
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoelectron spectroscopy of the deprotonated tryptophan anion: the contribution of deprotomers to its photodetachment channels†
Jemma A.
Gibbard
 *a,
Catherine S.
Kellow
a and
Jan. R. R.
Verlet
ab
*a,
Catherine S.
Kellow
a and
Jan. R. R.
Verlet
ab
aDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: jemma.gibbard@durham.ac.uk
bJ. Heyrovský Institute of Physical Chemistry, Czech Academy of Sciences, Dolejškova 3, 18223 Prague 8, Czech Republic
First published on 5th April 2024
Abstract
Photoelectron spectroscopy and electronic structure calculations are used to investigate the electronic structure of the deprotonated anionic form of the aromatic amino acid tryptophan, and its chromophore, indole. The photoelectron spectra of tryptophan, recorded at different wavelengths across the UV, consist of two direct detachment channels and thermionic emission, whereas the hν = 4.66 eV spectrum of indole consists of two direct detachment features. Electronic structure calculations indicate that two deprotomers of tryptophan are present in the ion beam; deprotonation of the carboxylic acid group (Trp(I)−) or the N atom on the indole ring (Trp(II)−). Strong similarities are observed between the direct detachment channels in the photoelectron spectra of tryptophan and indole, which in conjunction with electronic structure calculations, indicate that electron loss from Trp(II)− dominates this portion of the spectra. However, there is some evidence that direct detachment of Trp(I)− is also observed. Thermionic emission is determined to predominantly arise from the decarboxylation of Trp(I)−, mediated by the ππ* excited state near λ = 300 nm, which results in an anionic fragment with a negative electron affinity that readily autodetaches.
Introduction
The photoprocesses of biomolecules are often understood by considering the structure and dynamics of smaller photoactive chromophores that are part of the molecule. For example, the aromatic amino acid tryptophan, which has an indole chromophore, absorbs strongly in the UV with a similar absorption profile to indole in solution, as a result of excitation to an optically bright state with ππ* character (S1), as shown for the deprotonated anions in Fig. 1(a).1–4 However, tryptophan subsequently fluoresces at longer wavelengths (308–350 nm) with variable quantum yields, depending on the solvation of the chromophore, the local structure of the protein, the charge localisation within the molecule, and the presence of quenching.5,6 Therefore in order to inform our understanding of the complex behaviour of tryptophan within solution or a protein environment, it is instructive to understand the underlying structure and dynamics of the isolated tryptophan molecule in the gas phase.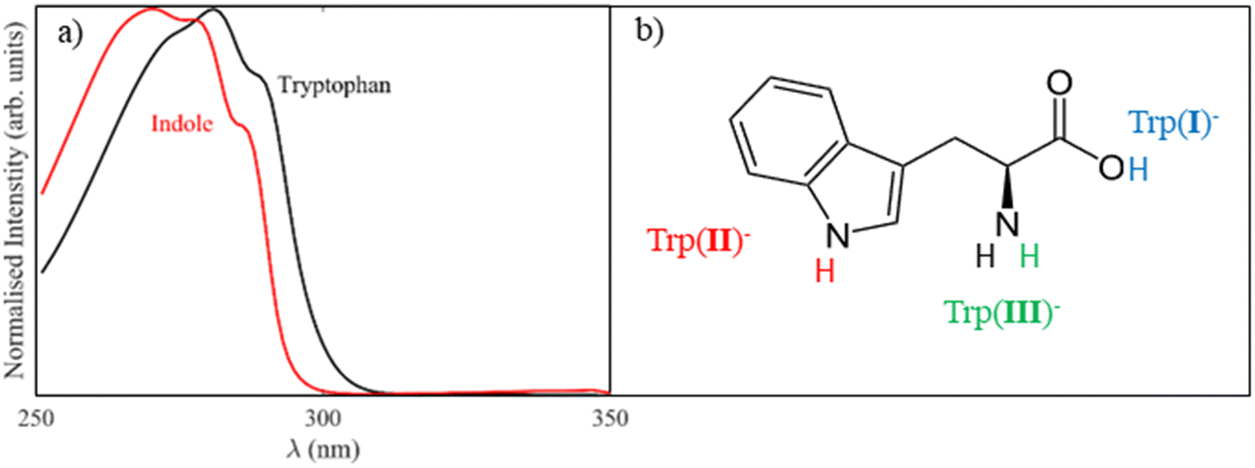 | ||
| Fig. 1 (a) The UV/vis absorption spectrum of the deprotonated tryptophan (black) and indole (red) anions demonstrates the similar absorption profile of the S1 states of the two molecules in solution. (b) Deprotonation of tryptophan can occur at three different sites: (I) on the carboxylic acid group (blue), (II) on the N atom of indole (red) and (III) on the amine group (green), leading to three distinct deprotomers of the tryptophan anion. | ||
The presence of a negative charge in the tryptophan molecule, i.e. from deprotonation, causes a redshift of the bright ππ* excited state by ∼25 nm to 315 nm, as reported by the gas-phase electron action spectrum of Compagnon et al..7 However, screening by the solvent minimises the observed redshift in solution (<3 nm), meaning that our UV/vis spectrum of the solution-phase deprotonated tryptophan anion (Fig. 1a) is very similar to the previously reported absorption spectrum of neutral tryptophan.1,8 Photoelectron spectroscopy, which is a powerful technique for studying the electronic and nuclear structure of anions, has been performed on a number of gas-phase amino acids, including the water clusters of tryptophan, and the chromophore of tryptophan (indole), but not on tryptophan itself.2,9–12 Recent photoelectron spectroscopy studies of deprotonated indole by Nelson et al. reported an electron affinity of 2.4315 eV for the neutral (radical) form, whilst Parkes et al. assigned a higher electron binding energy feature (eBE ∼3.2 eV) to the formation of neutral radical indole in an excited state (D1).9,10 In addition to direct detachment, photoelectron spectra recorded at wavelengths resonant with excitation to the S1 state showed autodetachment to the D0 neutral ground state, but no ground state recovery of the S0 anion state was observed.9
One complexity in studying tryptophan is that aromatic amino acids can be deprotonated at different sites leading to the formation of distinct deprotomer anions, which can have distinctive chemistry.11,13 The possible deprotomers of tryptophan are shown in Fig. 1(b). Evidence for the presence of multiple stable deprotomers of tryptophan in the gas phase is found in the neutral fragment spectrum of Nobel et al., which demonstrated the onset of electron loss near λ = 440 nm, i.e. at much longer wavelength (lower energy) than Compagnon et al. reported.13 Furthermore, the site of deprotonation in tryptophan determined the fate of the neutral after electron loss, as deprotonation of the N atom in the indole ring resulted in formation of a stable neutral after photodetachment, whereas deprotonation on the carboxylic acid group resulted in CO2 and electron loss.13 In the current work, we use photoelectron spectroscopy to study the electronic structure and electron detachment channels of deprotonated gas-phase indole and tryptophan anions, considering the roles of different deprotomers on the observed photoelectron spectra.
Methods
Photoelectron spectroscopy was performed using an apparatus which has been described in detail elsewhere.14,15 Briefly anions were produced via electrospray ionization of a 1 mM solution of tryptophan or indole in methanol, with several drops of ammonia. Gas-phase anions enter the apparatus via a capillary and are guided and trapped using a series of radio-frequency fields before acceleration using a Wiley–McLaren time of flight spectrometer.16 Mass selected anions of tryptophan and indole are intersected with nanosecond light of variable frequency, produced either via an optical parametric oscillator pumped using a Nd:YAG, which can access visible or UV wavelengths, or directly from the harmonics of a Nd:YAG. Photoelectrons are guided onto a position sensitive electron detector using a velocity map imaging setup. The resulting images are processed using a polar onion peeling algorithm, to extract electron kinetic energy (eKE) spectra and photoelectron angular distributions, characterised by an anisotropy parameter, –1< β2 < 2.17,18 The spectrometer was calibrated using the known photoelectron spectrum of iodide and has a resolution of ∼5% of the electron kinetic energy (eKE). Photoelectron imaging of tryptophan was performed at hν = 3.49 eV (λ = 355 nm), 3.80 eV (326 nm), 3.96 eV (313 nm), 4.13 eV (300 nm) and 4.66 eV (266 nm), whilst indole was studied at hν = 3.49 eV and 4.66 eV.Electronic structure calculations were performed using density functional theory at the B3LYP level of theory with the aug-cc-pVTZ basis set using the Gaussian 16 package.19–21 Geometry optimisations were performed, with the minima confirmed via vibrational analysis. The relative energetics were subsequently calculated, including zero-point energy corrections, and results tabulated as electron affinities (EA), vertical detachment energies (VDE) and dissociation asymptotes. Excited state calculations for S1 in Trp(I)− and Trp(II)− were also performed using time dependent density functional theory with the Tamm–Dancoff approximation in Gaussian 16, at several different levels of theory (B3LYP/aug-cc-pVTZ, B3LYP/6-311G++** and Cam-B3LYP/aug-cc-pVTZ).20–24 These computational methods were selected as they previously been used to study the excited states of deprotonated tryptophan or indole.7,9
To understand the electronic excited states and the deprotomers present of tryptophan, absorption spectra were recorded in the solution and gas phase. Solution phase absorption spectra were recorded using a Cary 5000 UV-vis-NIR spectrophotometer for 0.1 mM solutions of indole and tryptophan in methanol, with a large excess of ammonia, where anionic deprotonated forms dominate. Furthermore, a gas-phase action (absorption) spectrum for deprotonated tryptophan was recorded by measuring the electron yield as a function of photon energy using the photoelectron imaging spectrometer. The electron action spectrum is only reported at wavelengths where we had good signal to noise levels (292 nm < λ < 400 nm), as at longer wavelengths we had lower signal levels and at shorter wavelengths there was a significant contribution from electron noise.
Results
(a) Absorption spectra of tryptophan
To determine the location of the excited states of gas phase deprotonated tryptophan we recorded the electron action spectrum between hν = 3.10–4.16 eV (λ = 298–400 nm). Given that photodetachment is the dominant decay mechanism for deprotonated tryptophan in the gas-phase,13 this spectrum is a gas phase analogue to the solution phase absorption spectrum shown in Fig. 1(a), and for comparison both spectra are shown in Fig. 2. Electron loss occurs at all the wavelengths studied in the gas phase, but there is a significant increase in electron signal for hv > 3.70 eV (λ < 335 nm), which is still increasing in relative intensity at the highest photon energies investigated (4.16 eV). The spectrum also shows structure, with further maxima at hv = 3.96 and 3.82 eV (λ = 313 and 325 nm).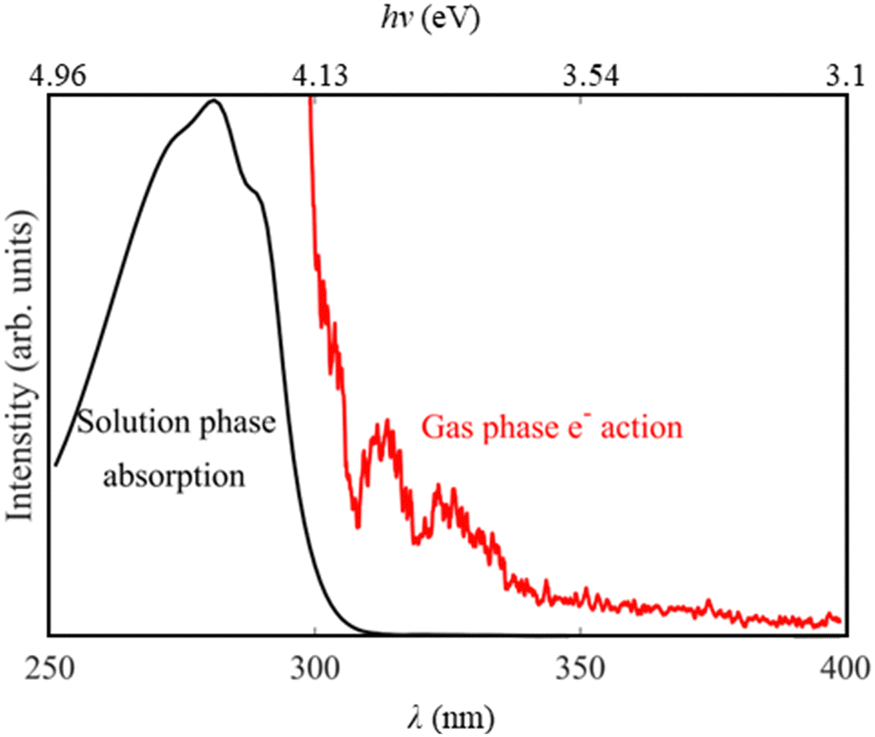 | ||
| Fig. 2 The solution phase absorption spectrum (black) and the gas-phase electron action spectrum (red) of deprotonated tryptophan. The spectra have been arbitrarily scaled and therefore the relative intensities between the two are not directly comparable. | ||
(b) Photoelectron spectroscopy of tryptophan
The photoelectron spectra of deprotonated tryptophan recorded at hν = 4.66, 4.13, 3.96, 3.80 and 3.49 eV (λ = 266, 300, 313, 326 and 355 nm) are shown in Fig. 3(a) and (b), on an electron kinetic energy (eKE) and electron binding energy (eBE) scale, respectively, where eKE + eBE = hν. There are three different spectral features observed in the photoelectron spectra, which are highlighted in Fig. 3: an intense feature at eKE = 0 eV (red), a broad band centred at eBE = 3.1 eV (blue), and another broad band centred at eBE = 3.9 eV (green). | ||
| Fig. 3 The photoelectron spectra of deprotonated tryptophan recorded at hν = 4.66 eV, 4.13 eV, 3.96 eV, 3.80 eV and 3.49 eV on an (a) electron kinetic energy and (b) electron binding energy scale. Three spectral features are highlighted (1) thermionic emission (red), (2) direct detachment at eBE =3.9 eV (green) and (3) direct detachment at eBE = 3.1 eV (blue). | ||
Two features in the spectra occur at a fixed eBE, as expected for a direct photodetachment process. The first has an adiabatic detachment energy (ADE) of 2.7 eV and a vertical detachment energy (VDE) of 3.1 eV, whilst the second band has an ADE of 3.5 eV and a VDE of 3.9 eV. Both bands are relatively broad (∼0.7 eV), structureless and are somewhat overlapping. At the lower hv, the lower eBE band dominates the spectra, whereas at higher hv, when both direct detachment pathways are energetically accessible, then the higher eBE band has a higher relative intensity.
The presence of photoelectrons at or near eKE = 0 eV is characteristic of thermionic emission.25,26 This occurs when electrons are lost via a statistical process from hot ground state anions: either directly from the parent species (in this case tryptophan), or from a photofragment. This process dominates the hν = 4.13 and 3.96 eV photoelectron spectra, but it is barely present at 3.49 eV and of a lower relative intensity at 4.66 eV. Given that the other spectral features arise from direct detachment (blue and green in Fig. 3), then the change in the relative intensity of the thermionic emission peak is a measure of the S0 → S1 photoexcitation cross section.27,28 Therefore we conclude that the S1 state lies around 300 to 315 nm, in good agreement with the previous gas-phase action spectroscopy of tryptophan.7 The contribution of the thermionic emission and direct detachment to the overall photoelectron spectrum of tryptophan is shown in Fig. S1 (ESI†), where the signal arising from thermionic emission is modelled using an exponential function fitted to the low eKE portion of the spectrum and the residual signal (spectrum – fit) is assigned to direct detachment.
(c) Photoelectron Spectroscopy of indole
To understand the electronic structure of the deprotonated tryptophan anion more fully, we also studied its isolated chromophore, the indole deprotonated anion (In−). The photoelectron spectra of the In−, recorded at hν = 4.66 eV and 3.49 eV, are shown in Fig. 4 on an eBE scale. Our In− spectrum at hν = 3.49 eV is in agreement with the previously reported spectra of Parkes et al. and Nelson et al.9,10 It consists of a poorly resolved vibrational progression starting at eBE = 2.4 eV, associated with direct detachment to the neutral ground state (D0), and a second low intensity feature centred at eBE = 3.2 eV assigned to direct detachment to the D1 excited state of the neutral.9,10 At higher photon energies, e.g., hν = 4.66 eV, the direct detachment feature forming the D1 state broadens out and increases in relative intensity, presumably as more of the Franck–Condon envelope for photodetachment to D1 becomes energetically accessible. The ADE = 2.8 eV and VDE = 3.3 eV for photodetachment to the D1 excited neutral state of In−. | ||
| Fig. 4 The photoelectron spectra of deprotonated indole recorded at hν = 4.66 eV (266 nm) and 3.49 eV (355 nm) on eBE scale. | ||
(d) Electronic structure calculations
Tryptophan can be deprotonated at three different sites: at the N atom on either the indole ring or the amine group, or at the O atom within the carboxylic acid group, leading to three distinct deprotomers, as shown in Fig. 1(b). We abbreviate the deprotonated tryptophan anion here as Trp(X)−, where X indicates the specific deprotomer. Electronic structure calculations, presented in Table 1, indicate that the carboxylate deprotomer, Trp(I)−, is the global anion minimum, but deprotonation on the indole group, Trp(II)−, is only 0.04 eV higher in energy, suggesting that both deprotomers are likely be present in the ion beam (where the internal energy of the ions is thermalised to ∼300 K). The amide deprotomer, Trp(III)−, is much less stable (1.79 eV higher in energy) and is, therefore, unlikely to be present in the experiments. The electron affinity (EA) of each of the deprotomers were calculated and found to be 2.76 eV and 3.49 eV for Trp(II) and Trp(I) respectively. These values match the experimental ADEs of the two direct detachment bands in the tryptophan photoelectron spectra (Fig. 3(b)).| Species | Relative E/eV | EA of neutral/eV | VDE/eV |
|---|---|---|---|
| Trp(I)− | 0 | 3.49 | 3.53 |
| Trp(II)− | 0.04 | 2.76 | 2.91 |
| Trp(III)− | 1.79 | 1.36 | 1.62 |
| Trp-CO2− + CO2 | 2.75 | −0.23 | — |
| In− | n/a | 2.36 | 2.47 |
Additionally, calculations were performed on the carboxylate deprotomer, Trp(I)−, as tryptophan has been shown to undergo dissociative photodetachment in previous work.13 Interesting, the CO2 loss channel is at a significantly lower energy than electron loss from Trp(I)−, by 0.74 eV. Finally, we note that the computed EA for Trp-CO2 is negative (as is that for CO2), suggesting that decarboxylation will lead to electron loss by rapid autodetachment.
The computed vertical excitation energies for Trp(I)− and Trp(II)− to S1, which are included in the ESI† (Table S1), varied significantly (∼0.4 eV) depending on the functional and basis set used. Similar large variations in computed VEE were reported by Parkes et al. for deprotonated indole, highlighting the challenges in performing accurate excited state calculations on aromatic anions.9,29 In such circumstances it is challenging to quantitatively rely upon the computed results, however consistently we find that the VEE for the Trp(I)− is larger than Trp(II)− and that excitation to the S1 excited state for both deprotomers will occur near 300 nm.
Discussion
Two deprotomers are likely to be present in the experiment given that the electronic structure calculations in Table 1 indicate that the Trp(I)−and Trp(II)− are very close in energy (∼0.04 eV). This is consistent with previous gas-phase spectroscopic studies of aromatic amino acids using electrospray ionization which have reported multiple deprotomers, including the work of Nobel et al. on tryptophan.11,13Direct evidence for the presence of multiple deprotomers in our ion beam arises from our electron action spectrum (Fig. 2). Previous work by Compagnon et al. has shown that the S1 (ππ*) excited state of Trp(I)− is redshifted by 25 nm in the gas phase, but that the effect is minimised in solution as a result of screening by the solvent.7,8 The comparision of our absorption and electron action spectra from the solution and gas phase (Fig. 2) may confirm this red shift, but it is hard to be certain as we do not reach the maxima of the gas phase action spectrum. However, our action spectrum seems to have the onset of electron loss at longer wavelengths than the photodetachment action spectrum of Compagnon et al., which provides evidence for the presence of two deprotomers in our anion beam.7 Our electron action spectrum is much closer to the combined stable and dissociative fragment action spectra of Nobel et al., which report the yield of Trp and Trp-CO2 as a function of wavelength.30 Two deprotomers were thought to be contributing to the fragment action spectra in that case, with Trp(II)− photodetaching at hv > 2.8 eV (λ < 440 nm), and a dissociative Trp(I)− channel switching on at hv > 3.8 eV (λ < 330 nm).13
However there are some differences between the structure of our electron action spectrum and the photofragment action spectra of Nobel et al., which may arise from a number of sources.13 Firstly, the deprotomer distribution within our ion beam is likely to be different from previous experiments. Secondly, whilst both deprotomers are likely to have a bright S1 excited state between hv ≈ 3.8–4.1 eV (λ ≈ 330–300 nm), the actual redshift of the S1 state is likely to depend upon the site of deprotonation and therefore the proximety of the negative charge to the π electron system. For example, the vertical excitation energy (VEE) of S1 (ππ*) in In−, is calculated to lie between hv = 3.8–4.0 eV (λ = 326–310 nm), whereas the S1 (ππ*) of deprotonated Trp(I)− is near hv ≈ 4.1 eV (λ ≈ 300 nm) from electron action spectroscopy, with both states assigned to ππ* character via electronic structure calculations.7,9 Our excited state calculations (ESI,† Table S1) also indicate that the VEE is higher for Trp(I)− than Trp(II)−. Thirdly, for a given deprotomer there may be structure in the photodetachment cross section for excitation to S1, as is observed in the solution phase absorption spectra (Fig. 1), where it is likely only Trp(I)− is present. Finally, changes in the laser fluence across the visible regime, which are charactersistic of our optical paramentric oscillator may contribute. Taken together these factors make it difficult to accurately assign all the features in our gas-phase electron action spectrum.
The photoelectron spectra for deprotonated tryptophan shown in Fig. 3 contain three spectral features: two direct photodetachment channels with ADE ∼2.7 eV (blue, Fig. 3(b)) and ∼3.5 eV (green, Fig. 3(b)), as well as thermionic emission (red, Fig. 3(a)). Given the likelihood that multiple isomers are present, one interpretation of the direct detachment channels (green and blue, Fig. 3) is that the two bands arise from different deprotomers i.e., the feature with ADE ∼2.7 eV (blue, Fig. 3) is from photodetachment of Trp(II)−, whereas the feature with ADE ∼3.5 eV (green, Fig. 3) arises from Trp(I)−. Support for this assignment of the spectrum comes from the fact that the calculated EAs and VDEs for the different deprotomers match the experimental ADEs and VDEs well.
In the case of the lower eBE channel (blue, Fig. 3), the assignment for direct detachment from S0 to D0 of the Trp(II)− deprotomer seems conclusive, as direct detachment from a carboxylate group would typically require removal of electrons with a significantly higher binding energy (∼3.5 eV).31 The assignment of the higher binding energy direct detachment channel (green, Fig. 3) is less certain though, despite the energetic onset matching well with the expected EA of Trp(I). Typically, direct detachment from a carboxylate group results in two electronic states, which are close in energy (∼0.5 eV).32 There may be some evidence for a further photodetachment feature in Fig. 3, but it is not clearly resolved and overlaps with the thermionic emission. However, there may be an alternative interpretation of the high eBE direct detachment channel. Consider the photoelectron spectrum of In− recorded at λ = 266 nm (Fig. 4): it contains two broad spectral bands of similar spacing (∼0.8 eV) to the tryptophan spectrum, suggesting that both direct detachment bands could arise from Trp(II)−, with photodetachment resulting in the neutral in either the D0 or D1 state of the radical. Hence, it is possible and indeed probable that the higher eBE feature observed (green, Fig. 3) arises from a combination of direct detachment of Trp(I)− to D0, and direct detachment of Trp(II)− to D1.
Thermionic emission (red, Fig. 3) occurs when ‘hot’ anions, which may be the parent species or a fragment, lose electrons via a statistical process. For parent anions, this may be mediated by an excited state, where excitation is followed by rapid internal conversion to the ground state and the formation of vibrationally excited anions. For tryptophan, the thermionic emission seems to be mediated by an excited state near 4 eV, which is likely to be the S1 state with indole ππ* character, given that the maximum relative intensity of thermionic emission is seen in the hν = 3.96 and 4.13 eV photoelectron spectra. However, as photoelectron imaging on In− reported that excitation to the S1 state resulted in autodetachment, rather than internal conversion to the ground state, it might be unlikely that tryptophan would undergo rapid internal conversion.9,10 As an alternative, thermionic emission might be originating from the formation of ‘hot’ fragments via a dissociative process, mediated by the S1 excited state. Such mechanisms have been observed, in particular for molecules that are the conjugate base of carboxylic acids, where the CO2 is a very good leaving group.31,33–37 Indeed, the CO2 dissociative process for Trp− is calculated to lie lower in energy than electron loss from Trp(I)− (at approximately the same energy for Trp(II)− (Table 1)) and Noble et al. reported the decarboxylation of tryptophan at hv > 3.8 eV (λ < 330 nm). We additionally calculated that the Trp-CO2 has a negative EA, and given that CO2 also has a negative EA, the thermionic emission observed is likely occurring alongside CO2 loss (i.e. dissociative photodetachment).13 Decarboxylation is a facile process for Trp(I)− being the anion where the proton on the carboxylic acid is removed, whereas for Trp(II)−, additional rearrangements are required. Hence, we conclude that the thermionic emission predominantly arises from Trp(I)−. A plausible mechanism that would enable the electron loss might be that excitation of the S1 excited state, predominantly located on the indole ring, would create a hole which the excess electron on the carboxylate group could migrate to fill. Following this process, the CO2 group would have significant carboxyl group character and decarboxylate readily, leaving the electronically unstable Trp-CO2− to lose an electron via autodetachment. Such a mechanism would be consistent with the formation of a vibrationally excited molecule, as expected from the low kinetic energy release (∼0.2 eV) observed by Noble et al.13 The various different photodetachment processes which contribute the photoelectron spectra of tryptophan are summarised in Fig. 5. For each process the arrow indicating the eKE of the photoelectron is coloured to match its corresponding spectral feature in Fig. 3, i.e., blue and green for the low and high eBE direct detachment channels and red for thermionic emission.
 | ||
| Fig. 5 The electron loss processes contributing to each of the different spectral features observed in the tryptophan photoelectron spectra (Fig. 3): low and high eBE direct detachment (blue and green) and thermionic emission (red). Anions are shown with solid lines, and neutrals with dashed lines. | ||
Ultimately this work demonstrates both the benefits and shortcomings of using the photoactive chromophore to understand the behaviour of a biomolecule. By considering tryptophan as a substituted indole, where some stabilisation of the anion by side chain is expected, it is possible to partially understand the direct detachment portion of the spectra and the character of the important electronic states. However, such a picture misses the potential for multiple deprotomers, and ultimately the possibility of fragmentation, rather than autodetachment, as an important decay pathway of the ππ* excited anion states in the gas phase.
Conclusions
The photoelectron spectra of tryptophan contain contributions from two distinct deprotomers in the ion beam: Trp(I)− and Trp(II)− which correspond to deprotonation at the carboxylic acid or indole ring, respectively. The lowest energy direct detachment band (eBE = 2.7 eV) is assigned to direct detachment of Trp(II)− to its D0 neutral state. The higher eBE direct detachment channel is more challenging to assign, but likely consists of contributions from direct detachment of Trp(II)− to its D1 neutral state (given the similarities with the indole photoelectron spectrum), and from direct detachment of Trp(I)− to its D0 neutral state (based on the agreement with the calculated electron affinity of this deprotomer). Thermionic emission, which peaks near λ ≈ 300 nm, is mediated by the S1 ππ* excited state, and arises from CO2 loss from Trp(I)− resulting in an unstable anionic fragment which autodetaches readily.Data availability statement
The data which support the findings of this study are available online at https://zenodo.org/doi/10.5281/zenodo.10907869.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Jemma A. Gibbard is grateful for the support of a Royal Society University Research Fellowship (URF/R1/221140). JRRV is grateful for funding from the OP JAK project No. CZ.02.01.01/00/22_008/0004649 (QUEENTEC).References
- E. R. Holiday, Spectrophotometry of proteins: Absorption spectra of tyrosine, tryptophan and their mixtures, Biochem. J., 1936, 30(10), 1795–1803 CrossRef CAS PubMed.
- R. Livingstone, O. Schalk, A. E. Boguslavskiy, G. Wu, L. Therese Bergendahl, A. Stolow, M. J. Paterson and D. Townsend, Following the excited state relaxation dynamics of indole and 5-hydroxyindole using time-resolved photoelectron spectroscopy, J. Chem. Phys., 2011, 135(19), 194307 CrossRef PubMed.
- J. Catalán, The first UV absorption band of L-tryptophan is not due to two simultaneous orthogonal electronic transitions differing in the dipole moment, Phys. Chem. Chem. Phys., 2016, 18(22), 15170–15176 RSC.
- J. Catalán, The first UV absorption band for indole is not due to two simultaneous orthogonal electronic transitions differing in dipole moment, Phys. Chem. Chem. Phys., 2015, 17(19), 12515–12520 RSC.
- J. T. Vivian and P. R. Callis, Mechanisms of Tryptophan Fluorescence Shifts in Proteins, Biophys. J., 2001, 80(5), 2093–2109 CrossRef CAS PubMed.
- A. Ghisaidoobe and S. Chung, Intrinsic Tryptophan Fluorescence in the Detection and Analysis of Proteins: A Focus on Förster Resonance Energy Transfer Techniques, Int. J. Mol. Sci., 2014, 15(12), 22518–22538 CrossRef CAS PubMed.
- I. Compagnon, A.-R. Allouche, F. Bertorelle, R. Antoine and P. Dugourd, Photodetachment of tryptophan anion: an optical probe of remote electron, Phys. Chem. Chem. Phys., 2010, 12(14), 3399 RSC.
- I. W. Sizer and A. C. Peacock, The Ultraviolet absorption of serum albumin and its constituent amino acids as a function of pH, J. Bio. Chem., 1947, 171(2), 767–777 CAS.
- M. A. Parkes, J. Crellin, A. Henley and H. H. Fielding, A photoelectron imaging and quantum chemistry study of the deprotonated indole anion, Phys. Chem. Chem. Phys., 2018, 20(22), 15543–15549 RSC.
- D. J. Nelson, A. M. Oliveira and W. C. Lineberger, Anion photoelectron spectroscopy of deprotonated indole and indoline, J. Chem. Phys., 2018, 148(6), 064307 CrossRef PubMed.
- Z. Tian, X.-B. Wang, L.-S. Wang and S. R. Kass, Are Carboxyl Groups the Most Acidic Sites in Amino Acids? Gas-Phase Acidities, Photoelectron Spectra, and Computations on Tyrosine, p-Hydroxybenzoic Acid, and Their Conjugate Bases, J. Am. Chem. Soc., 2009, 131(3), 1174–1181 CrossRef CAS PubMed.
- S. Xu, J. M. Nilles and K. H. Bowen, Zwitterion formation in hydrated amino acid, dipole bound anions: How many water molecules are required?, J. Chem. Phys., 2003, 119(20), 10696–10701 CrossRef CAS.
- J. A. Noble, J. P. Aranguren-Abate, C. Dedonder, C. Jouvet and G. A. Pino, Photodetachment of deprotonated aromatic amino acids: stability of the dehydrogenated radical depends on the deprotonation site, Phys. Chem. Chem. Phys., 2019, 21(42), 23346–23354 RSC.
- J. Lecointre, G. M. Roberts, D. A. Horke and J. R. R. Verlet, Ultrafast Relaxation Dynamics Observed Through Time-Resolved Photoelectron Angular Distributions, J. Phys. Chem. A, 2010, 114(42), 11216–11224 CrossRef CAS PubMed.
- L. H. Stanley, C. S. Anstöter and J. R. R. Verlet, Resonances of the anthracenyl anion probed by frequency-resolved photoelectron imaging of collision-induced dissociated anthracene carboxylic acid, Chem. Sci., 2017, 8(4), 3054–3061 RSC.
- W. C. Wiley and I. H. Mclaren, Time-of-Flight Mass Spectrometer with Improved Resolution, Rev. Sci. Instrum., 1955, 26(12), 1150–1157 CrossRef CAS.
- G. M. Roberts, J. L. Nixon, J. Lecointre, E. Wrede and J. R. R. Verlet, Toward real-time charged-particle image reconstruction using polar onion-peeling, Rev. Sci. Instrum., 2009, 80(5), 053104 CrossRef CAS PubMed.
- A. Sanov, Laboratory-Frame Photoelectron Angular Distributions in Anion Photodetachment: Insight into Electronic Structure and Intermolecular Interactions, Ann. Rev. Phys. Chem., 2014, 65(1), 341–363 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- R. A. Kendall, T. H. Dunning, Jr. and R. J. Harrison, Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions, J. Chem. Phys., 1992, 96(9), 6796–6806 CrossRef CAS.
- C. Adamo and D. Jacquemin, The calculations of excited-state properties with Time-Dependent Density Functional Theory, Chem. Soc. Rev., 2013, 42(3), 845–856 RSC.
- A. D. Mclean and G. S. Chandler, Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18, J. Chem. Phys., 1980, 72(10), 5639–5648 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393(1–3), 51–57 CrossRef CAS.
- B. Baguenard, J. C. Pinaré, C. Bordas and M. Broyer, Photoelectron imaging spectroscopy of small tungsten clusters: Direct observation of thermionic emission, Phys. Rev. A: At., Mol., Opt. Phys., 2001, 63, 2 CrossRef.
- B. Baguenard, J. C. Pinaré, F. Lépine, C. Bordas and M. Broyer, Thermionic emission in small carbon cluster anions, Chem. Phys. Lett., 2002, 352(3), 147–153 CrossRef CAS.
- A. Lietard, J. R. R. Verlet, S. Slimak and K. D. Jordan, Temporary Anion Resonances of Pyrene: A 2D Photoelectron Imaging and Computational Study, J. Phys. Chem. A, 2021, 125(32), 7004–7013 CrossRef CAS PubMed.
- G. A. Cooper, C. J. Clarke and J. R. R. Verlet, Low-Energy Shape Resonances of a Nucleobase in Water, J. Am. Chem. Soc., 2023, 145(2), 1319–1326 CrossRef CAS PubMed.
- T.-C. Jagau, Theory of electronic resonances: fundamental aspects and recent advances, Chem. Commun., 2022, 58(34), 5205–5224 RSC.
- G. A. Pino, R. A. Jara-Toro, J. P. Aranguren-Abrate, C. Dedonder-Lardeux and C. Jouvet, Dissociative photodetachment vs. photodissociation of aromatic carboxylates: the benzoate and naphthoate anions, Phys. Chem. Chem. Phys., 2019, 21(4), 1797–1804 RSC.
- J. A. Gibbard and R. E. Continetti, Photoelectron photofragment coincidence spectroscopy of carboxylates, RSC Adv., 2021, 11(54), 34250–34261 RSC.
- X.-B. Wang, J. B. Nicholas and L.-S. Wang, Intramolecular Coulomb repulsion and anisotropies of the repulsive Coulomb barrier in multiply charged anions, J. Chem. Phys., 2000, 113(2), 653–661 CrossRef CAS.
- C. J. Clarke, J. A. Gibbard, L. Hutton, J. R. R. Verlet and B. F. E. Curchod, Photochemistry of the pyruvate anion produces CO2, CO, CH3−, CH3, and a low energy electron, Nat. Commun., 2022, 13(1), 937 CrossRef CAS PubMed.
- C. W. West, J. N. Bull and J. R. R. Verlet, Charged Particle Imaging of the Deprotonated Octatrienoic Acid Anion: Evidence for a Photoinduced Cyclization Reaction, J. Phys. Chem. Lett., 2016, 7(22), 4635–4640 CrossRef CAS PubMed.
- J. A. Gibbard, E. Castracane, A. J. Shin and R. E. Continetti, Dissociative photodetachment dynamics of the oxalate monoanion, Phys. Chem. Chem. Phys., 2020, 22(3), 1427–1436 RSC.
- J. A. Gibbard and J. R. R. Verlet, Unraveling the decarboxylation dynamics of the fluorescein dianion with fragment action spectroscopy, J. Chem. Phys., 2023, 158(15), 154306 CrossRef CAS PubMed.
- J. A. Gibbard, E. Castracane, A. I. Krylov and R. E. Continetti, Photoelectron photofragment coincidence spectroscopy of aromatic carboxylates: benzoate and p-coumarate, Phys. Chem. Chem. Phys., 2021, 23(34), 18414–18424 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00309h |
| This journal is © the Owner Societies 2024 |