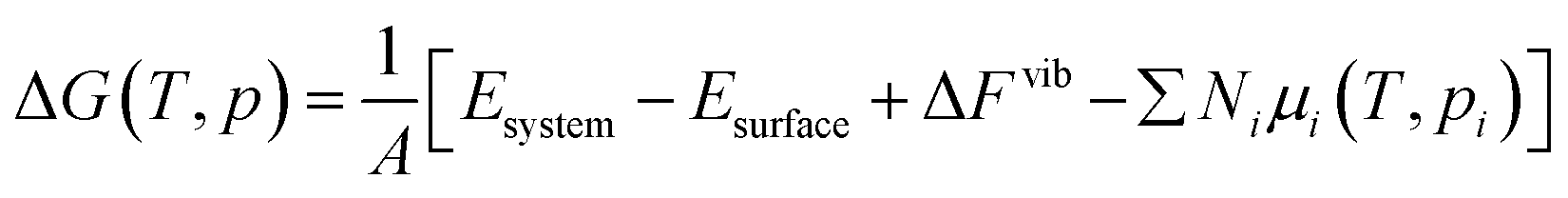 | (2) |
The chemical potential: μi(T,pi) in eqn (2) is defined as eqn (3), where Ei is the total energy produced by DFT calculation; EZPEi is the zero point vibrational energy, given there are no vibration-rotation interactions in the adsorption model, EZPEi is approximated as 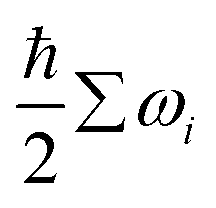 ; μi(T,p0) is defined as μi(T,p0) = H − T × S, where H represents the enthalpy, S represents the entropy, and values of H and S are freely available from the “NIST-JANAF Thermochemical Tables”,56 a copy of the values used in this study is attached in the ESI,† Table S1.
; μi(T,p0) is defined as μi(T,p0) = H − T × S, where H represents the enthalpy, S represents the entropy, and values of H and S are freely available from the “NIST-JANAF Thermochemical Tables”,56 a copy of the values used in this study is attached in the ESI,† Table S1.
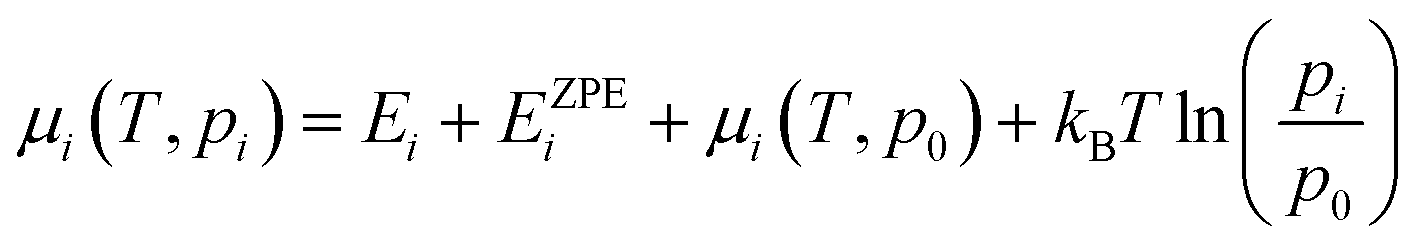 | (3) |
To investigate the influence of H2O on the reaction rate, we focused on NH3 adsorption at the Lewis acid site, a critical step in the SCR reaction mechanism. To further elucidate this, we employed transition state theory and proposed a rate constant in the Arrhenius form: RAds, as described as eqn (4), where R is the molar gas constant.
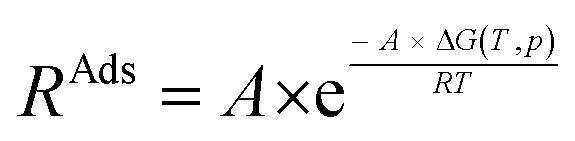 | (4) |
3. Results and discussion
3.1. Modeling of the sub-monolayer and monolayer V2O5 surface on a TiO2 support
A stable model with appropriate representation of reality is essential for studying water's influence on the catalyst surface. For the high loading supported V2O5/TiO2 system, the vanadium species was modeled with similar geometry as the anatase TiO2 (001) surface, and two layers of TiO2 support are allowed to relax, as described in the work of Calatayud et al.22 and Wilcox et al.23 The vanadium species in the sub-monolayer module are characterized by 4-fold coordination, including two adjacent exposed vanadium sites. These species form a linear crystalline structure on the surface, which is separated on the TiO2 surface to achieve approximately 7–8 VOx per nm2 coverage as observed by Raman spectroscopy.57 This model features three structurally distinct oxygen sites: O(1), the single coordinated terminal oxygen (V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); O(2), the bridging oxygen (V–O–V); and O(3), the anchoring oxygen (V–O–Ti) is coordinated with three atoms: two vanadium and one titanium atom. Representation of the sub-monolayer is shown in Fig. 1(a) and (b).
O); O(2), the bridging oxygen (V–O–V); and O(3), the anchoring oxygen (V–O–Ti) is coordinated with three atoms: two vanadium and one titanium atom. Representation of the sub-monolayer is shown in Fig. 1(a) and (b).
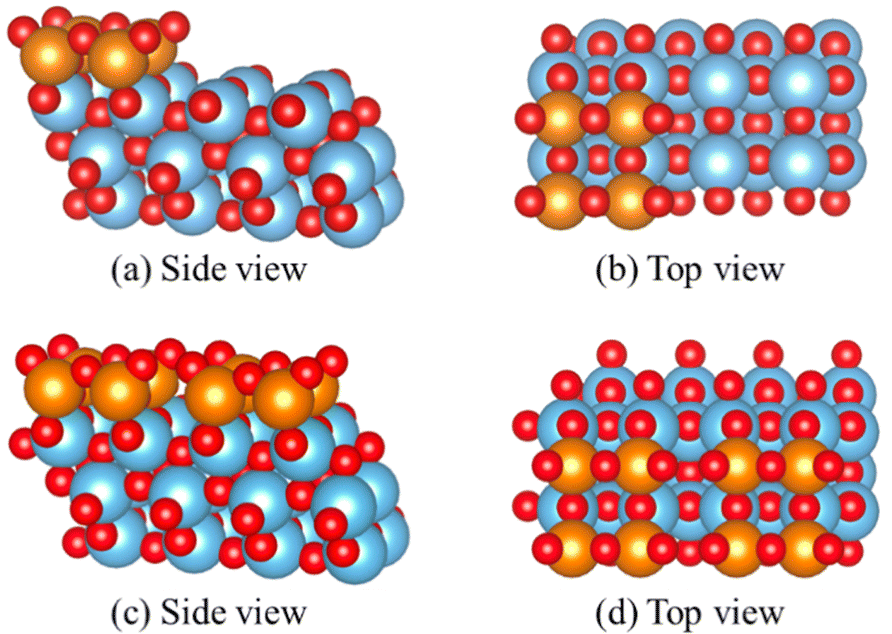 | ||
| Fig. 1 (a) Side view of the sub-monolayer model; (b) top view of sub-monolayer model; (c) side view of monolayer model; (d) top view of monolayer model. Red are oxygen atoms, blue are titanium atoms, and orange are vanadium atoms. | ||
A monolayer surface model for extreme high vanadium loading is modeled. It is reported that vanadium tends to form a thin film rather than a vertical growing crystalline structure on the anatase surface.17,19,57,58 The monolayer model was modeled with a similar approach as our sub-monolayer model, and the uninhabited TiO2 surface is all covered by V2O5 species in this configuration. There is no exposed titanium atom on the (001) surface. This model is compared with existing works and the contribution of TiO2 substrate during water adsorption is studied. To be noted, this model is widely used in other theocratical works, but the surface coverage of VOx exceeded the experimental observations.57,59
The bond distances of the supported dimers were compared with those from previous experimental and DFT studies. Despite the use of different methodologies, there is a reasonable agreement regarding the bond distances, as summarized in Table 1.
| Model | VO (Å) |
V–O bridge (Å) | V–O anchor (Å) |
|---|---|---|---|
| Sub-monolayer (this work) | 1.59 | 1.797 | 1.94 |
| monolayer (this work) | 1.58 | 1.77 | 1.94 |
| α-V2O5 (experimental)60 | 1.61 | 1.80 | — |
| V2O5 (experimental)59 | 1.58 | 1.80 | — |
| V2O5 (experimental)61 | 1.56 | — | — |
| V2O5 layer/TiO2 (DFT)62 | 1.58 | 1.78 | 1.76 |
| V2O5 polymeric (DFT)22 | 1.62 | 1.80 | — |
| V2O5 polymeric/TiO2 (DFT)22 | 1.59 | 1.82 | — |
| V2O5 dimer/TiO2 (DFT)27 | 1.60 | 1.80 | 1.76 |
| V2O5 layer/TiO2 (DFT)41 | 1.60 | 1.77 | 1.95 |
To verify the stability of the model, the formation energy was evaluated. This calculation followed similar steps to those outlined by Wilcox et al., Vittadini et al. and Gu et al.,41,62,63 and the formation energy was proposed as a key factor to understand the energy changes occurring during the surface formation process, particularly focusing on the energy variation associated with the inhabiting process of vanadium species. The energy of TiO2 surface reconstruction is denoted as Erecon, using the V2O5 bulk phase as a reference, EnVO2.5.
The formation energy Eform is defined in eqn (5):
| Eform = Esystem − [EnVO2.5 + ETiO2 + Erecon] | (5) |
The formation energy for the sub-monolayer model is calculated to be −1.84 eV, indicating strong and stable interaction between the TiO2 surface and vanadium species in this configuration. The formation energy for the monolayer model is calculated to be −1.23 eV. It is a stable surface structure but compared to the sub-monolayer model it is weaker.
The vibrational frequency was calculated and compared with experimental results. As noted in the literature, the VO bond exhibits a distinct frequency, typically ranging from 1020 cm−1 to 1030 cm−1, attributed to the stretching frequency of this bond.59,64,65 For both the sub-monolayer and monolayer systems, we calculated the stretching frequency of the VO bond. In the sub-monolayer model, this frequency corresponds to 1020 cm−1, whereas in the monolayer model, it is observed at 1093 cm−1. A similar increased frequency of the VO bond was reported by Wilcox et al.,23 who observed an increased frequency of the VO bond at 1091 cm−1 in their model, closely aligning with our results. Our hypothesis is that excessive vanadium loading on the surface reduces the electron negativity of terminal oxygen atoms, resulting in a minor decrease in VO bond strength. Consequently, this slightly weakened bond requires less energy to vibrate, leading to an increase in the calculated vibrational frequency.
3.2. Water adsorption process
The volume of water is usually around 10 vol% in the fuel gas, applying the ideal gas law in conjunction with Dalton's Law of partial pressures, and the number of water molecules in a finite region can be estimated through eqn (6), where nH2O is the number of water molecules in the simulation box, P is the atmospheric pressure, V is the volume of the simulation box (15.13 × 15.13 × 30 Å3), R is the ideal gas constant, T is temperature and NA is Avogadro's number.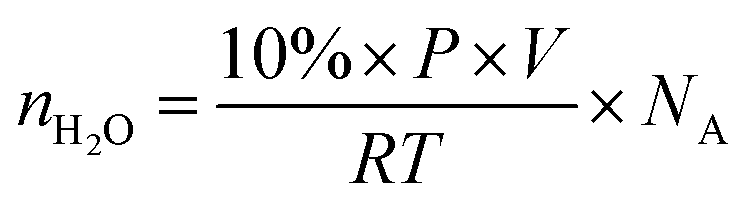 | (6) |
We estimated the quantity of potentially interacting water molecules at temperatures of 300 K, 400 K, and 500 K. The estimated numbers of water molecules in the simulation box are 1.65 × 10−3, 1.24 × 10−3, and 0.99 × 10−3, respectively. As indicated by the relatively small quantity, the deactivation process induced by water is gradual. Thus, for a more accurate representation of reality, H2O molecules were introduced to the surface sequentially.
The initial position of each H2O molecule was determined by a surface scanning, which used a mesh grid (30 × 30) located 3 Å above the surface, to identify the most likely locations for H2O molecule adsorption. After each scan, a water molecule was added to the identified position for adsorption. Once the energy of the system was minimized, this new configuration served as the base for the next scan. The H2O molecule is always added at the position with the overall highest adsorption energy, thereby avoiding adsorption at positions of local minima.
The scan results for the initial water molecule suggest a preference of adsorption at the interface between V2O5 and TiO2, as evidenced by the higher adsorption energy illustrated in Fig. 2(a). Additionally, the forces acting on the water molecules predominantly point towards the exposed TiO2 area. Considering the insights from the adsorption process calculations, the potential sites for water adsorption on the surface are limited. Each exposed titanium atom is capable of adsorbing one water molecule, and each V2O5 species can adsorb one water molecule as well. The surface coverage of water is defined as the ratio of adsorbed water molecules to the maximum adsorption capacity. In this simulation, the maximum capacity for water molecules is determined to be 12.
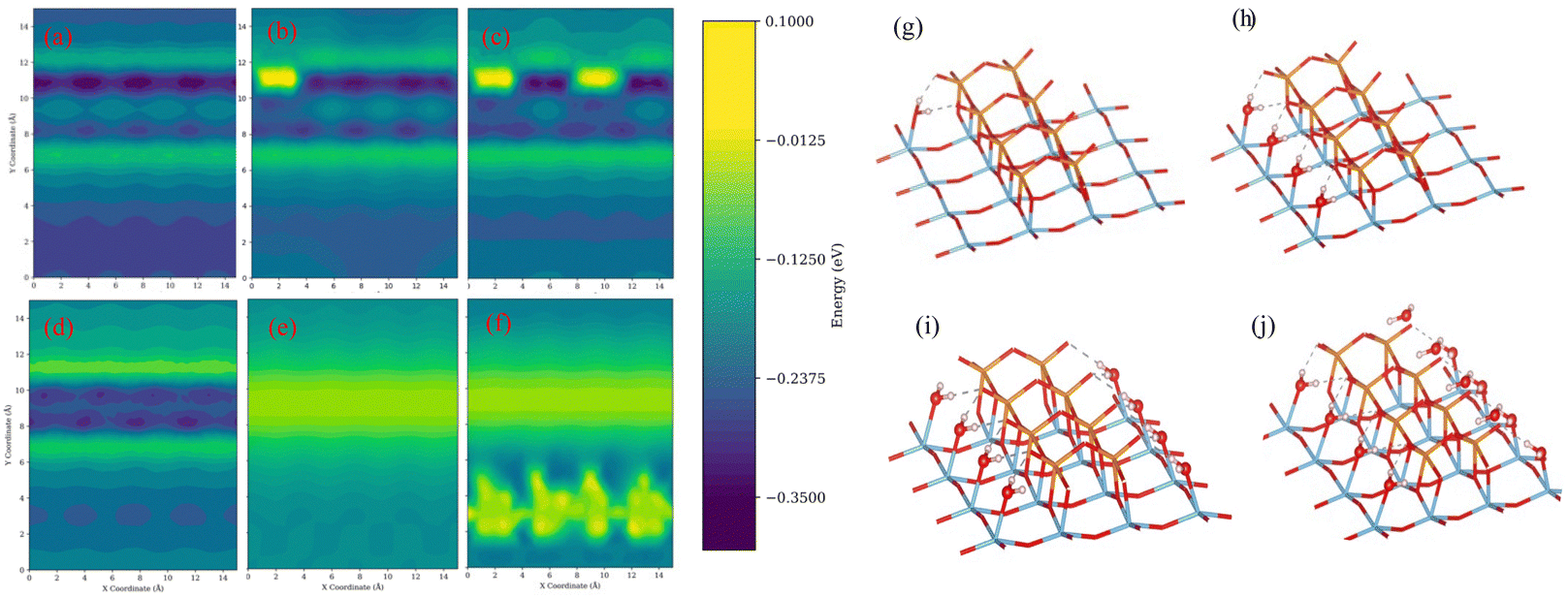 | ||
| Fig. 2 (a) to (f): The adsorption energy corresponding to models from iterations 0, 1, 2, 4, 8, and 12 (denote sub, sub-1-H2O, sub-2-H2O, sub-4-H2O, sub-8-H2O, and sub-12-H2O); (g) to (j): The adsorbed configurations corresponding to models from iterations 1, 4, 8, and 12; Red are oxygen atoms, blue are titanium atoms, orange are vanadium atoms, and light pink are hydrogen atoms; For clarity, 2 layers of TiO2 support are hidden and the vanadium polymeric stripe is centered. | ||
To have a clearer understanding of the adsorption process, we choose the representative adsorption configurations from the adsorption scan iterations 0, 1, 2, 4, 8, and 12 (denoted as sub, sub-1-H2O, sub-2-H2O, sub-4-H2O, sub-8-H2O, sub-12-H2O) for further study, and the corresponding adsorption energy and geometries are illustrated in Fig. 2. It was found that exposed titanium atoms have a strong interaction with the oxygen atoms in water molecules, and the hydrogens are attracted to the terminal oxygen atoms of vanadium species. After the adsorption of the first water molecule, the results from the second scan suggest a noticeable decrease in the adsorption energy around it, as shown in Fig. 2(b), and the corresponding geometry is shown in Fig. 2(g). Due to the repulsion between the terminal oxygen of vanadium species and the oxygen atoms in water molecules, water molecules have shown a preference towards the TiO2 area due to the repulsion between the terminal oxygen of vanadium species and oxygen atoms in water molecules. Fig. 2(d) and (i) illustrate the corresponding adsorption energy and a model in which 4 water molecules are adsorbed. All four water molecules are in similar configurations after geometry optimization: the oxygen atom in water is bonded with one undercoordinated titanium atom, and the hydrogens of water molecules are attracted to the terminal oxygen of vanadium dimer. The adsorption energy on the exposed TiO2 area is larger until it is fully covered by water molecules, as illustrated in Fig. 2(e) and (j). Compared to the clean surface, the overall average adsorption energy decreased compared to the clean surface. Subsequent adsorptions occur at the vanadium sites, as shown in Fig. 2(f) and (k). The interaction between the vanadium atom and the oxygen atom in water is not as strong as with titanium. The hydrogen bond between the terminal oxygen and the bridging oxygen of vanadium species contributes most significantly to the adsorption.
To further confirm the interaction between H2O molecule and TiO2, a molecular dynamic calculation was carried out at 373 K for 4000 fs with a timestep of 1 fs, using sub-1-H2O, sub-2-H2O, sub-4-H2O, and sub-8-H2O models. Dissociation of water molecules is evident in the sub-1-H2O and sub-2-H2O models, whereas the surface of sub-4-H2O and sub-8-H2O models maintains its stability, as illustrated in Fig. 3. The observation of water molecule dissociation and surface reconstruction occurring at lower water coverage levels align with the findings in existing works.22,23,62,66 The H2O molecule initially absorbs on the exposed titanium atom, with both H atoms interacting with neighboring terminal atoms from V2O5; The Ti–O bond is stretched leading to the formation of 2 Ti–OH groups as shown in Fig. 3(a) and (b). A comprehensive illustration of the dissociation behavior of H2O is presented in the ESI,† Fig. S1.
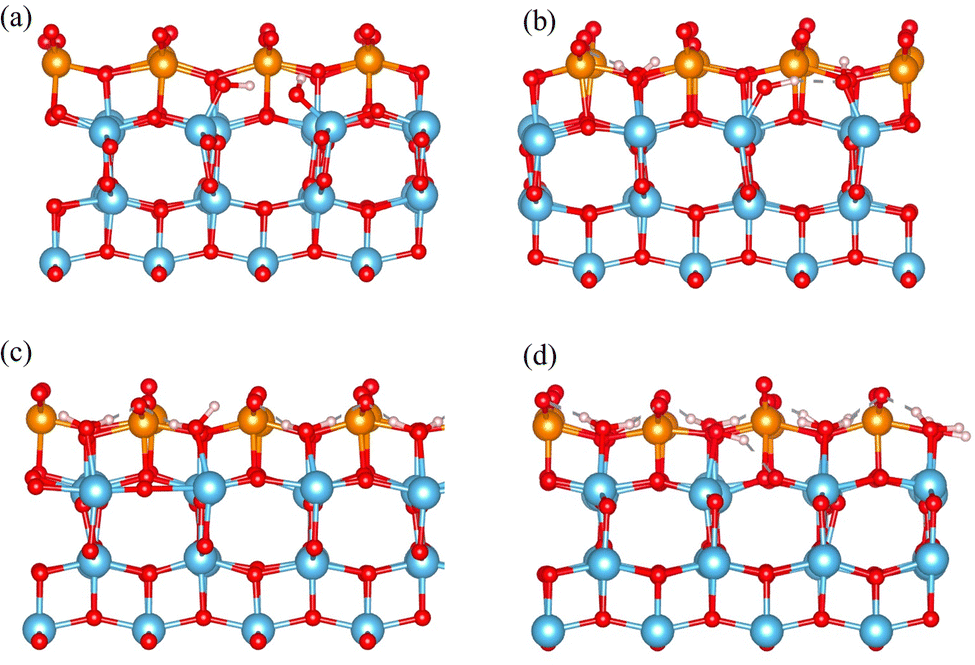 | ||
| Fig. 3 Molecule dynamic simulation of model sub-1-H2O, sub-2-H2O, sub-4-H2O and sub-8-H2O at 373 K for 4000 fs. (a)–(d) Side view of sub-1-H2O, sub-2-H2O, sub-4-H2O and sub-8-H2O after molecule dynamic simulation; red are oxygen atoms, blue are titanium atoms, orange are vanadium atoms, light pink are hydrogen atoms. | ||
Subsequent vibrational calculations for model sub-8-H2O were also carried out to verify the stability, and the frequencies assignable to the TiO2 anatase surface were 198 cm−1, 392 cm−1, 579 cm−1, 582 cm−1 and 661 cm−1. These peaks are slightly shifted but indicate a stable surface, as experimentally observed by Ohsaka et al.67
The difference in the interaction of water and the modeled V2O5/TiO2 surface observed in this study and existing works might be due to the slightly different vanadium model and different approaches. The model used in this study is more stable as indicated by the formation energy comparison and vibrational analysis. Also, the lateral influence and angle of water molecule can greatly affect the adsorption process as shown in Fig. 2(a) to (d), and the yellow stripe at y = 7 and y = 12 can be attributed to the lateral interaction between water molecules and vanadium sites.
The adsorption behavior of water suggests that the presence of exposed titanium atoms enhances water adsorption, with the interface between V2O5 and TiO2 acting as a reservoir for water molecules. Molecular dynamics calculations at 373 K confirm that at a low level of water coverage (one water molecule per surface), water molecules are dissociated upon adsorption; at higher levels of water coverage, the TiO2 support maintains its stability.
3.3. Electronic structure changes
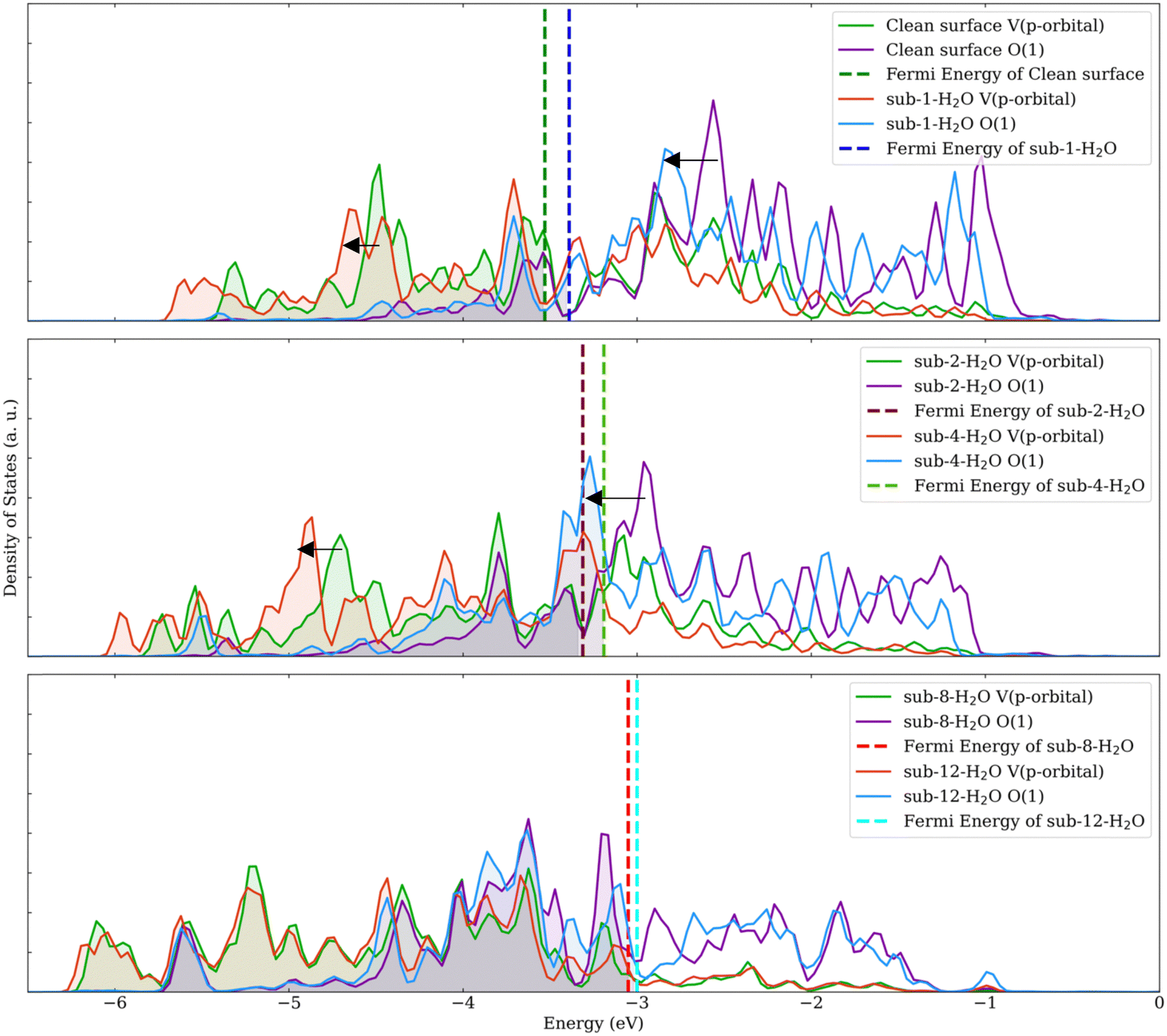 | ||
| Fig. 4 Projected density of states of models with different water coverage levels. Black arrows indicate the shift of peaks. | ||
The PDOS for the terminal oxygen atoms and vanadium atoms were analyzed, as depicted in Fig. 4. For configurations involving 1, 2, 4, and 8 water molecules adsorbed, a pronounced shift in the PDOS peaks is observed for both the oxygen atoms and the p orbitals of the vanadium atoms. However, this shift becomes less noticeable when the number of adsorbed water molecules increases from 8 to 12. This observation indicates that during the adsorption process, water molecules are likely to donate electrons to the surface. As free orbitals of the surface are saturated, the ability of the surface to adsorb H2O and NH3 decreases.
| Surface atom | O(1)–H | O(1)–H | Avg O(1)–H | O (bridge) |
|---|---|---|---|---|
| sub-1-H2O | −0.70 | −0.66 | −0.68 | −0.86 |
| sub-2-H2O | −0.76 | −0.99 | −0.87 | −0.88 |
| sub-3-H2O | −0.87 | −0.87 | −0.87 | −0.91 |
| sub-4-H2O | −0.8 | −0.93 | −0.87 | −1.03 |
| sub-5-H2O | −0.78 | −0.72 | −0.75 | −1.04 |
| sub-6-H2O | −0.79 | −0.82 | −0.81 | −1.03 |
| sub-7-H2O | −0.83 | −0.79 | −0.81 | −1.01 |
| sub-8-H2O | −0.71 | −0.97 | −0.84 | −1.01 |
| sub-9-H2O | −0.89 | −0.75 | −0.82 | −1.08 |
| sub-10-H2O | −0.72 | −0.88 | −0.80 | −1.10 |
| sub-11-H2O | −0.85 | −0.79 | −0.82 | −1.13 |
| sub-12-H2O | −0.73 | −0.92 | −0.83 | −1.09 |
In the water adsorption process for 1 to 4 molecules, the electronegativity of both bridging and terminal oxygen atoms at the adsorption sites increases. Specifically, the addition of the second water molecule markedly enhances the electronegativity of terminal oxygen atoms. Yet, as water coverage expands, terminal oxygen's electronegativity plateaus, while the bridging oxygen atoms continue to become more electronegative. In both the fifth and sixth models, the water molecules were absorbed on other exposed titanium atoms, resulting in an increase in the electronegativity of the terminal oxygen atoms at the V2O5/TiO2 interface. Meanwhile, the electronegativity of the bridging oxygen atoms remained stable. In configurations 9 to 12 (sub-9-H2O to sub-12-H2O), water molecules bind to the vanadium species, with one hydrogen of each molecule attracted to bridging oxygen and the other to terminal oxygen, affecting the electronegativity of bridging oxygen atoms. Combining these findings with thermodynamic analysis, the observed decrease in electronegativity indicates that the surface's adsorption capacity has been exceeded. The capacity to accept electrons from water molecules is crucial in this adsorption process.
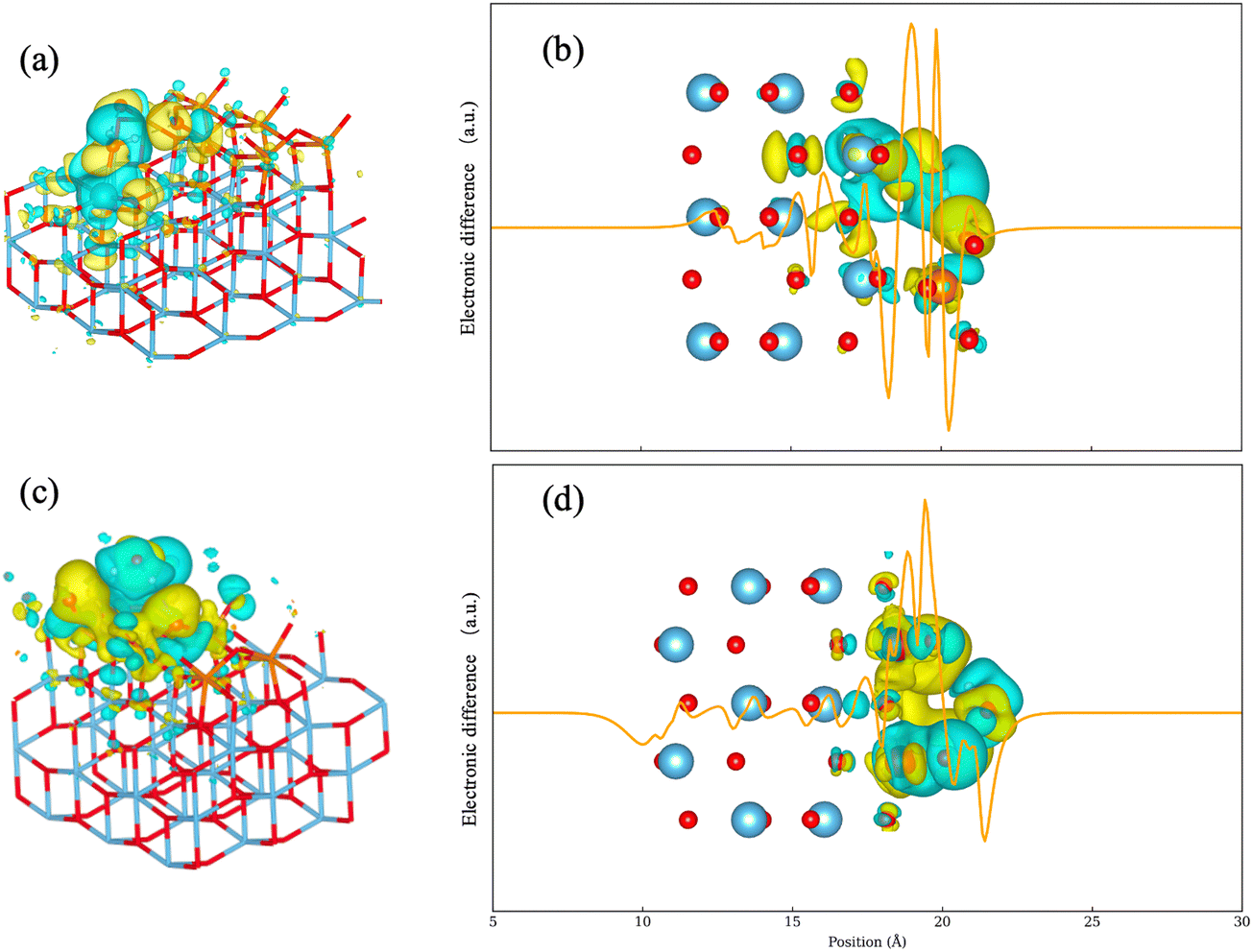 | ||
| Fig. 5 Isosurfaces of electron density difference and local integral curve; the electron density increases in the yellow regions, and decreases in the cyan regions. (a) Side view of model sub-1-H2O with isosurfaces of electron density difference; (b) local integral curve along the Z axis of the model sub-1-H2O with isosurfaces of electron density difference; (c) side view of model mono-1-H2O with isosurfaces of electron density difference; (d) local integral curve along the Z axis of model mono-1-H2O with isosurfaces of electron density difference. | ||
A significant difference in electronic density around the exposed titanium atom is observed. To better understand the contribution of the titanium support, a monolayer model with an adsorbed water molecule was employed. The effect of the exposed titanium atom is less evident in this model given the separation. Hydrogen atoms in the water molecule are attracted to two oxygen atoms: one bridging and one terminal. The electronic density change is confined to the monolayer V2O5 region. Although a decrease in electron density is still evident around the water molecule, it is less pronounced in the absence of an exposed titanium atom. This observation aligns with the lower adsorption energy of water when positioned vertically above V2O5 sites. These observations suggest that exposed titanium atoms play a major role in both enhancing water adsorption and weakening the acidity of Lewis acid sites when water is present.
The impact of adsorbed water molecules on the electronic structure was investigated. The PDOS analysis suggests notable increases of Fermi level are significant, suggesting the adsorbed H2O molecules saturated free orbitals of the V2O5/TiO2 surface. The AIM analysis found increases in electronegativity of the Lewis acid sites neighboring adsorbed H2O, indicating the acidity becomes weakened as the level of water coverage rises. The electron density difference revealed the role of TiO2 support during the adsorption of water. Exposed titanium atoms increase the electronegativity at Lewis acid sites, leading to a reduction in the acidity of Lewis sites in the presence of water.
3.4. Ab initio thermal dynamic analysis
This study employed a methodology similar to those described by Galimberti et al.55 and Wilcox et al.23 The vibrational contribution of the substrate is neglected, as Rogal et al.54 found that the impact of temperature on the vibrational contribution of metal oxide surfaces is minimal and fairly consistent, while the vibrational contribution of the gas species cannot be ignored.
The selective vibrational analysis was performed to obtain the vibrational contribution of the gas molecules. Fig. 6(a) illustrates the vibrational contributions for different numbers of adsorbed H2O molecules. As the temperature increases, there is an overall decrease in vibrational contribution. While more water molecules inhabit the surface, some low frequencies are emerging due to interaction between the surface and water molecules. In the context of the harmonic approximation, frequencies below 50 cm−1 are treated as 50 cm−1. The contribution of vibrational energy is a proxy to represent the effect of dissociation of water molecules for low water coverage models.
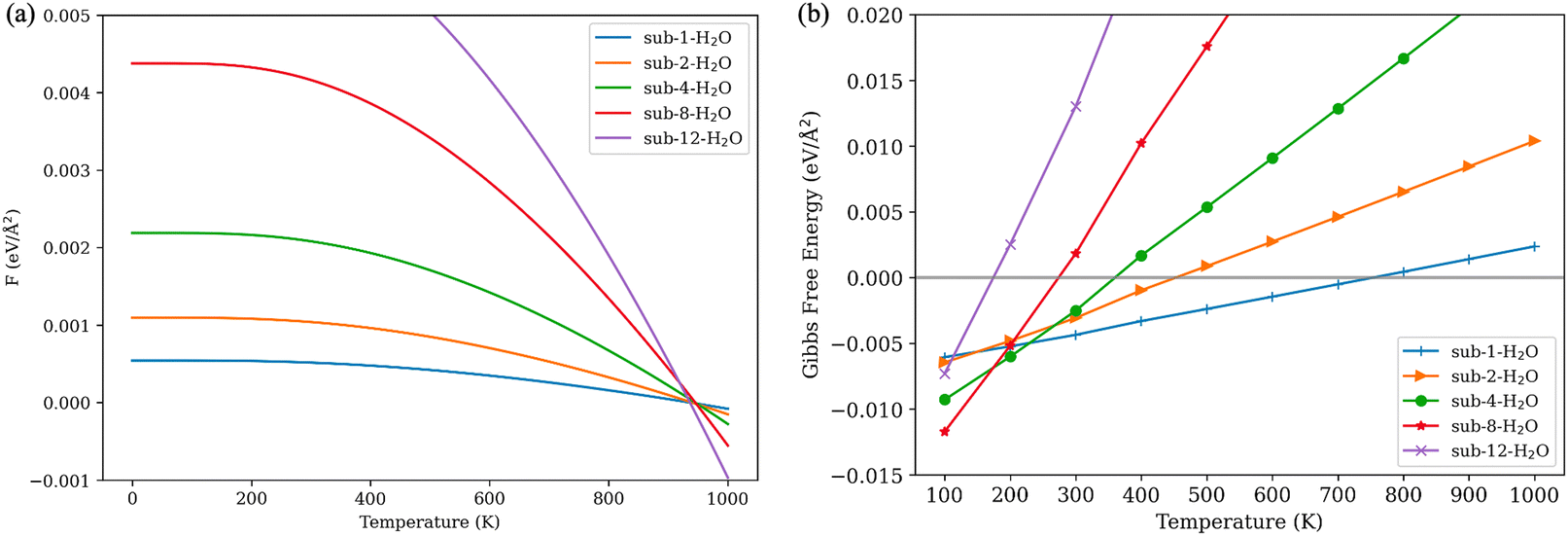 | ||
| Fig. 6 (a) Vibrational contributions for different numbers of adsorbed H2O molecules; (b) Gibbs free energy of adsorption for different numbers of adsorbed H2O molecules. | ||
Gibbs free energy of adsorption is calculated based on eqn (2). At temperatures below 200 K, even the models with extremely high coverage of water are thermodynamically favorable. However, as temperatures rise, these systems, particularly those with higher water loadings, become less thermodynamically stable. The system fully saturated with water (sub-12-H2O) becomes unfavorable below 200 K, indicating a water adsorption threshold. Water molecules are likely to be adsorbed at the interface of V2O5 and TiO2. Furthermore, the high Gibbs energy barrier renders this system impractical in real-world temperatures. In the sub-8-H2O model, titanium atoms and terminal oxygens of V2O5 are fully covered. This configuration remains thermodynamically favorable up to 280 K. Configurations with 4 and 2 adsorbed water molecules are thermodynamically favorable within the range of real-world working temperatures for SCR catalysts. Specifically, models with 4 water molecules remain favorable until 390 K, and the model with 2 water molecules until 440 K. The configuration with a single water molecule adsorbed to an undercoordinated titanium atom is the most thermodynamically stable and remains favorable up to 750 K, suggesting chemical adsorption and potential dissociation of a water molecule as observed in the molecular dynamic simulation.
The outcomes of thermodynamic calculations indicate that water-induced deactivation of the catalyst can be reversed by increasing the temperature of the gas stream to 400 K and above, as observations from existing experimental studies found.11,13,69,70 However, achieving complete desorption of water from the surface at application temperatures is nearly impossible, as suggested by the Gibbs free energy of adsorption yielded from the model sub-1-H2O. Prolonged exposure to a humid gas stream may lead to surface reconstruction of V2O5/TiO2 catalysts, potentially causing irreversible deactivation.70–72
The adsorption configuration is determined by surface scans of adsorption energy over the Lewis acid sites, for each configuration with different level water coverage. Fig. 7 displays the resulting adsorption configurations. The position of adsorbed NH3 molecule is only slightly influenced by the configuration of water molecules. As the coverage of water increases, the adsorption energy of NH3 decreases, accompanied by an increase in the distance between the NH3 molecule and the Lewis acid sites.
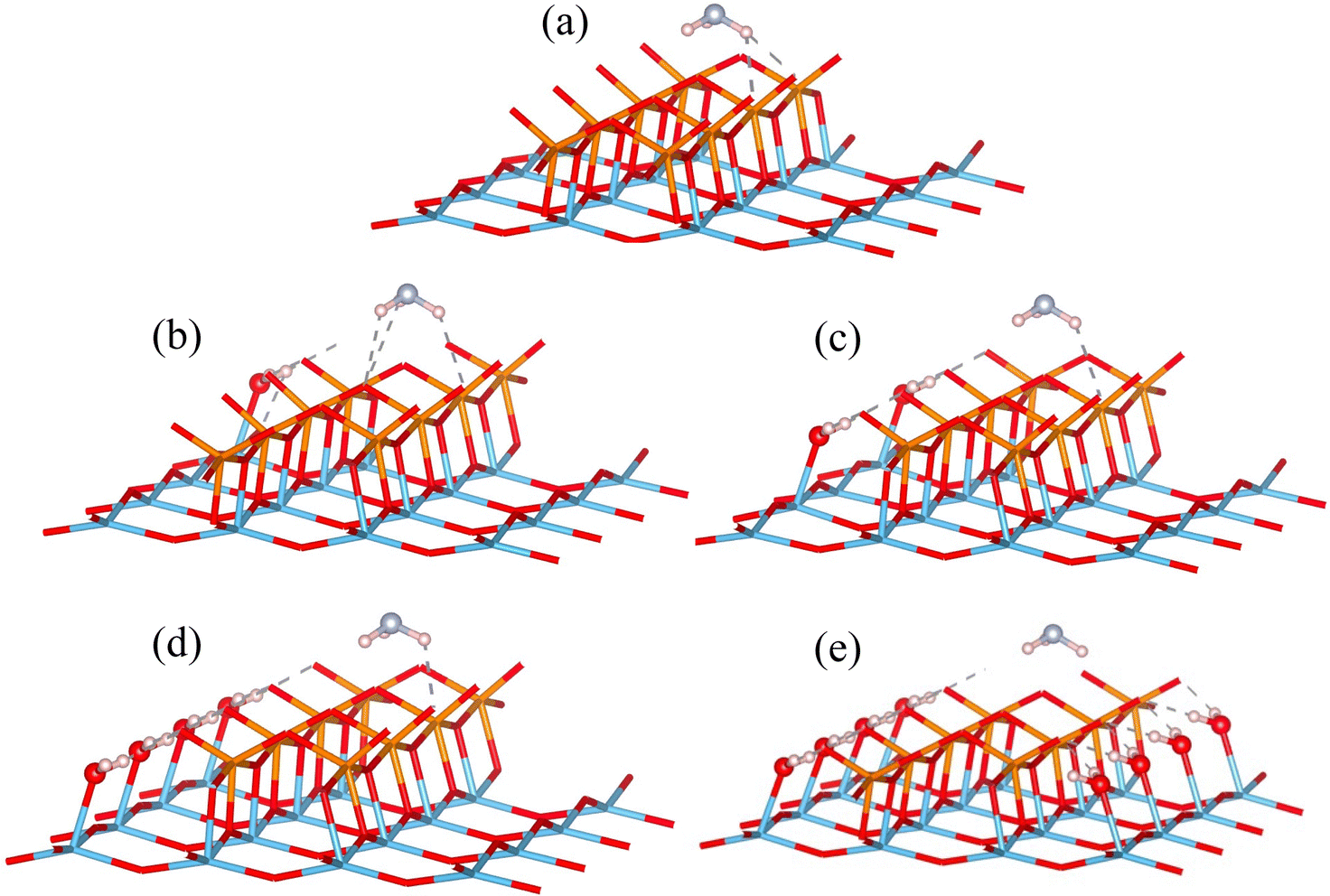 | ||
| Fig. 7 NH3 adsorption configurations at different water adsorbed levels. (a) to (e): 0, 1, 2, 4, and 8 H2O molecule(s) adsorbed. Red are oxygen atoms, blue are titanium atoms, orange are vanadium atoms, light pink are hydrogen atoms, and sky blue are nitrogen atoms; the 2 bottom layers of TiO2 support are hidden for clarity. | ||
The Gibbs free energy of adsorption of NH3 presents a complex picture. On a clean surface (sub-NH3), the Gibbs free energy of adsorption of NH3 is smaller than every co-sorption model with water adsorbed on the surface at 100 K, as shown in Fig. 8. However, 100 K is not a meaningful temperature for the NOx removal reactions. The adsorption of NH3 on the clean V2O5/TiO2 surface keeps being thermodynamically favorable until almost 500 K, truing less favorable slowly. Models with low water loading, particularly those with low water coverage (namely sub-1-H2O–NH3 and sub-2-H2O–NH3), demonstrate high stability as well. The temperature at which the Gibbs free energy of adsorption for the model sub-2-H2O–NH3 reaches zero (460 K) is slightly higher than that for the model without NH3 loading (sub-2-H2O, 430 K). Models with a higher degree of water coverage are less thermodynamically stable.
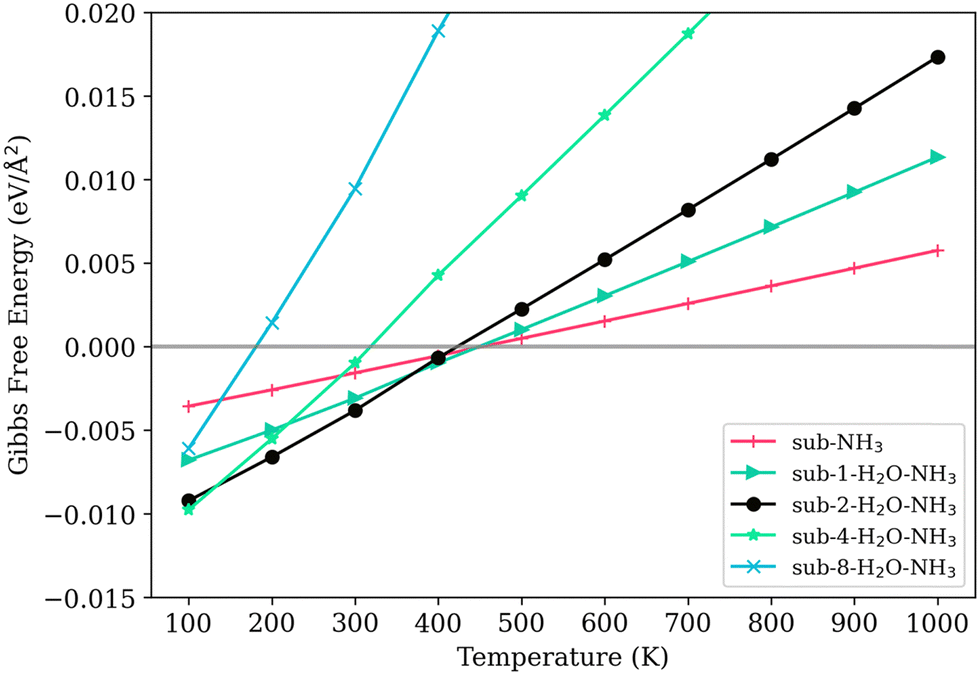 | ||
| Fig. 8 The Gibbs free energy of adsorption of NH3 on the surface with different levels of water adsorption. | ||
The models featuring 4 and 8 adsorbed water molecules, which cover half and the entirety of the exposed titanium area respectively (sub-4-H2O–NH3 and sub-8-H2O–NH3), is very thermodynamically unfavorable. The sub-4-H2O–NH3 model becomes thermodynamically unfavorable at 310 K, while the sub-8-H2O–NH3 model becomes unfavorable at a much lower temperature of 80 K. In contrast, their corresponding models without NH3, sub-4-H2O and sub-8-H2O, remain stable even when the temperature is raised to 400 K and 270 K, respectively. This distinct decrease can indicate that NH3 is less likely to be absorbed on the Lewis sites with a water coverage level higher than 35%, thus increases the energy barrier of the catalytic reaction, resulting in lower catalytic activity.
To further understand the effect of water coverage levels on reactivity, a rate constant in an Arrhenius form was proposed: RAds, as described in eqn (4). The purpose of this constant is to establish a link between the microscale model and the macroscale reaction dynamics. This rate constant can be effectively utilized for comparative analysis between different models that employ identical computational approaches, and it can also be used to explain changes in reactivity observed experimentally. We acknowledge that this constant has limited applicability for quantitative analysis of macroscale reaction rates.
As shown in Fig. 9, the adsorption rate for the model with no water (sub-NH3) decreases slowly as the temperature increases. For models with low levels of water coverage (<15%) (model sub-1-H2O–NH3 and sub-2-H2O–NH3), the presence of water initially increased the adsorption rate of NH3. The increase of adsorption rate can indicate an increase in activity at low temperatures under a low humid gas stream, aligning with the observations of Inomata et al..11,13 Interestingly, this enhanced adsorption rate contradicts the predictions made by the PDOS calculations. This discrepancy could be attributed to the potential dissociation of water molecules, an effect that is not directly reflected in PDOS but is captured in the zero-point vibrational energy calculations.
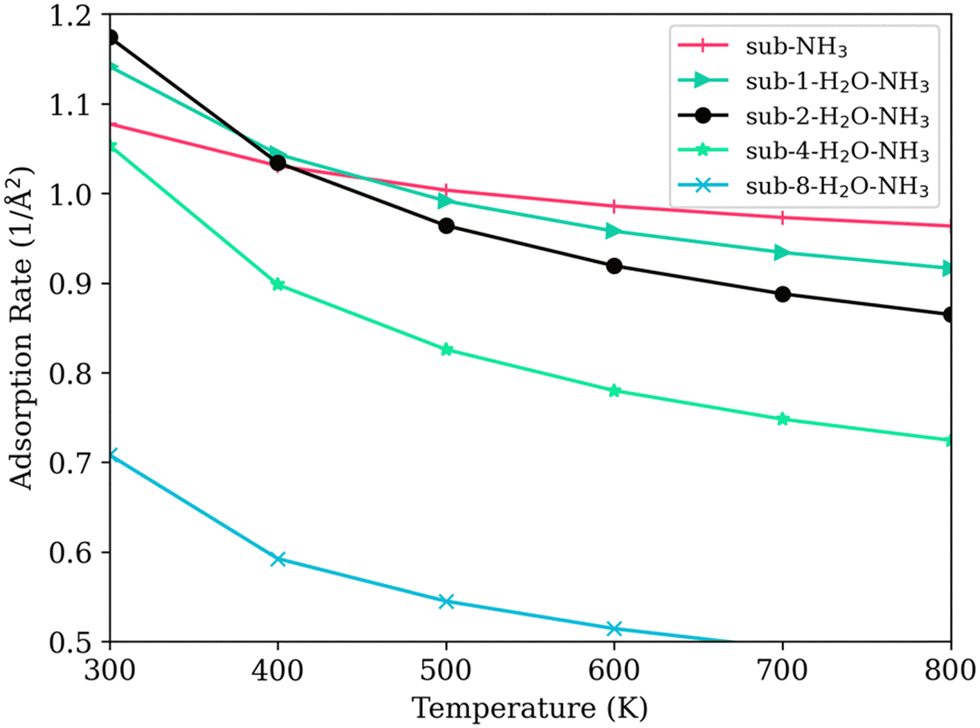 | ||
| Fig. 9 Temperature and the adsorption rate of NH3 on SCR surfaces with varying water coverage levels. | ||
As the number of adsorbed water molecules increases to four and eight, covering half and then the entire titanium surface (sub-4-H2O–NH3 and sub-8-H2O–NH3), the adsorption rate of NH3 decreases markedly at temperatures below 400 K. These decreases indicate a strong competitive adsorption effect: water molecules hinder the ability of Lewis acid sites to adsorb NH3.
Ab initio thermodynamic analysis of models with varying water coverages indicates that water-induced deactivation of the catalyst is reversible by increasing the gas stream temperature to 400 K or above, a well-established procedure in the industry. However, complete water desorption at application temperatures is difficult, and long-term exposure to humid gas streams might lead to irreversible deactivation. Interestingly, at low water coverage (≤35%), co-adsorption of NH3–H2O is more favorable than NH3 adsorption on a clean V2O5/TiO2 surface, thermodynamically. Low levels of water coverage (≤35%) can even enhance the NH3 adsorption rate at temperatures lower than 400 K.
3.5. Pathways to enhance water resistance
These findings suggest multiple pathways to mitigate water-induced deactivation. One approach is to use a support material with weaker interactions with water, improving water resistance while minimally affecting Lewis acid sites. Introducing species like tungsten oxides to the V2O5/TiO2 surface could shield exposed titanium atoms, limiting electronic changes during water adsorption. Alternatively, replacing vanadium with a different active element could increase the availability of Lewis acid sites, enhancing resistance to water. Finally, coating the TiO2 area with a highly non-polar species could significantly boost the acidity of Lewis acid sites and potentially create hydrophobicity.4. Conclusions
This work presented and validated stable models of the V2O5/TiO2 surface. The adsorption of water is enhanced by exposed titanium atoms, particularly at the V2O5/TiO2 interface. While low water coverage leads to dissociative adsorption, the TiO2 support maintains its stability at higher coverage levels. Water adsorption significantly alters the catalyst's electronic structure, including an increase in the Fermi level that suggests saturation of surface free orbitals. Additionally, water weakens the acidity of Lewis acid sites as coverage increases. Exposed titanium atoms play a key role in reducing Lewis acidity via adsorbed water. Ab initio thermodynamic analysis established the reversibility of water-induced catalyst deactivation at temperatures above 400 K, consistent with established industrial practices. Complete water desorption is difficult at typical operating temperatures. Low water coverage (≤35%) demonstrates a thermodynamically favorable co-adsorption of NH3–H2O compared to NH3 alone on a clean V2O5/TiO2 surface. However, prolonged exposure to humid conditions poses a risk of irreversible deactivation, as the TiO2 surface could be dissociated.Our findings suggest multiple pathways to mitigate water-induced deactivation in V2O5/TiO2 catalysts. Strategies include using a support material less prone to water interaction, shielding exposed titanium atoms with additives, employing an alternative active element to vanadium, or coating the TiO2 area with a highly non-polar species to enhance acidity and potentially achieve hydrophobicity.
Data availability
Data for this paper, including geometry data for gas species and crystallographic data for V2O5/TiO2, are available at https://github.com/ducksalted/v2o5tio2.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was sponsored by the National Natural Science Foundation of China (Grants No. 52204414), the National Energy-Saving and Low-Carbon Materials Production and Application Demonstration Platform Program (TC220H06N), the National Key R&D Program of China (Grants No. 2021YFC1910504), and the Fundamental Research Funds for the Central Universities (Grants No. FRF-TP-20-097A1Z).References
- J. S. Gaffney, G. E. Streit, J. H. Hall and W. D. Spall, Beyond acid rain. Do soluble oxidants and organic toxinsinteract with SO2 and NOx to increase ecosystem effects?, Environ. Sci. Technol., 1987, 21, 519–524 CrossRef CAS PubMed.
- H. He, Y. Wang, Q. Ma, J. Ma, B. Chu, D. Ji, G. Tang, C. Liu, H. Zhang and J. Hao, Mineral dust and NOx promote the conversion of SO2 to sulfate in heavy pollution days, Sci. Rep., 2014, 4, 4172 CrossRef PubMed.
- G. B. Marks, W. Ezz, N. Aust, B. G. Toelle, W. Xuan, E. Belousova, C. Cosgrove, B. Jalaludin and W. T. Smith, Respiratory Health Effects of Exposure to Low-NOx Unflued Gas Heaters in the Classroom: A Double-Blind, Cluster-Randomized, Crossover Study, Environ. Health Perspect., 2010, 118, 1476–1482 CrossRef CAS PubMed.
- J. L. Laughner and R. C. Cohen, Direct observation of changing NO x lifetime in North American cities, Science, 1979, 366, 723–727 CrossRef PubMed.
- J. K. Lai and I. E. Wachs, A Perspective on the Selective Catalytic Reduction (SCR) of NO with NH3 by Supported V2O5–WO3/TiO2 Catalysts, ACS Catal., 2018, 8, 6537–6551 CrossRef CAS.
- L. J. Muzio, G. C. Quartucy and J. E. Cichanowicz, Overview and status of post-combustion NOx control: SNCR, SCR and hybrid technologies, Int J. Environ. Pollut., 2002, 17, 4–30 CrossRef CAS.
- S. Djerad, M. Crocoll, S. Kureti, L. Tifouti and W. Weisweiler, Effect of oxygen concentration on the NOx reduction with ammonia over V2O5–WO3/TiO2 catalyst, Catal. Today, 2006, 113, 208–214 CrossRef CAS.
- Z. Ma, X. Wu, Y. Feng, Z. Si, D. Weng and L. Shi, Low-temperature SCR activity and SO2 deactivation mechanism of Ce-modified V2O5–WO3/TiO2 catalyst, Prog. Nat. Sci.: Mater. Int., 2015, 25, 342–352 CrossRef CAS.
- B. Ye, B. Jeong, M. Lee, T. H. Kim, S.-S. Park, J. Jung, S. Lee and H.-D. Kim, Recent trends in vanadium-based SCR catalysts for NOx reduction in industrial applications: stationary sources, Nano Convergence, 2022, 9, 51 CrossRef CAS PubMed.
- B.-L. Zhang, L.-F. Deng, B. Liu, C.-Y. Luo, M. Liebau, S.-G. Zhang and R. Gläser, Synergistic effect of cobalt and niobium in Co3-Nb-Ox on performance of selective catalytic reduction of NO with NH3, Rare Met., 2022, 41, 166–178 CrossRef CAS.
- Y. Inomata, H. Kubota, S. Hata, E. Kiyonaga, K. Morita, K. Yoshida, N. Sakaguchi, T. Toyao, K. Shimizu, S. Ishikawa, W. Ueda, M. Haruta and T. Murayama, Bulk tungsten-substituted vanadium oxide for low-temperature NOx removal in the presence of water, Nat. Commun., 2021, 12, 557 CrossRef CAS PubMed.
- Z. Lian, Y. Li, W. Shan and H. He, Recent Progress on Improving Low-Temperature Activity of Vanadia-Based Catalysts for the Selective Catalytic Reduction of NOx with Ammonia, Catalysts, 2020, 10, 1421 CrossRef CAS.
- Y. Inomata, S. Hata, M. Mino, E. Kiyonaga, K. Morita, K. Hikino, K. Yoshida, H. Kubota, T. Toyao, K. Shimizu, M. Haruta and T. Murayama, Bulk Vanadium Oxide versus Conventional V2O5/TiO2: NH3 –SCR Catalysts Working at a Low Temperature Below 150 °C, ACS Catal., 2019, 9, 9327–9331 CrossRef CAS.
- M. Qing, S. Su, K. Qian, L. Liu, Z. Yin, S. Hu, Y. Wang and J. Xiang, Insight into the catalytic performance and NH3 adsorption under high concentration of CO2 and/or H2O conditions on selective catalytic reduction of NO by NH3 over V2O5-WO3/TiO2 catalyst, Fuel, 2021, 286, 119478 CrossRef CAS.
- Y. Inomata, H. Kubota, S. Hata, E. Kiyonaga, K. Morita, K. Yoshida, N. Sakaguchi, T. Toyao, K. Shimizu, S. Ishikawa, W. Ueda, M. Haruta and T. Murayama, Bulk tungsten-substituted vanadium oxide for low-temperature NOx removal in the presence of water, Nat. Commun., 2021, 12, 557 CrossRef CAS PubMed.
- A. Vejux and P. Courtine, Interfacial reactions between V2O5 and TiO2 (anatase): Role of the structural properties, J. Solid State Chem., 1978, 23, 93–103 CrossRef CAS.
- M. Inomata, K. Mori, A. Miyamoto, T. Ui and Y. Murakami, Structures of supported vanadium oxide catalysts. 1. Vanadium(V) oxide/titanium dioxide (anatase), vanadium(V) oxide/titanium dioxide (rutile), and vanadium(V) oxide/titanium dioxide (mixture of anatase with rutile), J. Phys. Chem., 1983, 87, 754–761 CrossRef CAS.
- R. Saleh, The interaction of V2O5 with Ti02(anatase): Catalyst evolution with calcination temperature and O-xylene oxidation, J. Catal., 1986, 98, 102–114 CrossRef CAS.
- R. Kozlowski, R. F. Pettifer and J. M. Thomas, X-ray absorption fine structure investigation of vanadium(V) oxide-titanium(IV) oxide catalysts. 2. The vanadium oxide active phase, J. Phys. Chem., 1983, 87, 5176–5181 CrossRef CAS.
- Y. Wu, X. Zhou, T. Mi, M. Xu, L. Zhao and Q. Lu, Theoretical insight into the interaction mechanism between V2O5/TiO2 (0 0 1) surface and arsenic oxides in flue gas, Appl. Surf. Sci., 2021, 535, 147752 CrossRef CAS.
- G. C. Bond, Preparation and properties of vanadia/titania monolayer catalysts, Appl. Catal., A, 1997, 157, 91–103 CrossRef CAS.
- M. Calatayud, B. Mguig and C. Minot, A periodic model for the V2O5–TiO2 (anatase) catalyst. Stability of dimeric species, Surf. Sci., 2003, 526, 297–308 CrossRef CAS.
- A. Suarez Negreira and J. Wilcox, DFT Study of Hg Oxidation across Vanadia-Titania SCR Catalyst under Flue Gas Conditions, J. Phys. Chem. C, 2013, 117, 1761–1772 CrossRef CAS.
- Z. Lian, Y. Li, W. Shan and H. He, Recent Progress on Improving Low-Temperature Activity of Vanadia-Based Catalysts for the Selective Catalytic Reduction of NOx with Ammonia, Catalysts, 2020, 10, 1421 CrossRef CAS.
- A. Marberger, M. Elsener, D. Ferri and O. Kröcher, VOx Surface Coverage Optimization of V2O5/WO3-TiO2 SCR Catalysts by Variation of the V Loading and by Aging, Catalysts, 2015, 5, 1704–1720 CrossRef CAS.
- X. Lang, T. Wang, Z. Wang, L. Li, C. Yao and K. Cai, Reasonable design of a V2O5-x/TiO2 active interface structure with high polysulfide adsorption energy for advanced lithium-sulfur batteries, Electrochim. Acta, 2022, 403, 139723 CrossRef CAS.
- T. Mi, Y. Wu, X. Zhou, W. Li, L. Zhao, J. Liu and Q. Lu, Catalytic oxidation of CO over V2O5/TiO2 and V2O5-WO3/TiO2 catalysts: A DFT study, Fuel Process. Technol., 2021, 213, 106678 CrossRef CAS.
- L. Zheng, M. Casapu, M. Stehle, O. Deutschmann and J.-D. Grunwaldt, Selective catalytic reduction of NOx with ammonia and hydrocarbon oxidation over V2O5-MoO3/TiO2 and V2O5-WO3/TiO2 SCR catalysts, Top. Catal., 2018, 62, 129–139 CrossRef.
- J. P. Chen and R. T. Yang, Role of WO3 in mixed V2O5-WO3/TiO2catalysts for selective catalytic reduction of nitric oxide with ammonia, Appl. Catal., A, 1992, 80, 135–148 CrossRef CAS.
- J. K. Lai and I. E. Wachs, A Perspective on the Selective Catalytic Reduction (SCR) of NO with NH3 by Supported V2O5-WO3/TiO2 Catalysts, ACS Catal., 2018, 8, 6537–6551 CrossRef CAS.
- W. H. Weinberg, Eley−Rideal Surface Chemistry: Direct Reactivity of Gas Phase Atomic Hydrogen with Adsorbed Species, Acc. Chem. Res., 1996, 29, 479–487 CrossRef CAS.
- K. V. Kumar, K. Porkodi and F. Rocha, Langmuir–Hinshelwood kinetics – A theoretical study, Catal. Commun., 2008, 9, 82–84 CrossRef CAS.
- B. Zhang, S. Zhang and B. Liu, Effect of oxygen vacancies on ceria catalyst for selective catalytic reduction of NO with NH3, Appl. Surf. Sci., 2020, 529, 147068 CrossRef CAS.
- A. Marberger, D. Ferri, M. Elsener and O. Kröcher, The Significance of Lewis Acid Sites for the Selective Catalytic Reduction of Nitric Oxide on Vanadium-Based Catalysts, Angew. Chem., Int. Ed., 2016, 55, 11989–11994 CrossRef CAS PubMed.
- T. D. Kühne, M. Iannuzzi and M. Del Ben, et al., CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations, J. Chem. Phys., 2020, 152(19), 194103 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Y. Zhang and W. Yang, Comment on “Generalized Gradient Approximation Made Simple”, Phys. Rev. Lett., 1998, 80, 890 CrossRef CAS.
- D. Vilela Oliveira, J. Laun, M. F. Peintinger and T. Bredow, BSSE-correction scheme for consistent Gaussian basis sets of double- and triple-zeta valence with polarization quality for solid-state calculations, J. Comput. Chem., 2019, 40, 2364–2376 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. S. Negreira and J. Wilcox, Role of WO3 in the Hg oxidation across the V2O 5-WO3-TiO2 SCR catalyst: A DFT study, J. Phys. Chem. C, 2013, 117, 24397–24406 CrossRef.
- J. Li, Y. Peng, H. Chang, X. Li, J. C. Crittenden and J. Hao, Chemical poison and regeneration of SCR catalysts for NO x removal from stationary sources, Front. Environ. Sci. Eng., 2016, 10, 413–427 CrossRef CAS.
- Y. Peng, J. Li, W. Si, J. Luo, Q. Dai, X. Luo, X. Liu and J. Hao, Insight into deactivation of commercial SCR catalyst by arsenic: An experiment and DFT study, Environ. Sci. Technol., 2014, 48, 13895–13900 CrossRef CAS PubMed.
- D. Wang, J. Luo, Q. Yang, J. Yan, K. Zhang, W. Zhang, Y. Peng, J. Li and J. Crittenden, Deactivation Mechanism of Multipoisons in Cement Furnace Flue Gas on Selective Catalytic Reduction Catalysts, Environ. Sci. Technol., 2019, 53, 6937–6944 CrossRef CAS PubMed.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. R. Hoy and P. R. Bunker, A precise solution of the rotation bending Schrödinger equation for a triatomic molecule with application to the water molecule, J. Mol. Spectrosc., 1979, 74, 1–8 CrossRef CAS.
- T. N. Olney, N. M. Cann, G. Cooper and C. E. Brion, Absolute scale determination for photoabsorption spectra and the calculation of molecular properties using dipole sum-rules, Chem. Phys., 1997, 223, 59–98 CrossRef CAS.
- K. Yang, J. Zheng, Y. Zhao and D. G. Truhlar, Tests of the RPBE, revPBE, τ-HCTHhyb, ωB97X-D, and MOHLYP density functional approximations and 29 others against representative databases for diverse bond energies and barrier heights in catalysis, J. Chem. Phys., 2010, 132(16), 164117 CrossRef PubMed.
- K. Reuter, C. Stampf and M. Scheffler, Handbook of Materials Modeling, SpringerNetherlands, Dordrecht, 2005, pp. 149–194 Search PubMed.
- J. Rogal, Stability, composition and function of palladium surfaces in oxidizing environments: A first-principles statistical mechanics approach, PhD thesis, Freie Universität, Berlin, 2006.
- Y. Ikeda, B. Grabowski and F. Körmann, Ab initio phase stabilities and mechanical properties of multicomponent alloys: A comprehensive review for high entropy alloys and compositionally complex alloys, Mater. Charact., 2019, 147, 464–511 CrossRef CAS.
- B. Cheng, E. A. Engel, J. Behler, C. Dellago and M. Ceriotti, Ab initio thermodynamics of liquid and solid water, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1110–1115 CrossRef CAS PubMed.
- Joseph W. Ochterski, Thermochemistry in Gaussian, https://gaussian.com/wp-content/uploads/dl/thermo.pdf, (accessed 13 January 2024).
- J. Rogal and K. Reuter, Ab Initio Atomistic Thermodynamics for Surfaces: A Primer, Experiment, Modeling and Simulation of Gas-Surface Interactions for Reactive Flows in Hypersonic Flights, Educational Notes RTO-EN-AVT-142, Paper 2, RTO, Neuilly-sur-Seine, France, 2007, pp. 2-1–2-18, http://www.rto.nato.int/abstracts.asp Search PubMed.
- D. R. Galimberti and J. Sauer, Chemically Accurate Vibrational Free Energies of Adsorption from Density Functional Theory Molecular Dynamics: Alkanes in Zeolites, J. Chem. Theory Comput., 2021, 17, 5849–5862 CrossRef CAS PubMed.
- M. Chase, NIST-JANAF Thermochemical Tables, American Institute of Physics, 4th edn, vol. 1, 1998 Search PubMed.
- I. E. Wachs and B. M. Weckhuysen, Structure and reactivity of surface vanadium oxide species on oxide supports, Appl. Catal., A, 1997, 157, 67–90 CrossRef CAS.
- N. Y. Topsoe, J. A. Dumesic and H. Topsoe, Vanadia-Titania Catalysts for Selective Catalytic Reduction of Nitric-Oxide by Ammonia, J. Catal., 1995, 151, 241–252 CrossRef CAS.
- F. D. Hardcastle and I. E. Wachs, Determination of vanadium-oxygen bond distances and bond orders by Raman spectroscopy, J. Phys. Chem., 1991, 95, 5031–5041 CrossRef CAS.
- C. Wang, H. Li, X. Zhang, Y. Qiu, Q. Zhu, S. Xun, W. Yang, H. Li, Z. Chen and W. Zhu, Atomic-Layered α-V2O5 Nanosheets Obtained via Fast Gas-Driven Exfoliation for Superior Aerobic Oxidative Desulfurization, Energy Fuels, 2020, 34, 2612–2616 CrossRef CAS.
- S. Soyer, A. Uzun, S. Senkan and I. Onal, A quantum chemical study of nitric oxide reduction by ammonia (SCR reaction) on V2O5 catalyst surface, Catal. Today, 2006, 118, 268–278 CrossRef CAS.
- A. Vittadini and A. Selloni, Periodic Density Functional Theory Studies of Vanadia−Titania Catalysts: Structure and Stability of the Oxidized Monolayer, J. Phys. Chem. B, 2004, 108, 7337–7343 CrossRef CAS.
- Y.-T. Gu, Y.-M. Gu, Q. Tao, X. Wang, Q. Zhu and J. Ma, Machine learning for prediction of CO2/N2/H2O selective adsorption and separation in metal-zeolites, J. Mater. Inf., 2023, 3(3), 19 CAS.
- Y. Wu, G. Gao and G. Wu, Self-assembled three-dimensional hierarchical porous V2 O5/graphene hybrid aerogels for supercapacitors with high energy density and long cycle life, J. Mater. Chem. A, 2015, 3, 1828–1832 RSC.
- R. J. G. Nuguid, D. Ferri, A. Marberger, M. Nachtegaal and O. Kröcher, Modulated Excitation Raman Spectroscopy of V2O5/TiO2: Mechanistic Insights into the Selective Catalytic Reduction of NO with NH3, ACS Catal., 2019, 9, 6814–6820 CrossRef CAS.
- I. E. Wachs, T. Kim and E. I. Ross, Catalysis science of the solid acidity of model supported tungsten oxide catalysts, Catal. Today, 2006, 116, 162–168 CrossRef CAS.
- T. Ohsaka, F. Izumi and Y. Fujiki, Raman spectrum of anatase, TiO2, J. Raman Spectrosc., 1978, 7, 321–324 CrossRef.
- Th. Wolkenstein, Electron theory of catalysis on semiconductors, Adv. Catal., 1960, 12, 189–264 Search PubMed.
- J. A. Martín-Martín, M. Gallastegi-Villa, M. P. González-Marcos, A. Aranzabal and J. R. González-Velasco, Bimodal effect of water on V2O5/TiO2 catalysts with different vanadium species in the simultaneous NO reduction and 1,2-dichlorobenzene oxidation, Chem. Eng. J., 2021, 417, 129013 CrossRef.
- S. Li, W. Huang, H. Xu, T. Chen, Y. Ke, Z. Qu and N. Yan, Alkali-induced deactivation mechanism of V2O5-WO3/TiO2 catalyst during selective catalytic reduction of NO by NH3 in aluminum hydrate calcining flue gas, Appl. Catal., B, 2020, 270, 118872 CrossRef CAS.
- Q. Liang, J. Li, H. He, T. Yue and L. Tong, Effects of SO2 and H2O on low-temperature NO conversion over F-V2O5-WO3/TiO2 catalysts, J. Environ. Sci., 2020, 90, 253–261 CrossRef CAS PubMed.
- H. Chen, Y. Xia, R. Fang, H. Huang, Y. Gan, C. Liang, J. Zhang, W. Zhang and X. Liu, The effects of tungsten and hydrothermal aging in promoting NH3-SCR activity on V2O5/WO3-TiO2 catalysts, Appl. Surf. Sci., 2018, 459, 639–646 CrossRef CAS.
- G. Busca, G. Centi, L. Marchetti and F. Trifiro, Chemical and spectroscopic study of the nature of a vanadium oxide monolayer supported on a high-surface-area TiO 2 anatase, Langmuir, 1986, 2, 568–577 CrossRef CAS.
- M. Zhu, J.-K. Lai, U. Tumuluri, Z. Wu and I. E. Wachs, Nature of Active Sites and Surface Intermediates during SCR of NO with NH 3 by Supported V2O5–WO3/TiO2 Catalysts, J. Am. Chem. Soc., 2017, 139, 15624–15627 CrossRef CAS PubMed.
- H. W. Siaka, C. Dujardin, A. Moissette and P. Granger, Revisiting the Impact of Tungsten on the Catalytic Properties of Ammonia-SCR V2O5-WO3/TiO2 Catalysts: Geometric vs. Electronic Effects, Chemistry, 2023, 5, 294–313 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00893f |
| This journal is © the Owner Societies 2024 |