
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-dimensional Janus X2STe (X = B, Al) monolayers: the effect of surface selectivity and adsorption of small gas molecules on electronic and optical properties†
Y.
Zengin
and
Y.
Mogulkoc
 *
*
Department of Physics Engineering, Faculty of Engineering, Ankara University, Ankara, 06100, Turkey. E-mail: mogulkoc@eng.ankara.edu.tr
First published on 20th May 2024
Abstract
This investigation delves into the adsorption characteristics of CO, NO, NO2, NH3, and O2 on two-dimensional (2D) Janus group-III materials, specifically Al2XY and B2XY. The examination covers adsorption energies and heights, diverse adsorption sites, and molecular orientations. Employing first-principles analysis, a comprehensive assessment of structural, electronic, and optical properties is conducted. The findings highlight NO2 as a prominent adsorbate, emphasizing the Te surface of 2D Al2STe and B2STe materials as particularly adept for NO2 detection, based on considerations of adsorption energy, height, and charge transfer. Additionally, the study underscores the heightened sensitivity of work function changes in the B2STe material. The adsorption properties of all gas molecules, except for NO2, on both materials were determined to be physical. Upon adsorption of the NO2 gas molecule onto the B2STe Janus material, it was observed that the material exhibited weak chemical adsorption behavior, which was confirmed by the adsorption energy, larger band gap change, electron localization function, work function changes and charge transfer from the material. This research provides valuable insights into the gas-sensing potential of 2D Janus materials.
I. Introduction
Industrial processes, urbanization and vehicle emissions are significant factors contributing to the accumulation of harmful gases in the atmosphere, resulting in air pollution. Both environmentalists and individuals are deeply concerned about this situation.1–3 By measuring the concentration of harmful gases in the atmosphere, gas sensors determine air quality and help reduce health risks.4 Sensors are integral to sustainable urban planning, playing a crucial role in data collection for air pollution monitoring and environmental regulation.5 These devices provide real-time insights into air quality,6 aiding urban planners in informed decision-making. Additionally, the data they generate form the foundation for evidence-based environmental policies, contributing to the overall goal of creating ecologically sound urban environments. Furthermore, insights gleaned from environmental monitoring not only contribute to the evidence-based formulation of environmental policies but also align with research efforts on mitigating environmental pollution by enhancing energy efficiency.7 Within the domain of gas sensing, considerable attention has been directed towards 2D materials, propelled by their unique electronic characteristics and the prospect of exceptionally high theoretical specific surface applications.8,9Nitrogen dioxide (NO2), a highly toxic gas molecule, is a major atmospheric pollutant that causes various environmental pollution phenomena such as acid rain and photochemical smog.10,11 NO2, apart from being a highly detrimental environmental pollutant, also exerts adverse impacts on the human body.12 Elevated NO2 levels in the atmosphere have the potential to cause respiratory conditions such as chronic bronchitis.13 The detection of NO2 gas molecule is critical due to the serious consequences for human health, agricultural production, and climate change. Gas sensors demonstrate tangible utility in common societal contexts and play an indispensable role in the identification of unanticipated security incidents.14 Hence, developing gas-sensing materials with fast response times, full recovery and excellent selectivity has recently been a topic of interest to researchers.
The distinctive electronic properties15,16 and notably high theoretical specific surface areas17 of 2D materials have prompted substantial interest within the realm of gas sensing research. Additionally, with promising outcomes, the investigation into the adsorption of hazardous gas molecules onto two-dimensional materials has been a focal point of extensive research.18–25 In a recent investigation led by Zainal et al.,26 it was discerned that monolayers of silicene and germanene demonstrate heightened efficiency in the detection of CO and NO2, respectively. Furthermore, Abbasi et al.27 demonstrated that MoS2 monolayers possess a high capacity to adsorb gas molecules such as SO2, SO3, and O3, resulting in significant modifications to their electronic characteristics. Additional research has demonstrated that graphene possesses considerable potential for the identification and detection of gas molecules such as NO28 and SO2.29 The C9N4 material exhibits exceptional sensing capabilities when it adsorbs CO, NO, NO2, NH3, and H2S gas molecules.30 In a subsequent study, Yu et al.31 researched the process of adsorption of different atmospheric gas molecules on a two-dimensional polyimide material through hydrogen bonding. Additionally, 2D Janus transition metal chalcogenides (TMDs) and group-III chalcogenides are gaining attention in various research and technological fields due to their unique characteristics.32,33 These include adjustable electronic band gaps, excitonic and piezoelectric effects, and other notable properties.34–36 These studies collectively emphasize the capacity of two-dimensional group-III chalcogenide materials to attract and retain toxic gas molecules, which opens up possibilities for the advancement of sophisticated gas sensors.
The utilization of two-dimensional group-III chalcogenide compounds has garnered considerable attention across diverse applications, including but not limited to electronics, thermoelectric and photoelectric devices, photocatalysis, and chemical sensors, as highlighted in the literature.37–39 Nevertheless, the investigation into boron and aluminum-based chalcogenides is not as comprehensive as that of indium and gallium-based chalcogenide compounds. Considerable attention has been given to studying the mechanical, electrical, optoelectronic, and thermal properties of gallium and indium selenides. Boron and aluminum chalcogenides, characterized by their unique chemical compositions, have recently garnered significant attention in research. The majority of these studies have adopted a more theoretical approach. 2D aluminum-based materials have attracted significant interest due to their unique physico-chemical properties and wide range of potential applications.40 Their exceptional electrical, optical, mechanical, and thermal properties, as well as their high surface-to-volume ratio, make these materials very versatile and have many possible applications in fields as diverse as electronics, optoelectronics, and energy storage.41–43 The application of two-dimensional aluminum-based materials has the potential to yield manufacturing processes that are environmentally sustainable and economically efficient. The consideration of aluminum alloys as potential substitutes for key raw materials in electric vehicles supports this claim.44 Monolayers of 2D Janus Al2XY (X, Y = S, Se, Te; X ≠ Y) have shown remarkable electron mobility, making them well suitable for use in nanoscale electronic devices, as reported in the literature.45 Similarly, 2D Janus B2XY (X, Y = S, Se, Te; X ≠ Y) monolayers have recently emerged as innovative additions to the group-III chalcogenides for nanoelectronics, attributed to their exceptional carrier mobility at room temperature. However, the study of single layer B2XY is still in its infancy, with only structural, electronic, mechanical, transport39 and piezoelectric properties46 reported so far. Although their theoretical properties have been validated, further experimental investigation and validation are essential.47 The study of 2D Al2XY and B2XY Janus materials is an emerging field which, due to their unique properties, has significant potential to contribute to a wide range of practical applications.
In this research endeavor, the focal point is to scrutinize the surface selectivity of 2D monolayers derived from aluminum and boron, with a specific emphasis on their aptitude for adsorbing gas molecules, including CO, NO, NO2, NH3 and O2. For gas-sensing applications, the electrical properties of adsorbed systems are presently under investigation due to their potential to offer crucial information. Diverse adsorption arrangements are evaluated to ascertain the optimal adsorption locales and the associated adsorption energies.
II. Method
First-principles density functional theory (DFT) calculations were employed to conduct spin-polarized simulations of both structural and electronic characteristics. DFT was also used with the nudged elastic band (NEB) method to determine the minimal energy diffusion routes of various crystal phases.48 To execute the computational aspects of this study, the Vienna ab initio simulation package (VASP)49–52 was employed. The exchange–correlation potential was modeled utilizing a generalized gradient approximation (GGA) pseudopotential with the Perdew–Burke–Ernzerhof (PBE)53 functional for performing geometry relaxations and total energy calculations. To preclude any undesirable interactions between adjacent layers, a vacuum spacing of 20 Å along the z direction was maintained. The chosen plane wave cut-off energy was 500 eV. Throughout the optimization procedure, a 4 × 4 supercell was utilized, and the relaxation of all atoms continued until the residual force on each atom reached a level below 0.05 eV Å−1. The convergence criterion for ionic relaxation during the self-consistent computation was set to 10−5 eV for successive iterations. Integration over the Brillouin Zone (BZ) was conducted employing a 4 × 4 × 1 k-point mesh, utilizing the gamma-centered Monkhorst–Pack method.54 The DFT-D2 approach developed by Grimme55 was employed to consider van der Waals (vdW) interactions between two layers. An assessment of thermal stability was conducted through ab initio molecular dynamics (AIMD) analysis. The simulations were executed under canonical ensemble conditions (NVT) using an Andersen thermostat, with temperatures set at both 300 K and 600 K, over a duration of 5 ps. The scrutiny of charge transfer dynamics between the two layers was conducted through the application of the Bader charge analysis method, as endorsed by Henkelman et al.56–58 This methodological approach offers a comprehensive examination of the intricate processes governing the redistribution of charge across the interlayer interface. Visualization for electronic and structural analysis (VESTA) software was utilized to visualize the crystallographic representation and charge densities of the two-dimensional materials.59In delineating the relative stability of distinct structures following the adsorption of small gas molecules, one can articulate the adsorption energy Eads through the following expression, thereby elucidating the intricate interplay between molecular configurations and their corresponding energetic states utilizing the ensuing formulation:
| Eads = Etotal − (EX2STe + Emolecule) | (1) |
To express and compute the difference in charge density that exists between these molecule–monolayer systems, the following formula is utilized:
| Δρ = ρX2STe+molecule − ρX2STe − ρmolecule | (2) |
The analysis of recovery times for adsorbed structures was conducted employing the principles of transition state theory, as outlined in the work by Babariya et al.60 The recovery time (τ), a critical parameter in gas sensor performance, encapsulates the requisite duration for the sensor to restore both sensitivity and precision to optimal levels following exposure to gas, thereby delineating a key temporal aspect influencing its overall effect. This parameter can be calculated utilizing the following formula:
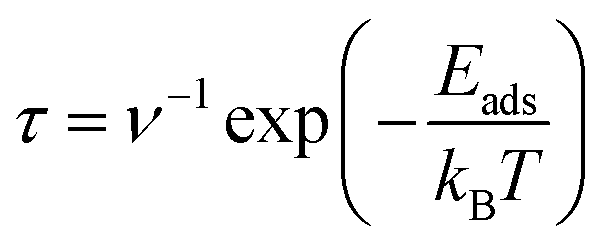 | (3) |
III. Results and discussion
The investigation deals with the adsorption behavior of small gas molecules on 2D Janus group III materials, specifically Al2XY and B2XY, where X ≠ Y and X, Y = S, Se, and Te. The target gases include CO, NO, NO2, NH3, and O2, each undergoing adsorption on both distinctive sides of the two-dimensional Janus materials. (The exploration encompasses all conceivable adsorption configurations, as elaborated in Fig. S1 of the ESI†.) Following the adsorption processes, structural optimization procedures are executed to characterize the stable configurations of the gas–molecule adsorbed Janus materials. After the optimization process, the gas sensing properties of aluminum and boron-based materials are examined by making calculations such as adsorption energies, adsorption heights, spin-polarized electronic band, density of states, charge density, Bader analysis, work function, and optical properties. It is determined that 2D Al2STe and B2STe Janus materials are more stable among these two material groups, and results of the materials belonging to the most stable state configuration are given (additional insights into various materials and molecules are provided in Fig. S2 of the ESI†). The figures provide a comprehensive visual representation of the adsorption configurations and structural optimizations undertaken for a wider range of materials and molecules beyond the previously mentioned Al2XY and B2XY systems. These supplementary details contribute to understanding how an adsorbent behaves and structural characteristics observed across diverse two-dimensional materials interacting with distinct gas molecules.A. Structural analysis
The computational analysis explores an extensive array of adsorption patterns with the aim of identifying the most stable configurations for small gas molecules adsorbed on 2D Al2STe and B2STe Janus materials. Notably, each of these materials possesses two distinct sides, prompting an investigation into appropriate adsorption configurations independently for each side. This meticulous approach involves the exploration of various adsorption arrangements, encompassing both vertical and parallel orientations, along with investigations into top, bridge, and hollow regions. By examining these different adsorption configurations, the study aims to elucidate the optimal configurations that govern the interaction between the studied gas molecules and the surfaces of the Al2STe and B2STe Janus materials.A comprehensive analysis of the most stable states of both materials in Fig. 1 elucidated that the structure made up of S and Te atoms is more stable. The surface selectivity of the Te surface exceeded that of the S surface, as indicated in Table 2. Consequently, the interaction of small gas molecules with the Te surface increased stability. Furthermore, a comparison of the most stable configurations of 2D Al2STe and B2STe Janus materials reveals that small gas molecules are stable in distinctly different orientations. In the 2D Al2STe material, the CO molecule is in the hole position, whereas in the B2STe material, the same molecule is in the bridge position. Additionally, in CO and NO gas molecules, the O atom is located further away from the 2D materials, while in the NO2 molecule, the opposite is the case and the O atom is most stable when it is closer to the materials. An analogous scenario applies to the NH3 molecule and the hydrogen (H) atom, presenting a similar set of considerations in their respective interactions with the investigated materials. Moreover, in the molecule-based comparison of both materials, it is seen that the small gas molecules are closer to the Al2STe material, whereas the same gas molecules are farther away from the B2STe material. Finally, the NO2 gas molecule is the molecule that shifted the most after the adsorption process in both materials. After optimization, the NO2 molecule in the bridge position in the Al2STe material shifted towards the Al atom, whereas the NO2 molecule in the gap position in the B2STe material shifted towards the Te atom.
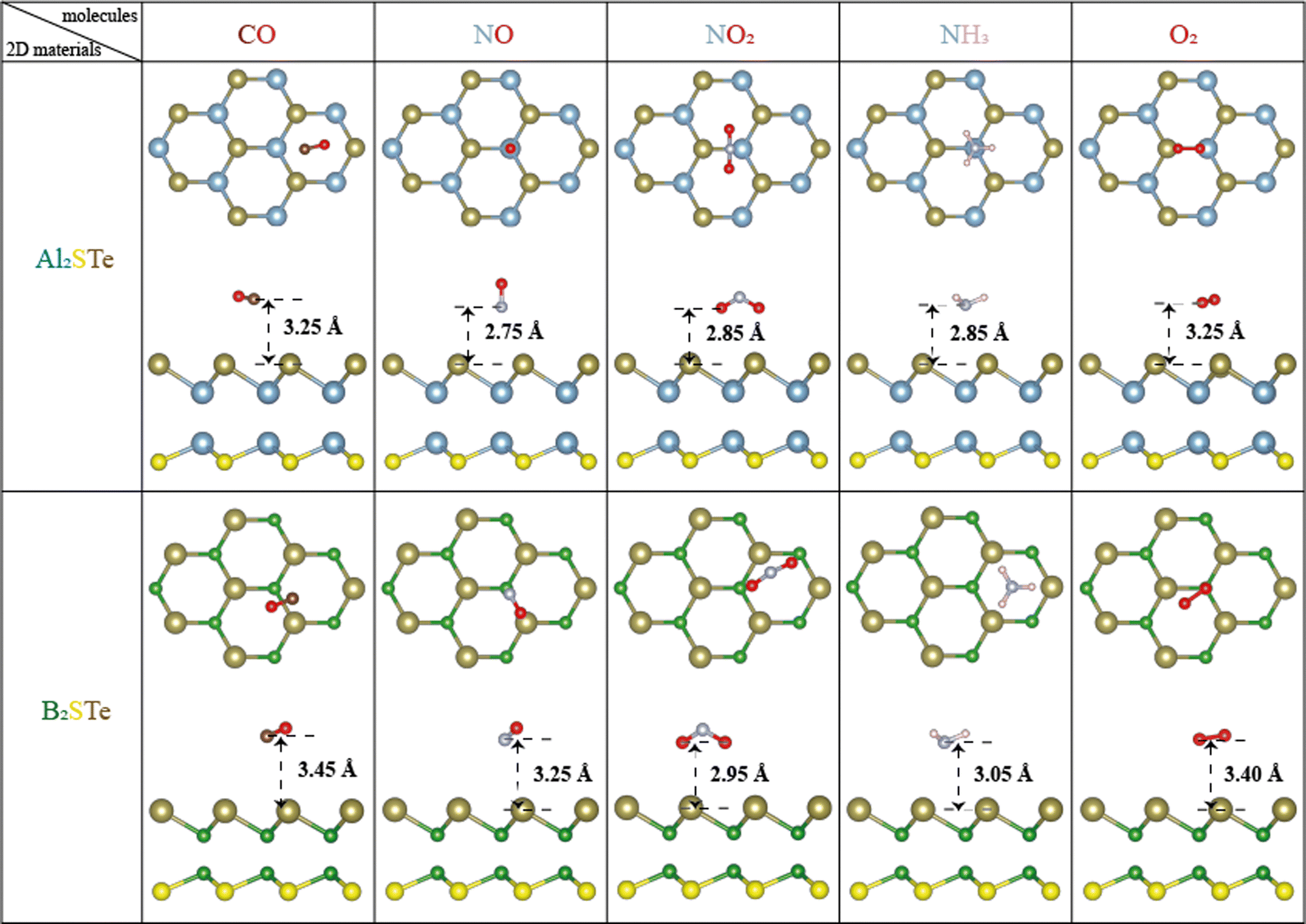 | ||
| Fig. 1 Optimal adsorption configurations of gas molecules (CO, NO, NO2, NH3, and O2) on Al2STe and B2STe monolayers, presented from both top and side views. | ||
Based on the adsorption energy values presented in Table 2, it can be observed that no chemical bonds are formed between the molecules and the 2D materials. As per the data presented in Table 2, the adsorption energies observed for CO, NO, NO2, NH3, and O2 molecules on aluminum and boron-based materials suggest the absence of chemical bond formations between the investigated molecules and the two-dimensional material. Both categories of materials exhibit an increased sensitivity to nitrogen-based molecules, which reflects the trends observed in the adsorption behaviour of the two-dimensional Janus SnSSe material, as reported in the work of Zengin et al.61 In addition, it has been observed that all molecules in aluminum and boron-based materials are more stable on the Te surface. In this case, the surface selectivity of the Te surface is higher than that of the S and Se surfaces in both material groups. Upon adsorption of NO, NO2 and O2 gases onto the surfaces of both Al2STe and B2STe 2D materials, the emergence of unpaired electrons initiates interactions within the monolayers, inducing a magnetic moment. In addition, a careful analysis of recovery times at room temperature (300 K) is carried out to elucidate the desorption kinetics of the adsorbed gases. The recovery times are determined employing the formula outlined in part II, and the obtained results are comprehensively presented in Table 2. The data reveal a significant correlation between the lower adsorption energies of gas molecules on Al2STe and B2STe monolayers and shorter recovery times. This trend is likely attributed to the distinct adsorption energy profiles observed for gas molecules interacting with Al2STe and B2STe monolayers. Furthermore, the recovery times, falling within the detectable range,62 strongly signify exceptionally rapid recovery dynamics.
To enhance comprehension of the interaction between 2D Janus materials and molecules, we calculate the adsorption energy of each small gas molecule by varying its distance from the material. Energy values are calculated by adjusting the distance between the centre of mass of the molecules and the top of the 2D materials from 1.75 to 5.00 Å. The determination of adsorption height involves identifying the height value corresponding to the lowest energy within the system. As depicted in Fig. 2, the adsorption heights of small gas molecules in the context of van der Waals (vdW) interactions span a range from 2.75 to 3.45 Å. The correlation between the adsorption height and adsorption energies involved can serve as a general measure of the intensity of interactions in adsorption systems. The adsorption of NO molecules onto Al2STe materials is an exceptional case. This may be attributed to the affinity of the nitrogen (N) atom within the NO2 molecule, which is located closer to the 2D Al2STe material and exhibits an attractive interaction with the aluminium atoms. Although the adsorption height can be used as a broad indicator of the intensity of interaction in adsorption systems, it is not universally true that the adsorption energy decreases along with the decrease in adsorption height.62 Nevertheless, the NH3 molecule exhibits lower adsorption energy compared to the NO2 molecule in the Al2STe material, even though both molecules possess identical adsorption heights. The primary factor contributing to this phenomenon can be attributed to the low electronegativity of the H atoms in the NH3 molecule.
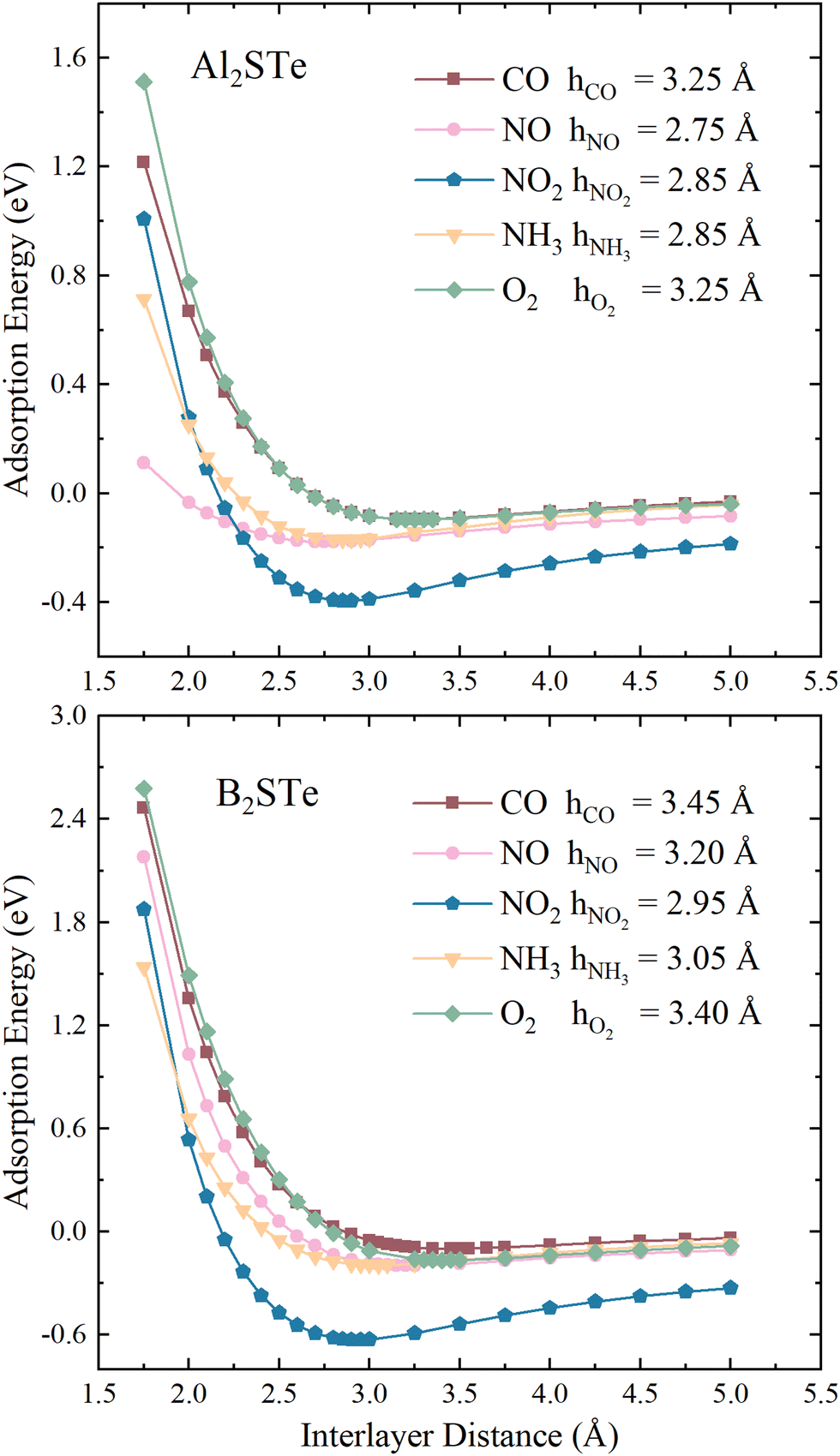 | ||
| Fig. 2 Variation of adsorption energies for toxic gas molecules on Al2STe and B2STe with respect to interlayer distance. | ||
The observations indicate that the gas molecule NO2 has a higher adsorption energy when the adsorption height is smaller in magnitude, whereas CO and O2 have lower adsorption energy when the adsorption height is larger in magnitude. Fig. 2 illustrates that when the distance between the molecule and the material is 5.00 Å, the curves approach the asymptotic value for all molecules. It is seen that this value is about 0 eV, as in 2D Al2STe and B2STe materials. However, the NO2 molecule approaches the asymptotic value more slowly. This is due to its stronger van der Waals bond with Al2STe and B2STe monolayers, as well as its higher adsorption energy compared to other gas molecules. Moreover, an asymmetric value of 0 eV signifies that no adsorption energy will be produced when the molecules are distant from the 2D material. This implies that there will be no interaction between the material and the molecules, thus the material will not be able to detect the molecules. It has been observed that when the molecules are brought close enough to the 2D Al2STe and B2STe Janus materials (such as 1.75 Å and 2.00 Å), the adsorption energy becomes positive. In this case, external energy must be supplied for the molecules to hold on to the material at close range. Exothermic physical reactions require external energy to occur spontaneously.
The thermal stabilities of small gas molecules adsorbed onto Al2STe and B2STe 2D Janus materials are examined using ab initio molecular dynamics (AIMD) simulation. The calculations are performed at temperatures of 300 K and 600 K with the Anderson thermostat in a canonical ensemble. The overall duration of the simulation is 5 ps with time intervals of 1 ps. Fig. 3 displays the variation in energy throughout the whole simulation and includes an image representing the energy at the end of 5 ps. The AIMD simulations demonstrate that the total energy of both monolayers remains constant throughout the simulation, implying the absence of any structural distortion in the material. Therefore, it can be concluded that both materials are thermodynamically stable at 300 K and 600 K. On the basis of the mentioned factors of adsorption energy, adsorption height and recovery time, it can be inferred that Al2STe and B2STe materials possess exceptional device characteristics as gas sensors for the detection of the NO2 gas molecule. Furthermore, this outcome aligns with research conducted on the empirical identification of the NO2 gas compound.63,64
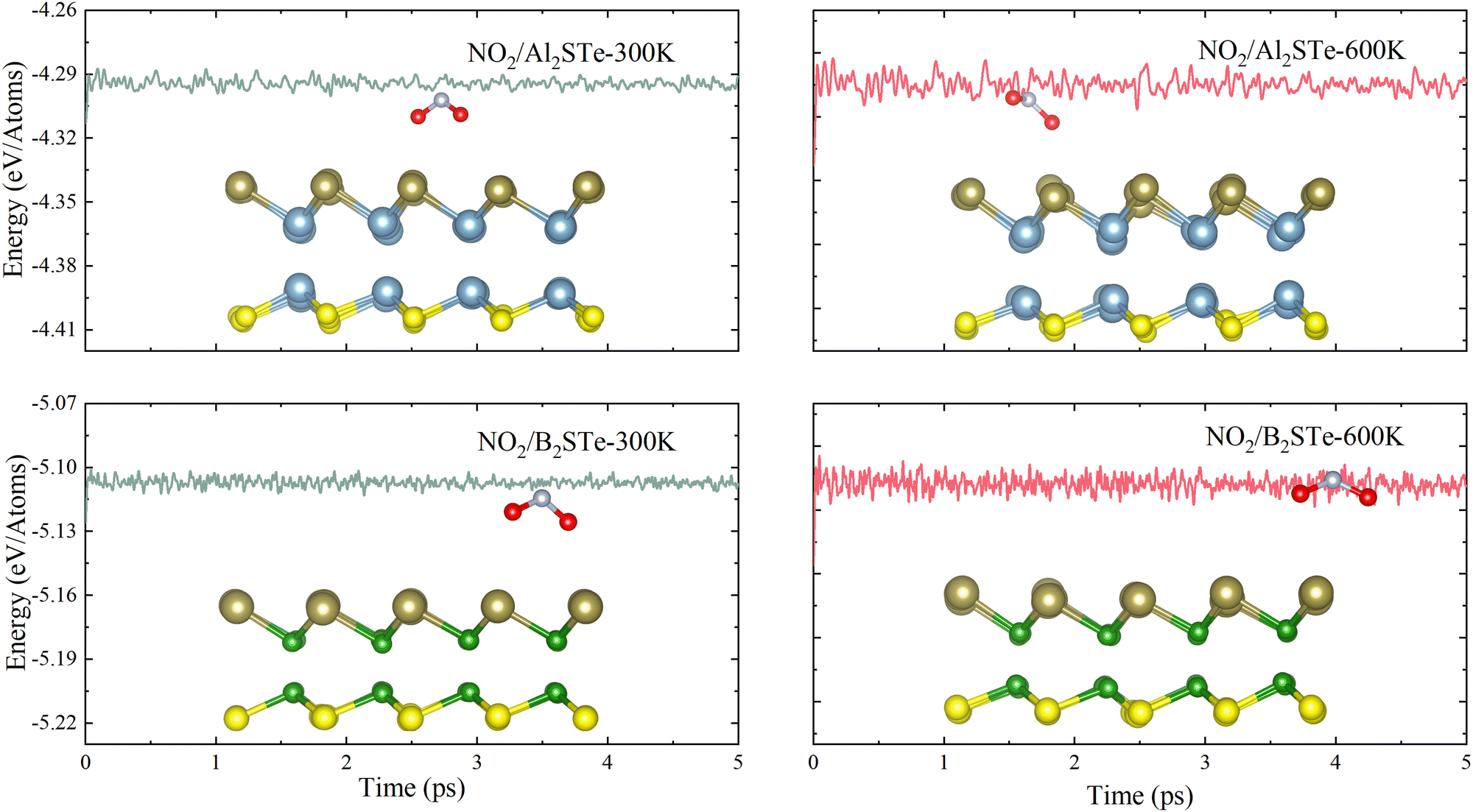 | ||
| Fig. 3 The energy changes for Al2STe and B2STe at temperatures of 300 and 600 K, respectively, and side view of the final snapshots obtained from ab initio molecular dynamics simulation. | ||
B. Electronic properties
To gain a better understanding of the interaction between gas molecules and 2D Janus materials, we conduct spin-polarized electronic band and density of state calculations after their adsorption onto the 2D material. Table 2 demonstrates that the adsorption of CO and NH3 molecules onto aluminium and boron-based materials has minimal impact on the band gaps and band transitions, which remains almost unchanged compared to pristine Al2STe and B2STe. Additionally, due to the spin-up and down symmetry of these two molecules, they are unable to generate magnetic moments. Comparing NO, NO2, and O2 molecules to their pristine state reveals alterations in band gap energies. The evident asymmetry in the spin-up and down states gives rise to the establishment of a magnetic moment in each of the three molecules. Moreover, the adsorption of NO, NO2, and O2 gases onto Al2STe and B2STe materials induces the formation of flat energy levels within their respective band structures. These findings align with the observations deduced from the partial density of states (PDOS) analysis of adsorbed NO, NO2, and O2 molecules on monolayers of Al2STe and B2STe. (For a more detailed exploration of the band structures, please refer to Fig. S3 and S4 in the ESI†.) The contributions of N and O atoms are discernible in the formation of these flat energy levels, as elucidated by the PDOS.Fig. 4 depicts the computational analysis of band structures and partial density of states (PDOS) for Al2STe and B2STe 2D materials in the presence of the NO2 molecule, identified as the most stable adsorption configuration. These calculations are conducted employing spin-polarized calculations to discern the impact of molecular adsorption on the electronic properties. Additionally, band structures and PDOS are computed for the pristine materials to facilitate a comparative analysis between the adsorbed and non-adsorbed states. The pristine Al2STe material manifests a direct transition with a band gap of 1.256 eV. Upon adsorption of the NO2 molecule, the material retains its direct transition nature, but there is a significant alteration in its electronic structure. This results in a notable reduction of the band gap to 0.12 eV. In contrast, the behavior observed in boron-based materials deviates slightly. Unlike Al2STe, the B2STe material acts as a semiconductor with an initial indirect transition and a band gap value of 1.034 eV before adsorption. Nevertheless, when the NO2 molecule is adsorbed, the material experiences a change in its semiconductivity, leading to the emergence metallic characteristics. The transition from a semiconductor to a conductive state has occurred in the adsorption material as a result of the displacement of the valence band above the Fermi level, as illustrated in Fig. 4d. The alteration in the band structures of aluminum and boron-based materials upon adsorption implies that the detection of NO2 is going to be facilitated when utilized as a gas sensor.
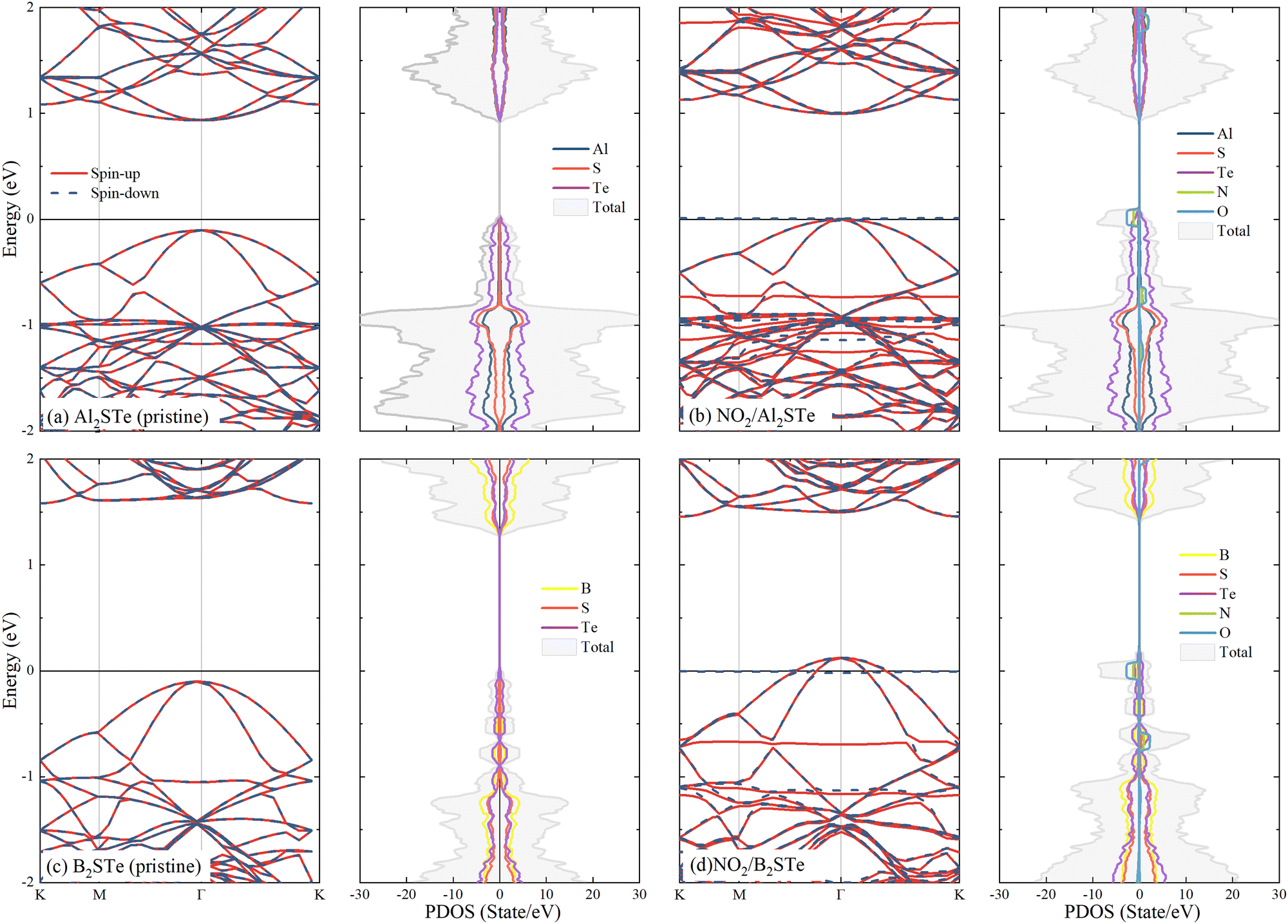 | ||
| Fig. 4 Display of band structures and partial density of states (PDOS) of molecules adsorbed to Al2STe and B2STe materials (using spin-polarized calculations with the Fermi level set to zero). | ||
Finally, the electronic band structure curves of 2D aluminum and boron-based materials reveal the presence of two spin-up flats positioned near the Fermi level. One band resides within the valence band, while the other is situated in the conduction band. Adsorption of the NO2 molecule on Al2STe and B2STe materials results in the formation of flat bands at the Fermi energy, as indicated by the PDOS results. The existence of flat bands can be attributed to the strong oxidizing properties and orbital couplings of the NO2 gas molecule. Moreover, this is associated with the significant charge transfer of the NO2 molecule as compared to other molecules. Therefore, the generation of flat bands around the Fermi energy is expected to improve the electrical transmission performance. These findings seem to align with previous research on the NO2 molecule that has been published in the literature.14 Additionally, previous investigations22,27,65 have observed that comparing the expected partial density of states (PDOS) of a system pre- and post-adsorption could provide insight into the nature of the adsorption. The figure illustrates a modification in the PDOS distribution of the system subsequent to the adsorption of the NO2 gas molecule onto the B2STe material. More precisely, the PDOS curve has an upward shift, whereas the total curve expands significantly. The adsorption of the NO2 gas molecule has a noticeable impact on the electron state density in the system. All of these observations validate the characteristics of chemisorption.
The transfer of charge between gas molecules being adsorbed and 2D materials is a key factor affecting the performance of gas sensors. The resistivity of the system can be altered due to the charge transfer that occurs as a result of adsorption. Resistivity variability is an important indicator of gas sensor performance and can be evaluated by experimental testing. Fig. 5 illustrates the variations in charge density observed between NO2 gas molecules and 2D Al2STe and B2STe materials.
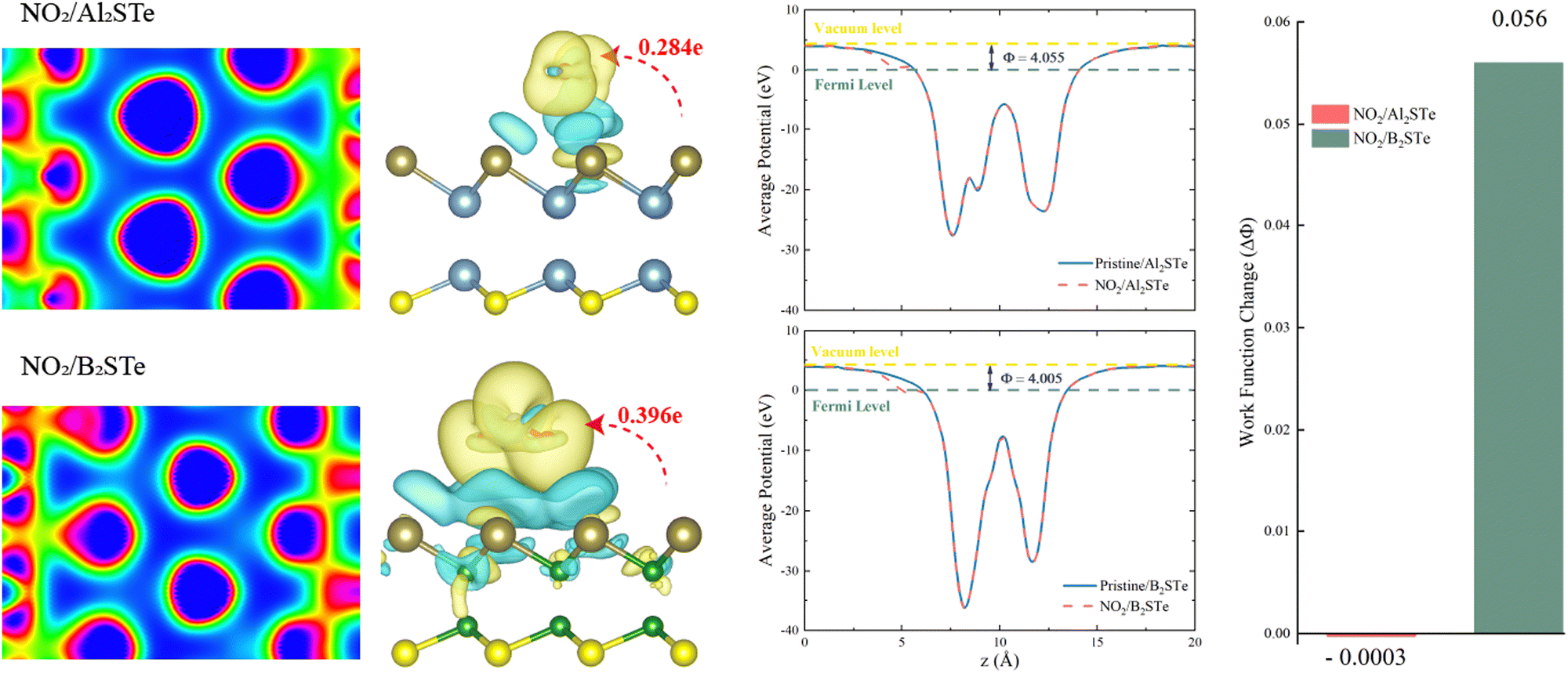 | ||
| Fig. 5 Electron localization function (ELF), charge density differences, averaged potential, and work function change of NO2 gas molecule plots for Al2STe and B2STe monolayers. Charge accumulation (depletion) is shown by the yellow (blue) region. The value of the isosurface is calculated as 5 × 10−4 e Å−3. The direction and amount of the charge transfer are also indicated. | ||
The yellow region is representative of the accumulation of charges, while the blue region is representative of the depletion of charges. Compared to other molecules, the charge distributions around atoms in NO2 gas molecules are higher for both 2D material groups. The alteration in charge distribution results in a more robust interaction between the surface of the adsorbent and the NO2 gas molecule. The adsorption energies, adsorption heights, and charge transitions that are presented in Table 2 are consistent with the result that is obtained from the Bader analysis. Positive or negative charge transfers by gas molecules indicate the admission or donation of electrons to the monolayer. Furthermore, the Bader analysis is performed to determine the magnitude of charge transfer exhibited by gas molecules upon adsorption onto monolayers composed of aluminum and boron. The aforementioned data is illustrated in Fig. 5. The results indicate that the NO2 molecule functions as an electron donor in both categories of materials. A considerable degree of charge migration is detected among monolayers of NO2 molecules, indicating an elevated level of sensitivity. Moreover, the findings indicate a significant interaction between the NO2 molecule and the two-dimensional B2STe material. After conducting comprehensive analysis of the adsorption energy between NO2 and B2STe, it is evident that a weak chemical bond is established.62 The improved effectiveness of the B2STe material in detecting the NO2 gas molecule can be ascribed to the existence of this chemical bond. Furthermore, it is evident from the findings from the analysis of charge density and the Bader charge analysis with electron localization function (ELF) values, which are 0.63 for Al2STe and 0.70 for B2STe. The Al2STe material exhibits a lower ELF value compared to the B2STe material due to its weaker charge density. Hence, it can be inferred that the NO2 gas molecule exhibits a higher degree of interaction with the B2STe material. This finding highlights that the B2STe material exhibits a greater capacity to form chemical bonds with the NO2 molecule compared to the Al2STe material. The observed chemical adsorption behavior can be attributed to the exceptional electron acceptance/donation behavior exhibited by the B2STe material through its interaction with the NO2 gas molecule.
The work function, a significant parameter in solid-state applications, is defined as the amount of energy needed to move an electron from its Fermi level to its vacuum level on a solid surface. The work function can be expressed using the following formula:
| Φ = Evac − EF | (4) |
Furthermore, the change in work function is defined as the following:
| ΔΦ = ΦX2STe+gas − ΦX2STe | (5) |
Fig. 5 demonstrates that the work function of Al2STe and B2STe materials is nearly identical. The work function remains nearly constant even after the adsorption of NO2 gas molecules. However, the B2STe material demonstrates a more significant change in work function upon adsorption, despite its initially lower work function. It has been demonstrated that the adsorption of the NO2 gas molecule onto the Al2STe material has negligible effects on the work function. The noticeable work function change for the system in which the NO2 gas molecule adsorbs onto the B2STe material is due to the short adsorption height and the significant charge transfer between the molecule and the material. The response of the NO2 gas molecule in terms of work function indicates that the B2STe material has selectivity, and the change in work function significantly improves the probability of gas molecule detection.
To enhance the analysis of the gas sensing abilities of the materials being studied, the sensitivity (S) is determined by analyzing the conductivity parameter. The fundamental equations required to compute sensitivity are listed below.67
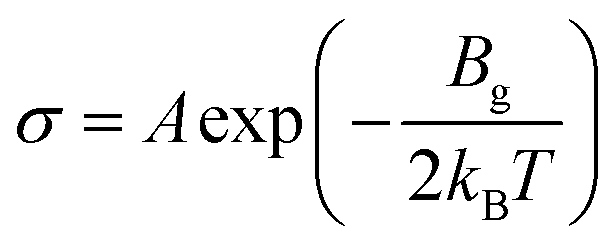 | (6) |
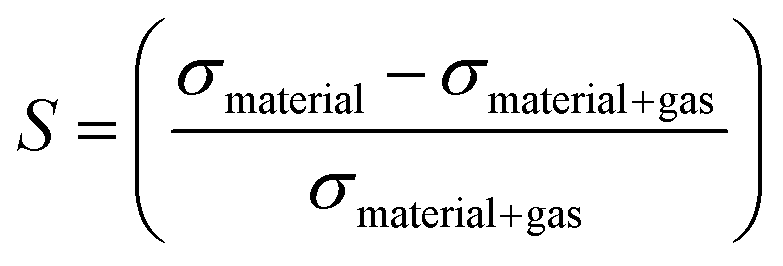 | (7) |
Based on the findings presented in Table 1, it is evident that the sensitivity for the identification of CO, NO, NO2, NH3, and O2 gas molecules can attain a maximum value of −100.0% for NO, NO2, and O2 gas molecules. Furthermore, the obtained sensitivity results are consistent with the load transfer values. Al2Ste and B2STe materials exhibit a notable level of sensitivity in detecting these gases. NO, NO2, and O2 molecules have a substantial impact on the conductivity of pure materials when compared to other molecules adsorbed onto the two-dimensional Janus material. Therefore, it can be inferred that both materials exhibit potential as a viable option for a resistance-type sensor in the identification of NO, NO2, and O2 gas molecules. Furthermore, the selective detection of these three molecules can be accomplished by leveraging the varying magnitudes of conductivity changes.65
| Material | Gas | Site | Interface | h (Å) | E ads (eV) | M (μB) | Δ (|e|) | τ (ns) | S (300 K) | S (600 K) | E gap (eV) | Symmetry |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Al2STe | CO | H-Site | Te | 2.95 | −0.089 | 0.00 | −0.025 | 0.041 | −19.16 | −10.10 | 1.023 | Γ–Γ |
| NO | T-Site | Te | 2.75 | −0.102 | 1.00 | −0.045 | 0.079 | −100.00 | −100.00 | 0.223 | M′–Γ | |
| NO2 | B-Site | Te | 2.50 | −0.286 | 0.78 | −0.284 | 362.1 | −100.00 | −100.00 | 0.056 | Γ–Γ | |
| NH3 | T-Site | Te | 2.60 | −0.163 | 0.00 | 0.028 | 0.417 | −10.95 | −5.64 | 1.028 | Γ–Γ | |
| O2 | T-Site | Te | 3.10 | −0.084 | 1.94 | −0.086 | 0.020 | −100.00 | −100.00 | 0.101 | Γ–Γ | |
| B2STe | CO | B-Site | Te | 3.45 | −0.099 | 0.00 | −0.028 | 0.046 | −7.44 | −3.80 | 1.379 | Γ–Γ |
| NO | H-Site | Te | 3.20 | −0.196 | 1.08 | −0.103 | 1.957 | −100.00 | −100.00 | 0.017 | M′–Γ | |
| NO2 | B-Site | Te | 2.95 | −0.629 | 0.64 | −0.396 | 3.7 × 107 | −100.00 | −100.00 | — | Metal | |
| NH3 | H-Site | Te | 3.05 | −0.195 | 0.00 | 0.005 | 1.883 | 8.04 | 3.94 | 1.387 | Γ–Γ | |
| O2 | T-Site | Te | 3.40 | −0.166 | 1.86 | −0.161 | 0.613 | −100.00 | −100.00 | — | Metal | |
| Material | d N–O (Å) | A (°) | μ (Å) | |
|---|---|---|---|---|
| Pre-adsorption | NO2/Al2STe | 0.9998 | 119.9871 | −0.5689 |
| NO2/B2STe | 0.9998 | 119.9871 | 0.3221 | |
| Post-adsorption | NO2/Al2STe | 1.2317 | 127.1356 | −0.9186 |
| NO2/B2STe | 1.2389 | 124.4851 | −0.2457 | |
The presence of a dipole moment in systems signifies the existence of an internal electric field. In this study, the internal electric fields of two-dimensional Al2STe and B2STe Janus materials are measured to be −0.5689 e Å and 0.3221 e Å, respectively, prior to the adsorption of the NO2 molecule. The negative sign denotes the orientation of the internal electric field, signifying its movement from Te to S. Upon the adsorption of the NO2 molecule, the dipole moment values exhibit an increase to −0.9186 e Å and −0.2457 e Å, respectively. The total dipole moment of Al2STe and B2STe materials is enhanced by the presence of NO2 gas molecules. The Al2STe material exhibits a more pronounced alteration in the magnitude of the dipole moment. In addition, the NO2 molecule in the B22Te material alters the orientation of the dipole moment. The total dipole moment and the charge transfer between molecules and materials are in the same direction following adsorption.
C. Optical properties
The impacts on the optical characteristics of Al2STe and B2STe that have been adsorbed with the NO2 toxic gas molecule are calculated to examine the potential future uses of 2D Janus Al2STe and B2STe materials as an optical gas-sensitive material. As illustrated in Fig. 6, the final objective of this study is to compare the optical characteristics of the pristine and adsorbed states of 2D Al2STe and B2STe Janus materials about their dielectric function, absorption coefficient, and reflection. In all cases, the adsorption of the NO2 gas molecule has not resulted in any noticeable changes to the observed peaks on the graph.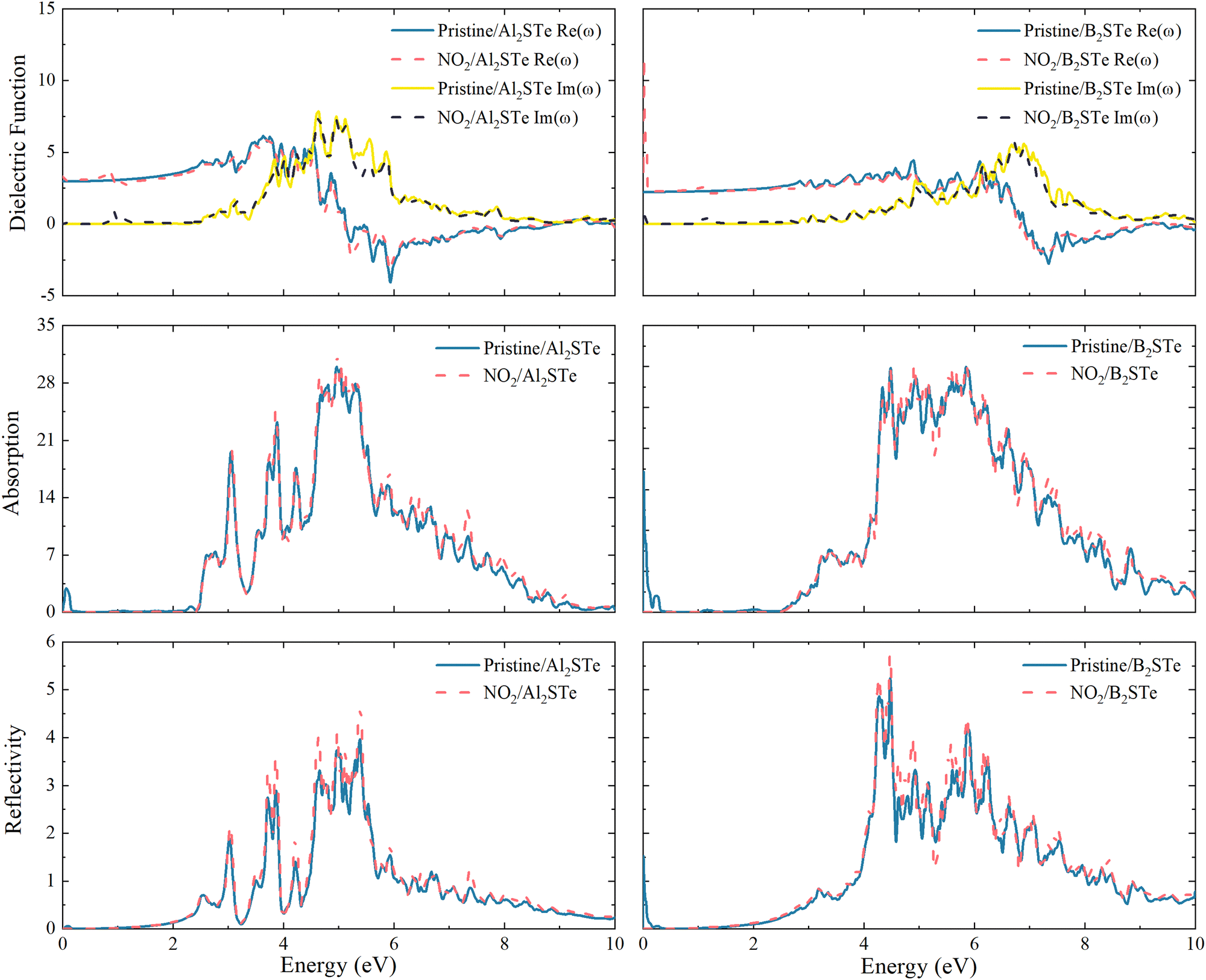 | ||
| Fig. 6 Real part, imaginary part, adsorption coefficient and reflectivity of Al2STe and B2STe monolayers before and after adsorption. | ||
The dielectric function is a crucial optical parameter that describes the optical properties of materials in terms of their ability to absorb and polarise light. It explains the way in which light propagates through a given material. The function is a complex number with real and imaginary parts, typically dependent on frequency. The real part of the material describes how it transmits or reflects light, while the imaginary part indicates how much energy the material absorbs. In Fig. 6 the peaks of both the real and imaginary parts of the dielectric function are prominent in the ultraviolet region of the electromagnetic spectrum. The imaginary component of the dielectric constant becomes zero at energy levels of around 8 and 12 eV for 2D Al2STe and B2STe materials, respectively. This indicates that these materials become transparent above these energies.
The absorption graph of Fig. 6 demonstrates that 2D Al2STe and B2STe materials absorb in the ultraviolet spectrum but not at low energies (infrared). The highest points observed in this area represent transitions between bands. In the absorption graph in Fig. 6, which is compatible with the dielectric graph, the absorption edges of the pristine state of the 2D Al2STe Janus material and the adsorbed state of the NO2 molecule correspond to almost the same region. The highest absorption coefficient for both its pristine state (29.98 cm−1 at 5.04 eV) and its adsorbed state (31.42 cm−1 at 4.97 eV) is found in the ultraviolet region, and in both cases, the optical absorption ranges are constant to the same width. The phenomenon concerning the absorption edge applies to the B2STe molecule as well. The highest absorption coefficient of the B2STe material in its pristine state (29.98 cm−1 at 5.04 eV) and in the NO2 molecule adsorbed state (31.42 cm−1 at 4.97 eV) is found in the ultraviolet region, and in both cases, the optical absorption ranges are constant to the same width. The absorption plot in Fig. 6 depicts the absorption characteristics of the 2D Al2STe and B2STe materials, revealing their ability to absorb ultraviolet light while exhibiting no absorption at low energy levels (infrared). The highest points observed in this area represent transitions between bands.
Reflective qualities relate to the ability of a substance to reflect light or radiation. Fig. 6 clearly shows the reflection (R) curve of the NO2 gas molecule adsorbed on aluminum and boron-based 2D Janus materials. For each of these pristine materials, the most prominent reflection peaks are observed at energy levels of 14.12 eV and 20.1 eV, respectively. Observations indicate a small rise in the peak points following the adsorption of the NO2 gas molecule. Both pristine and adsorbed materials have two different peaks in the ultraviolet (UV) spectra, with energy levels of around 5.40 eV and 4.80 eV, respectively. The UV spectrum retains the strongest reflection peaks even after gas molecules are adsorbed. The reflection curve has a comparable pattern to the absorption curve.
IV. Conclusion
Briefly, in this study, the adsorption behavior of CO, NO, NO2, NH3 and O2 small gas molecules on 2D Janus Group III Al2XY and B2XY (X ≠ Y and X, Y = S, Se and Te) materials has been determined. The study investigates the adsorption energies and heights of the interaction between the materials and gas molecules. To determine stable configurations, various adsorption sites and molecule orientations are considered. This study based on first principles is employed to examine the structural, electrical, and optical properties of gas molecules following their adsorption onto materials. The study, which included the vdW interactions between materials and molecules, observed that the toxic gas molecule NO2, which is widely acknowledged as the most crucial greenhouse gas, yields more notable outcomes in comparison to other molecules. Furthermore, it is determined that the Te surface of 2D Al2STe and B2STe materials could detect NO2 gas molecules more easily in terms of adsorption energy, adsorption height, and charge transfer than other materials and surfaces. The weak chemisorption behavior of NO2 gas molecules onto the B2STe Janus material has been observed and confirmed by charge transfer, adsorption energy, larger band gap change, and electron localization function. Upon examination of the work function changes of Al2STe and B2STe materials, it is evident that the B2STe material is capable of more sensitive measurements.Author contributions
Yasin Zengin: data curation, conceptualization, writing – original draft, visualization, methodology, formal analysis, conceptualization and validation. Yesim Mogulkoc: project administration, funding acquisition, conceptualization, writing – original draft, writing – review and editing, supervision, validation and software.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is supported within the scope of the Ankara University Scientific Research Project with code number FBA-2023-2800. The computations are carried out at the High Performance and Grid Computing Center (TR-Grid e-Infrastructure) located at TUBITAK ULAKBIM.References
- D. Upadhyay, B. Roondhe, A. Pratap and P. K. Jha, Two-dimensional delafossite cobalt oxyhydroxide as a toxic gas sensor, Appl. Surf. Sci., 2019, 476, 198–204, DOI:10.1016/j.apsusc.2019.01.057.
- D. Pandey, C. Kamal, R. Dutt and A. Chakrabarti, Improved gas adsorption on functionalized aluminene surface: a first-principles study, Appl. Surf. Sci., 2020, 531, 147364, DOI:10.1016/j.apsusc.2020.147364.
- M. Jyothi, V. Nagarajan and R. Chandiramouli, Adsorption studies of 2,3-butanedione and acetic acid on ζ-phosphorene sheets based on the first-principles study, Comput. Theor. Chem., 2022, 1208, 113548, DOI:10.1016/j.comptc.2021.113548.
- Y. Mao, L. Long, J. Yuan, J. Zhong and H. Zhao, Toxic gases molecules (NH3, SO2 and NO2) adsorption on GeSe monolayer with point defects engineering, Chem. Phys. Lett., 2018, 706, 501–508, DOI:10.1016/j.cplett.2018.06.061.
- X. Chen, C. K. Wong, C. A. Yuan and G. Zhang, Nanowire-based gas sensors, Sens. Actuators, B, 2013, 177, 178–195 CrossRef CAS.
- T. Alaa Hussein, N. M. Shiltagh, W. Kream Alaarage, R. R. Abbas, R. A. Jawad and A. H. Abo Nasria, Electronic and optical properties of the BN bilayer as gas sensor for CO2, SO2, and NO2 molecules: a DFT study, Results Chem., 2023, 5, 100978, DOI:10.1016/j.rechem.2023.100978.
- J. J. Escobar, J. Ortega, M. Damas, R. S. Kızıltepe and J. Q. Gan, Energy-Time Analysis of Convolutional Neural Networks Distributed on Heterogeneous Clusters for EEG Classification, Adv. Comput. Intelli., 2019, 11507, 895–907, DOI:10.1007/978-3-030-20518-8_74.
- N. A. Tukadiya, S. K. Jana, B. Chakraborty and P. K. Jha, C24 Fullerene and its derivatives as a viable glucose sensor: DFT and TD-DFT studies, Surfaces Interfaces, 2017, 41, 103220, DOI:10.1016/j.surfin.2023.103220.
- S. Y. Xia, L. Q. Tao, T. Jiang, H. Sun and J. Li, Rh-doped h-BN monolayer as a high sensitivity SF6 decomposed gases sensor: a DFT study, Appl. Surf. Sci., 2017, 536, 147965, DOI:10.1016/j.apsusc.2020.147965.
- L. Liu, S. Li, J. Zhuang, L. Wang, J. Zhang, H. Li, Z. Liu, Y. Han, X. Jiang and P. Zhang, Improved selective acetone sensing properties of Co-doped ZnO nanofibers by electrospinning, Sens. Actuators, B, 2011, 155, 782–788, DOI:10.1016/j.snb.2011.01.047.
- Y. Qin and Z. Zhang, Co-modulation of graphene by N hetero-doping & vacancy defects and the effect on NO2 adsorption and sensing: First-principles study, Phys. E, 2020, 116, 113737, DOI:10.1016/j.physe.2019.113737.
- A. Abbasi and J. J. Sardroodi, Exploration of sensing of nitrogen dioxide and ozone molecules using novel TiO2/Stanene heterostructures employing DFT calculations, Appl. Surf. Sci., 2018, 442, 368–381, DOI:10.1016/j.apsusc.2018.02.183.
- J. Kong, N. R. Franklin, C. Zhou, M. G. Chapline, S. Peng, K. Cho and H. Dai, Nanotube molecular wires as chemical sensors, Science, 2000, 287, 622–625, DOI:10.1126/science.287.5453.622.
- Z. Cui, K. Yang, Y. Shen, Z. Yuan, Y. Dong, P. Yuan and E. Li, Toxic gas molecules adsorbed on intrinsic and defective WS2: gas sensing and detection, Appl. Surf. Sci., 2023, 613, 155978, DOI:10.1016/j.apsusc.2022.155978.
- D. Kecik, E. Durgun and S. Ciraci, Stability of single-layer and multilayer arsenene and their mechanical and electronic properties, Phys. Rev. B, 2016, 94, 205409, DOI:10.1103/PhysRevB.94.205409.
- V. Nagarajan and R. Chandiramouli, Twisted bilayer arsenene sheets as a chemical sensor for toluene and M-xylene vapours – A DFT investigation, J. Mol. Graphics Modelling, 2021, 109, 108034, DOI:10.1016/j.jmgm.2021.108034.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Atomically thin MoS2: a new direct-gap semiconductor, Phys. Rev. Lett., 2010, 105, 136805, DOI:10.1103/PhysRevLett.105.136805.
- A. Abbasi and J. J. Sardroodi, Exploration of sensing of nitrogen dioxide and ozone molecules using novel TiO2/Stanene heterostructures employing DFT calculations, Appl. Surf. Sci., 2018, 442, 368–381, DOI:10.1016/j.apsusc.2018.02.183.
- A. Abbasi, A. Abdelrasoul and J. J. Sardroodi, Adsorption of CO and NO molecules on Al, P and Si embedded MoS2 nanosheets investigated by DFT calculations, Adsorption, 2019, 25, 1001–1017, DOI:10.1007/s10450-019-00121-6.
- A. Abbasi and J. J. Sardroodi, Investigation of the adsorption of ozone molecules on TiO2/WSe2 nanocomposites by DFT computations: Applications to gas sensor devices, Appl. Surf. Sci., 2018, 436, 27–41, DOI:10.1016/j.apsusc.2017.12.010.
- A. Abbasi and J. J. Sardroodi, Theoretical study of the adsorption of NOx on TiO2/MoS2 nanocomposites: a comparison between undoped and N-doped nanocomposites, J. Nanostruct. Chem., 2016, 6, 309–327, DOI:10.1007/s40097-016-0204-3.
- A. Abbasi, Tuning the structural and electronic properties and chemical activities of stanene monolayers by embedding 4d Pd: a DFT study, RSC Adv., 2019, 9, 16069–16082, 10.1039/C9RA01472A.
- A. Abbasi and J. J. Sardroodi, The adsorption of sulfur trioxide and ozone molecules on stanene nanosheets investigated by DFT: Applications to gas sensor devices, Phys. E, 2016, 108, 382–390, DOI:10.1016/j.physe.2018.05.004.
- A. Abbasi and J. J. Sardroodi, Density functional theory investigation of the interactions between the buckled stanene nanosheet and XO2 gases (X = N, S, C), Comput. Theor. Chem., 2018, 1125, 15–28, DOI:10.1016/j.comptc.2017.12.010.
- A. Abbasi, Theoretical investigation of the interaction between noble metals (Ag, Au, Pd, Pt) and stanene nanosheets: a DFT study, J. Inorg. Organomet. Polym. Mater., 2019, 29, 1895–1915, DOI:10.1007/s10904-019-01151-x.
- M. A. Zainal, C. K. Tim, H. Zainuddin, N. M. Shah and R. O. C. Heng, First principles study of toxic gas molecules adsorption on Group IVA (C, Si, Ge) 2-dimensional materials, Sains Malaysiana, 2023, 52, 625–639, DOI:10.17576/jsm-2023-5202-23.
- A. Abbasi, Adsorption of phenol, hydrazine and thiophene on stanene monolayers: a computational investigation, Synth. Met., 2019, 247, 26–36, DOI:10.1016/j.synthmet.2018.11.012.
- H. Gau and Z. Liu, DFT study of NO adsorption on pristine graphene, RSC Adv., 2017, 7, 13082–13091, 10.1039/C6RA27137E.
- Q. Zhou, W. Ju, X. Su, Y. Young and X. Li, Adsorption behavior of SO2 on vacancy defected graphene: a DFT study, J. Phys. Chem. Solids, 2017, 109, 40–45, DOI:10.1016/j.jpcs.2017.05.007.
- Y. Hu, L. Liu, S. Zhang and X. Chen, Investigating the adsorption performance of CO, NO, NO2, NH3, and H2S molecules on C9N4 through DFT for potential gas sensor applications, Colloids Surf., A, 2024, 689, 133716, DOI:10.1016/j.colsurfa.2024.133716.
- S. Yu, D. Zhang, W. Pan and J. Zeng, Adsorption of atmospheric gas molecules (NH3, H2S, CO, H2, CH4, NO, NO2, C6H6 and C3H6O) on two-dimensional polyimide with hydrogen bonding: a first-principles study, New J. Chem., 2021, 45, 5240–5251, 10.1039/D0NJ06013E.
- M. Modarresi, A. Kakoee, Y. Mogulkoc and M. R. Roknabadi, Effect of external strain on electronic structure of stanene, Comput. Mater. Sci., 2015, 101, 164–167, DOI:10.1016/j.commatsci.2015.01.039.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Two-dimensional gas of massless Dirac fermions in graphene, Nature, 2005, 438, 197–200, DOI:10.1038/nature04233.
- X.-L. Li, T.-J. Lou, X.-M. Sun and Y.-D. Li, Highly sensitive WO3 hollow-sphere gas sensors, Inorg. Chem., 2004, 43, 5442–5449, DOI:10.1021/ic049522w.
- H. Zhu, Y. Wang, J. Xiao, M. Liu, S. Xiong, Z. J. Wong, Z. Ye, Y. Ye, X. Yin and X. Zhang, Observation of piezoelectricity in free-standing monolayer MoS2, Nat. Nanotechnol., 2015, 10, 151–155, DOI:10.1038/nnano.2014.309.
- M. M. Alyörük, Y. Aierken, D. Çakur, F. M. Peeters and C. Sevik, Promising piezoelectric performance of single layer transition-metal dichalcogenides and dioxides, J. Phys. Chem. C, 2015, 119, 23231–23237, DOI:10.1021/acs.jpcc.5b06428.
- M. Wasala, H. I. Sirikumara, Y. Raj Sapkota, S. Hofer, D. Mazumdar, T. Jayasekera and S. Talapatra, Recent advances in investigations of the electronic and optoelectronic properties of group III, IV, and V selenide based binary layered compounds, J. Mater. Chem. C, 2017, 5, 11214–11225, 10.1039/C7TC02866K.
- S. Demirci, N. Avazl, E. Durgun and S. Cahangirov, Structural and electronic properties of monolayer group III monochalcogenides, Phys. Rev. B, 2017, 95, 115409, DOI:10.1103/PhysRevB.95.115409.
- V. Kumar and J. Jung, Two-dimensional janus group-iii ternary chalcogenide monolayer compounds B2XY, Al2XY, and BAlX2 (X, Y = S, Se, Te) with high carrier mobilities, Bull. Korean Chem. Soc., 2022, 43, 138–146, DOI:10.1002/bkcs.12440.
- W. Wang, M. Pei, X. Zhao, C. Xia, T. Wang, X. Dai and S. Wei, Interface-dependent electronic structure and optical properties of staggered Al2STe/Ga2STe van der Waals heterostructures, Phys. Status Solidi RRL, 2024, 2300362, DOI:10.1002/pssr.202300362.
- P. Kumbhakar, J. S. Jayan, A. S. Madhavikutty, P. R. Sreeram, A. Saritha, T. Ito and C. S. Tiwary, Prospective applications of two-dimensional materials beyond laboratory frontiers: A review, iScience, 2023, 26(5), 1–24, DOI:10.1016/j.isci.2023.106671.
- M. C. Lemme, D. Akinwande, C. Huyghebaert and C. Stampfer, 2d materials for future heterogeneous electronics, Nat. Commun., 2022, 13, 1392, DOI:10.1038/s41467-022-29001-4.
- G. S. Khosa, S. Tripathi, A. Alshaikhi, S. Gupta and R. Kumar, Janus Al2STe monolayer: a prospective thermoelectric material, Solid State Commun., 2022, 341, 114579, DOI:10.1016/j.ssc.2021.114579.
- Y. Wei, X. Tang, J. Shang, L. Ju and L. Kou, Two-dimensional functional materials: from properties to potential applications, Int. J. Smart Nano Mater., 2020, 11, 247–264, DOI:10.1080/19475411.2020.1790055.
- T. V. Vu and N. N. Hieu, Novel janus group iii chalcogenide monolayers Al2XY2 (X/Y = S, Se, Te): first-principles insight onto the structural, electronic, and transport properties, J. Phys.: Condens. Matter, 2021, 34, 115601, DOI:10.1088/1361-648X/ac4401.
- X.-J. Yan, W.-Y. Li, X. Zou, L. li Liu, S.-F. Wang, Y. Wei, C.-M. Yang, Y.-F. Sun and L. Hu, Janus B2XY (X, Y = S, Se, Te) monolayers as piezoelectric materials: a first-principle study, Chem. Phys. Lett., 2022, 806, 140007, DOI:10.1016/j.cplett.2022.140007.
- M. Bhavyashree, S. R. Rondiya and K. Hareesh, Exploring the emerging applications of the advanced 2-dimensional material borophene with its unique properties, RSC Adv., 2022, 12, 12166–12192, 10.1039/d2ra00677d.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 12166–12192, DOI:10.1063/1.1329672.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561, DOI:10.1103/PhysRevB.47.558.
- G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269, DOI:10.1103/PhysRevB.49.14251.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50, DOI:10.1016/0927-0256(96)00008-0.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186, DOI:10.1103/PhysRevB.54.11169.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192, DOI:10.1103/PhysRevB.13.5188.
- S. Grimme, Semiempirical gga-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799, DOI:10.1002/jcc.20495.
- W. Tang, E. Sanville and G. Henkelman, A grid-based bader analysis algorithm without lattice bias, J. Phys.: Condens. Matter, 2009, 21, 084204, DOI:10.1088/0953-8984/21/8/084204.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, Improved grid-based algorithm for bader charge allocation, J. Comput. Chem., 2007, 28, 899–908, DOI:10.1002/jcc.20575.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360, DOI:10.1016/j.commatsci.2005.04.010.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276, DOI:10.1107/S0021889811038970.
- B. Babariya, D. Raval, S. K. Gupta and P. N. Gajjar, Selective and sensitive toxic gas-sensing mechanism in a 2D janus MoSSe monolayer, Phys. Chem. Chem. Phys., 2022, 24, 15292–15304, 10.1039/D2CP01648F.
- Y. Zengin, R. Caglayan and Y. Mogulkoc, Adsorption of small gas molecules onto the two-dimensional janus SnSSe monolayer, Comput. Condens. Matter, 2023, 36, e00815, DOI:10.1016/j.cocom.2023.e00815.
- J.-H. Li, J. Wu and Y.-X. Yu, DFT exploration of sensor performances of two-dimensional WO3 to ten small gases in terms of work function and band gap changes and I–V responses, Appl. Surf. Sci., 2021, 546, 149104, DOI:10.1016/j.apsusc.2021.149104.
- S. Xue, S. Cao, S. Huang, D. Yang and G. Zhang, Improving gas-sensing performance based on MOS nanomaterials: a review, Materials, 2021, 14, 4263, DOI:10.3390/ma14154263.
- H. J. Choi, S. H. Knov, W. S. Lee, K. G. Im, T. H. Kim, B. R. Noh, S. Park, S. Oh and K. K. Kim, Ultraviolet Photoactivated Room Temperature NO2 Gas Sensor of ZnO Hemitubes and Nanotubes Covered with TiO2 Nanoparticles, Nanomaterials, 2020, 10, 4263 Search PubMed.
- G. Guo, J. Min, Y. Xu, Y. Zhou and G. Xu, Gas Sensing Properties of Pd-Decorated GeSe Monolayer toward Formaldehyde and Benzene Molecules: A First-Principles Study, Langmuir, 2023, 40, 997–1006, DOI:10.1021/acs.langmuir.3c03221.
- S. Yang, C. Jiang and S.-H. Wei, Gas sensing in 2D materials, Appl. Phys. Rev., 2017, 4, 021304, DOI:10.1063/1.4983310.
- H. Cui, X. Zhang, Y. Li, D. Chen and Y. Zhen, First-principles insight into Ni-doped InN monolayer as a noxious gases scavenger, Appl. Surf. Sci., 2019, 494, 859–866, DOI:10.1016/j.apsusc.2019.07.218.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00380b |
| This journal is © the Owner Societies 2024 |