
Enhanced intramolecular charge transfer and near-infrared fluorescence in 4-dimethylamino-chalcone analogues through extended conjugation: synthesis, photophysical properties, and theoretical modelling†
Balqees S.
Al-Saadi
,
A. Ramadan
Ibrahim
,
John
Husband
*,
Ahmed H.
Ismail
,
Younis
Baqi
 * and
Osama K.
Abou-Zied
*
* and
Osama K.
Abou-Zied
*
Department of Chemistry, College of Science, Sultan Qaboos University, P.O. Box 36, Postal Code 123, Muscat, Sultanate of Oman. E-mail: johnh@squ.edu.om; baqi@squ.edu.om; abouzied@squ.edu.om
First published on 9th April 2024
Abstract
The distinctive characteristics of near-infrared fluorescent organic molecules render them indispensable across diverse applications, from energy harvesting to bioimaging and sensing technologies. In this work, we continue our investigation on the chalcone derivative, 4-dimethylamino-2′-hydroxychalcone (nDHC, n = 1; where n is the number of olefinic bonds), by expanding the number of central double bonds (n = 2 (2DHC) and n = 3 (3DHC)). Additionally, we also synthesized the structurally related chalcones lacking the OH group (DC, 2DC, 3DC) in order to obtain a comprehensive understanding of their effects on the intramolecular charge transfer (ICT). The results show remarkable bathochromic shifts in absorption and fluorescence peaks in solution as n increases. These shifts, 20 nm and 35 nm for absorption and 100 nm and 200 nm for fluorescence in 2DHC and 3DHC, respectively, signify enhanced ICT and a significant increase in the excited state's dipole moment. The presence of OH groups notably amplifies these shifts due to additional electron donation, influencing solute–solvent interactions in solution. Femtosecond fluorescence upconversion and transient absoprtion techniques unraveled distinct dynamics in these derivatives, exhibiting the dominance of vibrational cooling, solvation, and intramolecular motions, particularly in the larger conjugated systems 3DHC and 3DC. The observed changes in the femtosecond transinet absorption spectra suggest the existence of new active states in extended conjugation systems, indicating diverse intramolecular conformational states contributing to their relaxation dynamics. The results of this study provide invaluable insights into excited-state spectroscopy, offering a roadmap for tailoring chalcone derivatives for specific applications.
1. Introduction
In various chemical and biological processes, photoinduced reactions often involve proton and/or electron transfer between chemical species. These reactions find crucial applications in diverse fields such as photosynthesis, artificial light harvesting, photocatalysis, and bioimaging.1–5 The molecular understanding of photoinduced charge transfer processes has received significant attention due to their impact on photophysical properties, influenced by intramolecular torsional dynamics.6,7 When an organic molecule bearing electron donor (D) and acceptor (A) groups is photoexcited, extensive intramolecular charge transfer (ICT) occurs, sometimes involving twisting known as twisted intramolecular charge transfer (TICT).8,9 The relaxation of excited states through twisting bonds induces qualitative charge separation, making the ICT process irreversible and facilitating efficient chemical energy conversion. However, most organic (D–π–A) molecules with strong charge transfer character exhibit an ICT without undergoing TICT relaxation.10,11 The photophysical properties, including fluorescence quantum yield, lifetime, and band gap, show a strong correlation with the push–pull abilities between donor and acceptor substitutes and/or π-conjugated linkers.12–14 This knowledge is vital for advancing fields like photoinduced catalysis and energy conversion.The compounds under investigation in this paper are part of the chalcone compound family (see Fig. 1; where R1 = R2 = R3 = H, n = 1), a group of naturally occurring compounds widely used in medicinal chemistry.15,16 By attaching a dimethyl amino group in ring B (R1 = N(CH3)2) and an OH group in ring A (R2 = OH), a molecular charge transfer mechanism was induced, resulting in strong red fluorescence in the crystalline state with a high quantum yield (ϕF = 0.32).17
 | ||
| Fig. 1 Structural backbone of chalcone molecules. | ||
This specific chalcone derivative, 4-dimethylamino-2′-hydroxychalcone (DHC), exhibited red amplified spontaneous emission (ranging from 650 to 710 nm) depending on crystal size and packing configuration. Our recent study of DHC revealed femtosecond dynamics that highlighted an efficient excited-state intramolecular proton transfer (ESIPT) process, responsible for the significant red shift and high fluorescence quantum yield in the J-like aggregates within the crystalline form.18 However, in solution, a twisting motion around the hydroxyphenyl ring (A) inhibits ESIPT, leading the excited molecule back to the ground state via a conical intersection.18,19 Different chalcone derivatives, including those with OH groups or potent Lewis acids such as boron difluoride attached to the phenyl ring, have found applications as photosensitizers in dye-sensitized solar cells20,21 or as sensors for water in solvent mixtures22,23 and quantification of proteins.24 Our investigation involving chalcone derivatives bearing a carboxylic group attached to ring A (R3 = CO2H) revealed enhanced electron injection from the dyes to TiO2 photoanode, compared to derivatives lacking CO2H.25 This positions these novel chalcone derivatives as potential organic dyes for solar energy harvesting and catalytic reactions. In our recent pursuit of enhancing the fluorescence properties of DHC in solution, we examined the impact of various electron-donating groups on the ICT process in three hydroxychalcone derivatives.26 A more efficient ICT was observed by substituting N(CH3)2 with a compact pyrrolidine ring (R1 in Fig. 1) due to reduced hydrogen bonding with solvent. Introducing NH2 into the structure (R3 in Fig. 1) induced greater planarity, resulting in a higher fluorescence quantum yield and a narrower fluorescence peak. This was validated through femtosecond transient absorption, where the ultrafast intramolecular twisting process observed in other derivatives was absent in the presence of NH2.26
To this end, we expand our investigation of chalcones in the current work by examining the impact of enlarging the central double bond spacer (n in Fig. 1) to include two and three double bonds (Fig. 2). A larger spacer introduces two significant effects in the chemical structure: it elongates the π-conjugation and generates additional sites for twisting. The former effect is anticipated to enhance the ICT mechanism, whereas the latter is likely to create more nonradiative pathways.
 | ||
| Fig. 2 Chemical structures of the chalcone derivatives under investigation. | ||
Our objective is to discern which effect predominantly governs the molecular return to the ground state and to investigate the role played by the OH group in the R2 position. Our findings demonstrate that increasing the number of double bonds leads to a notable red shift in the fluorescence peak, transitioning from green to the near-infrared region. Our investigation incorporates steady-state and femtosecond time-resolved spectroscopic methods, alongside density functional theory (DFT) and time-dependent DFT. This work serves as a valuable contribution, offering insights into and customization of the excited-state spectroscopy of chalcone derivatives, thus facilitating their application across diverse fields.
2. Experimental and theoretical methods
2.1 General
All chemicals and solvents were used as purchased from Sigma-Aldrich, Alfa Aesar, and Enamine. TLC aluminium sheets (pre-coated with silica gel 60 F254) were purchased from Merck KgaA Company. The UV lamp (UVGL-58 Handheld, λ = 254/365 nm) was used to monitor the UV active spots in TLC. Chemical shifts of 1H- and 13C-NMR data in parts per million (ppm) are reported relative to DMSO-d6 deuterated solvent (δ1H: 2.49 ppm, 13C: 39.7 ppm) and recorded using Bruker Avance 500 MHz (1H) and 126 MHz (13C), respectively. Coupling constants (J value) are given in Hertz and spin multiplicities are given as s (singlet), d (doublet), t (triplet), dd (doublet of doublet), td (triplet of doublet) and m (multiplet). The mass spectra were acquired and recorded by HPLC-(UV)-ESI-MS using previously described procedure.27 The melting point of the products were measured via Gallenkamp (SMP10) digital apparatus.2.2 Synthesis of chalcone derivatives 2DHC, 2DC, 3DHC, and 3DC
The two chalcone derivatives DHC and DC were synthesized as we previously described.18,25 The other chalcones (2DHC, 2DC, 3DHC, and 3DC) were obtained via aldol condensation reaction in basic media between the aldehyde derivative (4-dimethylaminocynnamaldehyde, or (2E,4E)-(5-(4-dimethylamino)phenyl)penta-2,4-dienal) and acetophenone or its derivative (1-(2-hydroxy-phenyl)-ethanone) in (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mole ratio, furnishing the chalcone products (see Scheme 1) in isolated yields of 42–75% within (48–96) hours. The two reactants, 1 mmol each, were dissolved in absolute ethanol (10 mL) in 50 mL round-bottomed flask under stirring in ice-bath, followed by dropwise addition of piperidine (1 mmol). The reaction temperature was gradually raised to room temperature over 3–6 hours based on the reaction progress which was monitored by TLC using ethylacetate/hexane (5–20%) as eluent. The reactions were worked-up by neutralizing the reaction mixture to the pH 7 through dropwise addition of diluted hydrochloric acid (0.5 M). The neutralized mixture was poured in a beaker charged with a small amount of ice. The precipitate was left to settle down for a few hours, then filtered off using a Büchner funnel. Vacuum was used to dry the solid products. 3DC solid was washed with diethyl ether (3 × 2 mL) after isolation.
:1) mole ratio, furnishing the chalcone products (see Scheme 1) in isolated yields of 42–75% within (48–96) hours. The two reactants, 1 mmol each, were dissolved in absolute ethanol (10 mL) in 50 mL round-bottomed flask under stirring in ice-bath, followed by dropwise addition of piperidine (1 mmol). The reaction temperature was gradually raised to room temperature over 3–6 hours based on the reaction progress which was monitored by TLC using ethylacetate/hexane (5–20%) as eluent. The reactions were worked-up by neutralizing the reaction mixture to the pH 7 through dropwise addition of diluted hydrochloric acid (0.5 M). The neutralized mixture was poured in a beaker charged with a small amount of ice. The precipitate was left to settle down for a few hours, then filtered off using a Büchner funnel. Vacuum was used to dry the solid products. 3DC solid was washed with diethyl ether (3 × 2 mL) after isolation.
 | ||
| Scheme 1 Chemical synthesis pathway of the chalcone compounds 2DHC, 2DC, 3DHC, and 3DC. * Synthesis pathway was described in ref. 18 and 25. | ||
2.3 Spectral data of 2DHC, 2DC, 3DHC, and 3DC
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). LC-MS (m/z): 294.0 [M+H+]. Purity by HPLC-UV (254 nm)-ESI-MS: 98.0%.
O). LC-MS (m/z): 278.1 [M+H+]. Purity by HPLC-UV (254 nm)-ESI-MS: 98.0%.
O). LC-MS (m/z): 320.1 [M+H+]. Purity by HPLC-UV (254 nm)-ESI-MS: 96.6%.
O). LC-MS (m/z): 304.3 [M+H+]. Purity by HPLC-UV (254 nm)-ESI-MS: 90.9%.
O). LC-MS (m/z): 294.0 [M+H+]. Purity by HPLC-UV (254 nm)-ESI-MS: 98.0%.
O). LC-MS (m/z): 278.1 [M+H+]. Purity by HPLC-UV (254 nm)-ESI-MS: 98.0%.
O). LC-MS (m/z): 320.1 [M+H+]. Purity by HPLC-UV (254 nm)-ESI-MS: 96.6%.
O). LC-MS (m/z): 304.3 [M+H+]. Purity by HPLC-UV (254 nm)-ESI-MS: 90.9%.
2.4 Steady-state spectroscopic measurements
Absorption and fluorescence experiments were performed using an Agilent 8453 spectrophotometer and a Duetta spectrometer (Horiba Scientific), respectively. All spectroscopic measurements in solution were conducted at 23 ± 1 °C in a quartz cell of 1 cm path length with fixed concentration (0.02 mM). Fluorescence quantum yield of the compounds dissolved in methanol (MeOH) was estimated using a|e software (https://FluorTools.com), relative to DCM28 or IR780 iodide29 dyes as standards.2.5 Time-resolved spectroscopic measurements
Two femtosecond laser setups, previously described in detail18,30 and included in the ESI,† were utilized: pump–probe transient absorption and fluorescence upconversion. These methods allowed us to observe dynamics within the time range of 100 femtoseconds to 5 nanoseconds. All measurements involving time-resolved spectroscopy were performed using solution samples prepared in 2 mm thick fused silica cuvettes. To prevent photodegradation, the samples were continuously stirred during the experiments, which were conducted at a constant temperature of 22.0 ± 0.5 °C. Data analysis was performed using the Surface Xplorer software.The measured transients were fitted to a multiexponential function (eqn (1)), convoluted with the system's response function (IRF), as depicted by:
 | (1) |
In this expression, the αi values denote the relative amplitudes (pre-exponential factors) and the τi values represent the fluorescence lifetimes. Σiαi is normalized to unity and indicates the relative contribution of the lifetime values to the overall transient.  , where IRF is the width of the instrument response function (full width at half maximum) and t0 is time zero.
, where IRF is the width of the instrument response function (full width at half maximum) and t0 is time zero.
This function allows fitting of a kinetic trace for the selected wavelength using a sum of convoluted exponentials.
2.6 Density functional theory calculations
Density functional theory (DFT) calculations were performed at the CAM-B3LYP/6-311+g (2d,p) level as implemented in Gaussian09.31 Solvent effects were included via the polarizable continuum model (PCM) with parameters appropriate for methanol. Starting structures for geometry optimizations were drawn “by hand” and the nature of all stationary states verified by frequency calculation. Time-dependent (TD-DFT) calculations, at the same level of theory, were used to approximate electronic absorption spectra by calculating vertical excitation energies to the first 100 singlet excited-states at the optimized ground state geometry, while emission energies were approximated by calculating vertical excitation energies at the optimized geometry of the first excited state.3. Results and discussion
3.1 Steady-state absorption and fluorescence spectra
Fig. 3 illustrates the absorption and fluorescence spectra of the six chalcone derivatives when dissolved in MeOH. These derivatives include 2DHC (2DC) and 3DHC (3DC), which were synthesized with two and three olefinic bonds, respectively (as depicted in Fig. 2). The purpose of this synthesis was to investigate the impact of expanding the π-bridge conjugation on the spectroscopic characteristics, in comparison to DHC (DC). Table 1 provides a summary of the spectroscopic parameters for these compounds.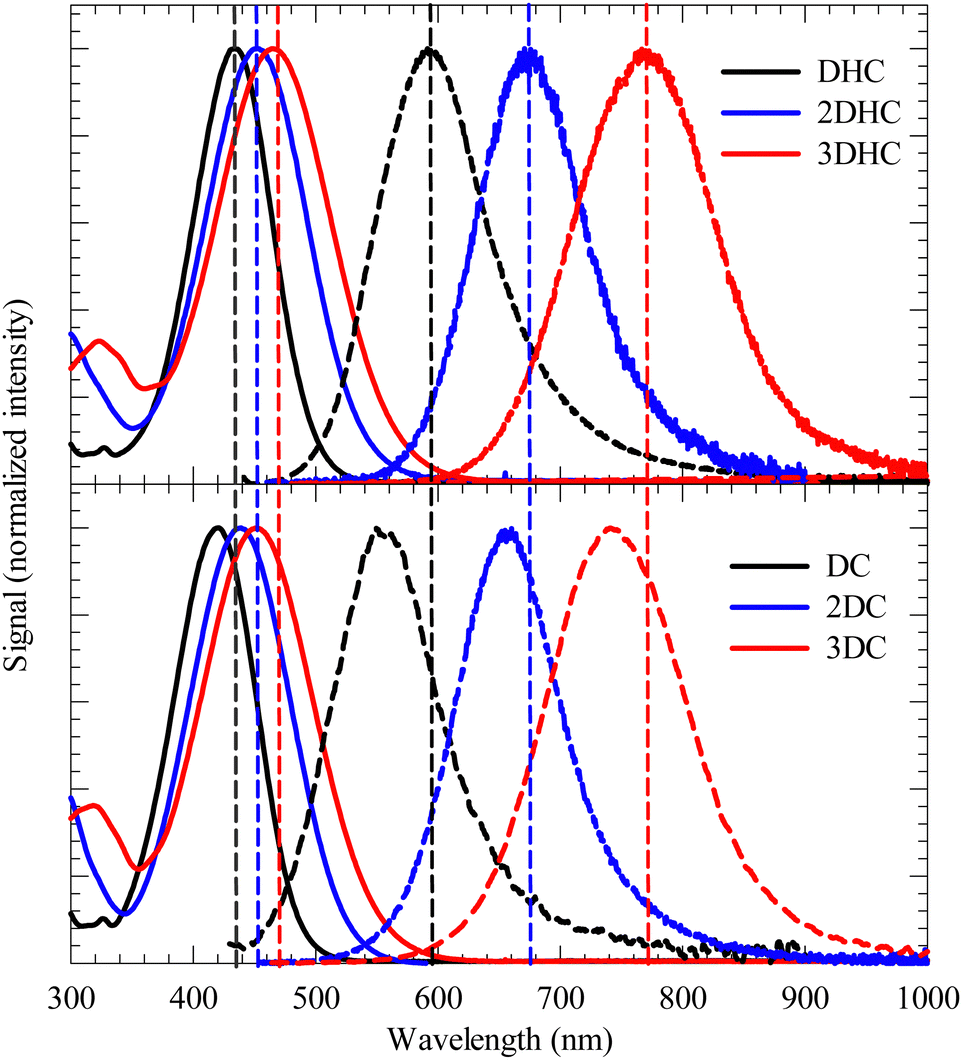 | ||
| Fig. 3 Steady-state absorption (solid lines) and fluorescence (dashed lines) spectra of the chalcone derivatives dissolved in MeOH. Fluorescence was detected after excitation at λmax,abs shown in Table 1. | ||
| λ max,abs (nm) | λ max,flu (nm) | ε (M−1 cm−1) | Stokes shift (cm−1) | Quantum yield (% ϕF) | |
|---|---|---|---|---|---|
| DHC | 435 | 591 | 42650 |
5597.2 | 0.5 |
| 2DHC | 455 | 676 | 36475 |
7185.1 | 0.5 |
| 3DHC | 470 | 772 | 45230 |
8439.6 | 6 |
| DC | 420 | 555 | 25000 |
5791.5 | 0.5 |
| 2DC | 437 | 658 | 44949 |
7229.7 | 2 |
| 3DC | 454 | 743 | 45500 |
8713.9 | 12 |
In a comparative analysis, it is observed that 2DHC and 3DHC display a red-shift of 20 nm and 35 nm, respectively, in their absorption peaks compared to DHC. Similarly, the fluorescence peaks of 2DHC and 3DHC exhibit red-shifts of 100 nm and 200 nm, respectively, in relation to DHC. These significant bathochromic shifts observed in both the absorption and fluorescence spectra indicate that the expanded donor–acceptor conjugation framework facilitates ICT, leading to a more efficient stabilization of the excited state. Moreover, the notably larger red shift in the fluorescence peaks suggests a substantial increase in the dipole moment upon excitation which stabilizes the excited state, consistent with the increase in the measured Stokes shift (Table 1). This trend was also observed for the DC, 2DC, and 3DC compounds, as outlined in Table 1. In all the six compounds, the increase in the number of double bonds is accompanied by more broadening in the absorption and fluorescence peaks as a result of more accessible vibrational states (see Table S1, ESI,† for FWHM details).
Furthermore, the presence of OH groups significantly enhances the red shift in the absorption and fluorescence peaks. This enhancement can be attributed to the introduction of an additional electron-donating group in DHC, 2DHC, and 3DHC (D–A–D), in contrast to only D–A in DC, 2DC, and 3DC. We have shown that tautomerization in DHC (Ph–OH⋯OC) → (PhO⋯HO–C) in the presence of MeOH is not possible due to a twisted phenyl ring.18 A torsional angle of this ring was calculated to be 45–60° which prevents the initially prepared enol from undergoing ESIPT.19 The OH group and the keto oxygen are thus expected to form intermolecular hydrogen bonds with MeOH. Such solvation mechanism does not lead to intermolecular proton transfer which was confirmed by comparing the steady state spectra in MeOH with those in the crystalline state of DHC. The latter shows clear tautomerization in the ground and excited states.18 For the current four derivatives, the similarity between the Stokes shift for 2DHC (3DHC) and 2DC (3DC) implies the absence of ESIPT in the former derivatives.
The fluorescence quantum yield measurements are presented in Table 1, revealing that the quantum yield increases when the OH group is replaced with H, despite being relatively small. This consistent trend can be attributed to the influence of solute–solvent interaction on the relaxation of the excited state. The presence of the OH group enhances the interaction with a protic solvent like MeOH. Consequently, and as mentioned above, the protic solvent can effectively solvate both the OH group and the CO group, while also interacting with the lone pair of electrons in nitrogen. Furthermore, an apparent increase in quantum yield is observed with an increase in the number of double bonds, which can be attributed to the heightened stability of the excited state, as mentioned earlier. It is worth noting that the relatively small values of ϕF indicate the possibility of internal twisting motions, as documented in our previous work18,25,33 and by other researchers.22
3.2 Ultrafast dynamics
To gain a comprehensive understanding of the impact of the π-spacer length on the behaviour of the excited state, we employed femtosecond fluorescence upconversion technique to measure the ultrafast dynamics of our systems. Table 2 summarizes the results. For DC and DHC, the data are taken from our previous work18,25 and included here for comparison.| τ 1 fs | α 1 | τ 2 ps | α 2 | τ 3 ps | α 3 | τ 4 ps | α 4 | 〈τ〉 ps | k r ns−1 | k nr ns−1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Taken from ref. 18 and 25. | |||||||||||
| DHC | 400 ± 100 | 0.14 | 4.8 ± 1.0 | 0.13 | 16.8 ± 1.4 | 0.68 | 350 ± 30 | 0.05 | 30 | 0.17 | 34 |
| 2DHC | 600 ± 100 | −0.28 | 5.5 ± 0.6 | 0.12 | 15.5 ± 0.8 | 0.17 | 103 ± 2 | 0.43 | 48 | 0.10 | 21 |
| 3DHC | 4.1 ± 0.7 | 0.56 | 13.9 ± 1.2 | 0.29 | 209 ± 2 | 0.15 | 38 | 1.59 | 25 | ||
| DC | 2.2 ± 0.3 | −0.44 | 18.4 ± 1.0 | 0.56 | 11 | 0.44 | 88 | ||||
| 2DC | 400 ± 100 | −0.10 | 3.1 ± 0.2 | −0.34 | 19.7 ± 1.9 | 0.17 | 114 ± 4 | 0.44 | 55 | 0.37 | 18 |
| 3DC | 650 ± 100 | −0.20 | 8.0 ± 0.6 | 0.43 | 19.9 ± 2.0 | 0.21 | 210 ± 2 | 0.16 | 42 | 2.89 | 21 |
Fig. 4 illustrates the fluorescence transients of all derivatives dissolved in MeOH during the initial 20 ps after excitation at 450 nm, accompanied by the best-fit functions for detection at 700 nm. The contribution from each lifetime component to the overall transient (α) is indicated as a fraction in Table 2. Negative values indicate buildup components (rise times). These transients were measured within the time frame of molecular decay up to 1 ns (refer to Fig. S1, ESI†). The prompt increase in intensity observed within the instrument response signifies the direct formation of the excited state from the ground state species.
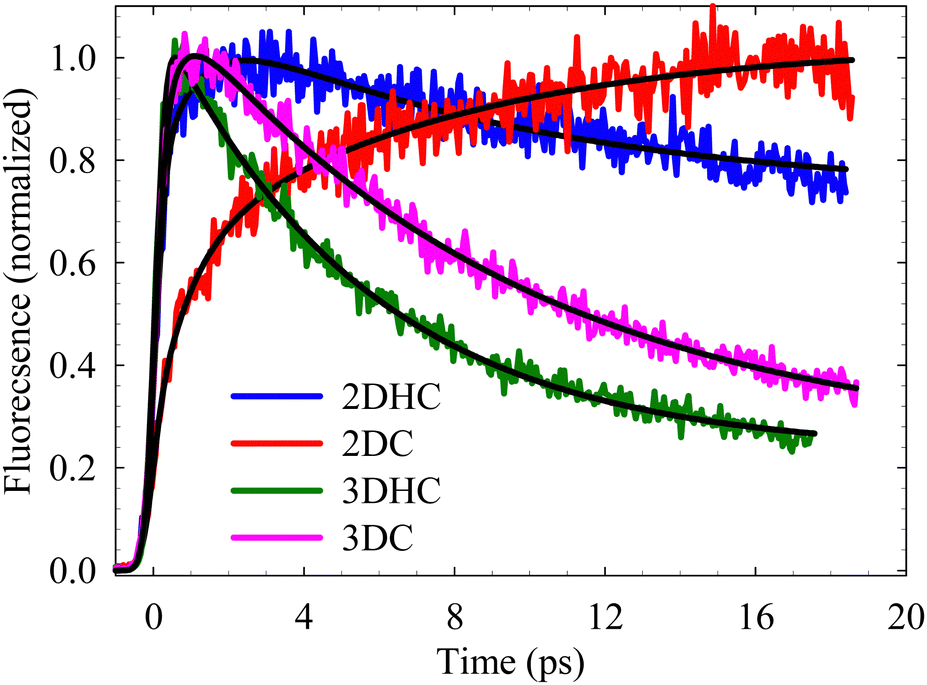 | ||
| Fig. 4 Fluorescence upconversion transients of the chalcone derivatives dissolved in MeOH. λex = 450 nm, λdetection = 700 nm. Best fits are shown by solid lines. | ||
In the four new derivatives, the fitting of a multiexponential pattern reveals four distinct contributions. The fastest component (τ1 = 400–650 fs) corresponds to the typical time constant associated with vibrational cooling of the initially prepared Franck–Condon (FC) state, which relaxes the molecule in the ICT excited state. This cooling process occurs within the instrumental time resolution in DC and 3DHC, which hinders our ability to make an accurate estimation of its value.25 A better resolution in the femtosecond transient absorption spectra allowed us to detect the vibrational cooling process in both derivatives to be 200 fs (vide infra). The second component (τ2) falls within the range of the average solvation dynamics of MeOH (2–8 ps), which correlates well with the reported dynamics for other chalcone derivatives in different solvents.32,33 During this period, the solvent molecules (MeOH) attain an equilibrium structure around the excited solute. To validate this assignment, we measured the transients in acetonitrile (with an average solvation time of 200 fs) and did not observe any fast component, which, if present, should be shorter than the instrument response function (IRF) (see Fig. S2, ESI†). We note here the contribution of this lifetime component as a rise time in DC and 2DC, as shown in Table 2. This effect arises because the detection wavelength at 700 nm resides on the red side of the fluorescence peaks (refer to Fig. 3), causing this component to manifest as a gradual increase in intensity during the solvent relaxation process. Conversely, for the other derivatives, the detection wavelength was positioned at the blue side of the fluorescence peak (for DHC and DC, detection was at 580 nm),18,25 which results in a decay component. A similar pattern is expected for τ1; however, its extremely short lifetime approaches the IRF limit, sometimes complicating its differentiation from the prompt buildup caused by excitation. Fig. S3 (ESI†) shows transients recorded at three different emission wavelengths.
We ascribe the last two components of the lifetime to dynamic intramolecular twisting and torsional motions (τ3), followed by a slower relaxation of the ICT state to the ground state (τ4). Notably, the latter component is absent in DC (Table 2). It is worth noting that the contribution of τ3 to the overall decay transient in DC is 56%, implying the major role of the twisting motions that lead the relaxation pathway to the ground state.25 This is also observed in DHC (68%), whereas τ4 only contributes 5% to the overall decay transient. It becomes evident that in the two systems containing one central double bond (DHC and DC), twisting and torsional motions overwhelmingly govern the relaxation pathway of the excited state. The minor contribution of τ4 in DHC may be attributed to the presence of the intramolecular hydrogen bond, which induces a marginal restriction on the twisting of the hydroxyphenyl ring. Conversely, the significance of the τ4 component in the overall decay is much more pronounced in 2DHC (2DC) and 3DHC (3DC). This observation could be attributed to the influence of extended conjugation, which stabilizes the planarity of the molecular structure. This point will be discussed later in the light of the DFT/TD-DFT calculations.
Using the measured quantum yield of fluorescence (Table 1) and the average fluorescence lifetime (〈τ〉 in Table 2), we calculated the radiative (kr) and nonradiative (knr) rate constants for all the chalcone derivatives, as presented in Table 2. Overall, the nonradiative rate constants surpass their corresponding radiative rate constants by more than an order of magnitude, underlining the significant influence of nonradiative processes in solution. Upon closer examination of kr and knr for each derivative, it becomes evident that the radiative rate constants are notably high for 3DHC and 3DC, thereby reducing the value of the nonradiative rate constants for these derivatives.
Further insights into the dynamics of the excited state can be garnered through femtosecond transient absorption (TA) spectroscopic measurements. The snapshots presented in Fig. 5 show the spectral changes of 2DHC, 2DC, 3DHC, and 3DC recorded at varying time intervals subsequent to excitation at 450 nm. Within the 550–800 nm range, a negative band that matches the spectral position of the steady-state fluorescence in all derivatives is attributed to stimulated emission (SE). The marked dynamic red shift and broadening of the SE band indicate the involvement of multiple vibrational states during the initial relaxation mechanism. This mechanism encompasses solvation and other relaxation processes occurring within a similar timeframe. The initial relaxation, occurring within a sub-picosecond period, is primarily attributed to vibrational relaxation, with the fastest component fitted to a time constant of 200 fs for DC (Fig. S5, ESI†) and 3DHC (Fig. S6, ESI†). All other observed lifetime components align with those derived from the fluorescence measurements, signifying the absence of further dynamics involved in the excited state relaxation.
 | ||
| Fig. 5 Transient absorption spectra of the chalcone derivatives dissolved in MeOH. λex = 450 nm. Raman scattering from the solvent in shown by * in the case of 2DC which is used to determine the IRF. Fig. S4, ESI† includes spectra for extended delay times between the pump and probe pulses. | ||
Concurrently, a distinctive excited-state absorption (ESA) band peaking at 500–550 nm is consistently detected across all derivatives (observed previously for DHC18 and for DC (Fig. S5, ESI†)). This spectral feature signifies a transition from the S1 state to higher electronically excited states. Notably, in 2DHC and 2DC, an isosbestic point between the ESA and SE bands is evident, suggesting that both bands evolve over time due to the same transient species. However, the spectral dynamics in 3DHC and 3DC exhibit a significant divergence, manifesting the appearance of an additional ESA peak around 600–610 nm (see Fig. 5).
In the case of larger conjugated systems like 3DHC and 3DC, the elongated spacer group linking the donor (N(CH3)2) and acceptor (CO) centers is anticipated to offer multiple potential twisting sites that could lead to various active intramolecular conformational states. It is evident from the spectra in Fig. 5 that the peak at 600–610 nm gains its intensity at the expense of that at 500–550 nm in 3DHC and 3DC. This observation strongly suggests the potential existence of new states, the population of which takes place during the initial relaxation process subsequent to excitation.
3.3 Theoretical modelling
The TD-DFT spectroscopic data are summarized in the ESI† (Fig. S7, S8 and Tables S2, S3). The observed red-shifts on addition of OH and upon each additional double-bond spacer, are clearly reproduced in these calculations. The S1 ← S0 vertical excitation energies closely match those of the measured absorption bands. However, the observed Stokes shifts are drastically underestimated by the corresponding adiabatic energies (calculated from the relaxed S1 state). In all cases the S1 state results from a HOMO to LUMO transition, (90% contribution for DC, reducing to 84% for 3DHC) which moves electron density from the donor side of the molecule to the acceptor side (Fig. S9 and S10, ESI†) with a concurrent increase in dipole moment (Table S1, ESI†). Relaxation in the excited state lengthens the CC double bonds of the π-bonded bridging group, while simultaneously reducing the C–C single bond lengths (Fig. S11, ESI†). Relaxation in this coordinate is a likely contributor to τ1. The Ph–C(O) single bond undergoes only modest shortening in the S1 state for DC and DHC and is essentially unchanged in the larger conjugated systems; conversely the CO bond is lengthened by a similar degree to the CC double bonds, highlighting the contribution of the carbonyl bond to the electron delocalization in these systems. Similarly, the nominal Ph–N single bond is shortened in the excited state, and this effect increases with the number of double bonds in the spacer unit. An overall enhancement of electron delocalization across the spacer group and through to the donor is thus observed with an accompanying increase in molecular rigidity. These results rule out the likelihood of TICT formation, instead revealing a clear CT character, as indicated by the significant rise in the excited state dipole moment (Table S2, ESI†). To investigate the potential for ESIPT in 2DHC and 3DHC, computations initially began with a linear keto structure (PhO⋯HO–C), subsequently reverted to the enol structure (Ph–OH⋯OC) upon optimization in both the S0 and S1 states. Therefore, ESIPT is not supported in these calculations.
Based on our experimental and computational results, we propose the following model for the evolution and relaxation of the excited state (Fig. 6). In this model, both the ground and excited states exhibit characteristics of ICT. Excitation at 450 nm prepares the hot S1 state. Following rapid relaxation from the FC region within time intervals τ1 (vibrational relaxation) and τ2 (solvent relaxation), the initially planar system undergoes numerous skeletal oscillations over time constant τ3. Within this period, there is a redistribution of vibrational energy from the stretching modes to the torsional modes. This energy distribution is likey facilitated by an energy plateau over which torsional motions about the planar equilibrium structure can occur with little energy cost. Such plateaus are common to excited state energy surfaces of extended conjugated systems.34 Consequently, the trajectory gradually broadens along the energy plateau, primarily via central bond torsions, before eventually converging towards a planar configuration, reflecting the fully relaxed equilibrium structure in S1. The N(CH3)2 group, in extended conjugation with the carbonyl group, affords substantial mesomeric stabilization to the planar ICT state as the number of olefin bonds increases. Relaxation from the planar ICT state is characterized by the time constant τ4. In the case of MeOH solvent, τ4 was not observed in DC, indicating direct population of the ground state via twisting motions.25 Conversely, τ4 was observed when DC was dissolved in acetonitrile (350–380 ps).32 This difference can be attributed to the higher solute–solvent hydrogen bonding in MeOH, a protic solvent, compared to acetonitrile, an aprotic solvent. Unlike acetonitrile, MeOH can form hydrogen bonds with the lone pair of electrons on the nitrogen atom of N(CH3)2 and with those on the oxygen atom of the carbonyl group, thus interfering with ICT and yielding less overall molecular polarity.26,35,36 The observation of τ4 in larger conjugated systems dissolved in MeOH solidifies the pivotal role of extended conjugation through central olefin bonds in preserving the ultimate planarity of the molecular structure via robust ICT.
 | ||
| Fig. 6 Schematic depiction of the photophysical model, outlining the dynamic processes observed in the investigated systems. Energies (eV) relative to S0 are shown for each compound. | ||
4. Conclusions
The behaviour of six chalcone derivatives in solution was investigated through steady-state and time-resolved spectroscopy, along with theoretical modelling via DFT. The results revealed significant insights into how extending π-conjugation and introducing OH groups affect their spectroscopic properties. Notably, as the number of central double bonds increased, substantial red-shifts were observed in both absorption and fluorescence spectra, highlighting the enhanced ICT within the expanded donor–acceptor conjugation network. Furthermore, variations in fluorescence quantum yield emphasized the impact of interactions between the solute and solvent, as well as the length of conjugation, on the stability of the excited state.Femtosecond techniques were used to uncover various relaxation pathways, highlighting how the length of the π-spacer impacts the dynamics of the excited state. Notably, smaller conjugated systems like DHC and DC revealed the absence of specific relaxation components, indicating the dominant influence of intramolecular twisting motions. In contrast, larger conjugated systems such as 2DHC and 3DHC displayed notable relaxation components due to their extended conjugation, which stabilizes the molecular structure. The femtosecond transient absorption spectra unveiled distinct spectral features in 3DHC and 3DC, manifesting as new ESA peaks, hinting at the emergence of new states during the initial relaxation processes. The present study provides insight into the complex relationship between molecular structure, conjugation length, and the behaviour of the excited state, offering valuable knowledge for advanced applications in photophysics and molecular design.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Sultan Qaboos University for its support through the Internal Research Grant (IG/SCI/CHEM/24/05) and the International Co-Funding Program (CL/SQU\QU/SCI/24/01). This work is also supported by The Research Council of Oman (TRC) through a Graduate Research Project. Y. B. Acknowledges the Arab-German Young Academy of Sciences and Humanities (AGYA) grants (01DL16002 and 01DL20003).References
- S. J. Formosinho and L. G. Arnaut, J. Photochem. Photobiol., A, 1993, 75, 21 CrossRef CAS.
- G. E. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 1 CrossRef.
- Y. Zhou, L. Zhang, X. Zhang and Z. J. Zhua, J. Mater. Chem. B, 2019, 7, 809 RSC.
- F. Ricci, B. Carlotti, B. Keller, C. Bonaccorso, C. G. Fortuna, T. Goodson III, F. Elisei and A. Spalletti, J. Phys. Chem. C, 2017, 121, 3987 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS.
- G. Ramakrishna, A. Bhaskar and T. Goodson, J. Phys. Chem. B, 2006, 110, 20872 CrossRef CAS PubMed.
- S. Yang, J. Liu, P. Zhou and G. He, J. Phys. Chem. B, 2011, 115, 10692 CrossRef CAS PubMed.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899 CrossRef PubMed.
- M. Glasbeek and H. Zhang, Chem. Rev., 2004, 104, 1929 CrossRef CAS PubMed.
- S. Arzhantsev, K. A. Zachariasse and M. Maroncelli, J. Phys. Chem. A, 2006, 110, 3454 CrossRef CAS PubMed.
- S. Techert and K. A. Zachariasse, J. Am. Chem. Soc., 2004, 126, 5593 CrossRef CAS PubMed.
- S. Roquet, A. Cravino, P. Leriche, O. Alévêque, P. Frère and J. Roncali, J. Am. Chem. Soc., 2006, 128, 3459 CrossRef CAS PubMed.
- X. H. Zhang, B. J. Chen, X. Q. Lin, O. Y. Wong, C. S. Lee, H. L. Kwong, S. T. Leeand and S. K. Wu, Chem. Mater., 2001, 13, 1565 CrossRef CAS.
- O. P. Varnavski, J. C. Ostrowski, L. Sukhomlinova, R. J. Twieg, G. C. Bazan and T. Goodson III, J. Am. Chem. Soc., 2002, 124, 1736 CrossRef CAS PubMed.
- B. Zhou and C. Xing, Med. Chem., 2015, 5, 388 Search PubMed.
- C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762 CrossRef CAS PubMed.
- X. Cheng, K. Wang, S. Huang, H. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 8369 CrossRef CAS PubMed.
- N. I. Zahid, M. S. Mahmood, B. Subramanian, S. M. Said and O. K. Abou-Zied, Phys. Chem. Lett., 2017, 8, 5603 CrossRef CAS PubMed.
- M. Dommett and R. Crespo-Otero, Phys. Chem. Chem. Phys., 2017, 19, 2409 RSC.
- K. Y. Teo, M. H. Tiong, H. Y. Wee, N. Jasin, Z. Q. Liu, M. Y. Shiu, J. Y. Tang, J. K. Tsai, R. Rahamathullah, W. M. Khairul and M. G. Tay, J. Mol. Struct., 2017, 1143, 42 CrossRef CAS.
- S. Chambon, A. D’Aléo, C. Baffert, G. Wantz and F. Fages, Chem. Commun., 2013, 49, 3555 RSC.
- A. Bhattacharyya, S. C. Makhal and N. Guchhait, J. Phys. Chem. A, 2019, 123, 6411 CrossRef CAS.
- X. Jin, L. Dong, X. Di, H. Huang, J. Liu, X. Sun, X. Zhang and H. Zhu, RSC Adv., 2015, 5, 87306 RSC.
- Y. Li, K. Li and J. He, J. Lumin., 2015, 31, 905 CrossRef PubMed.
- A. R. Ibrahim, B. S. Al-Saadi, J. Husband, A. H. Ismail, Y. Baqi and O. K. Abou-Zied, J. Mol. Struct., 2023, 1271, 134012 CrossRef CAS.
- R. J. Abdel-Jalil, A. R. Ibrahim and O. K. Abou-Zied, Chem. Phys. Lett., 2023, 813, 140299 CrossRef CAS.
- Y. Baqi and C. E. Müller, Nat. Protoc., 2010, 5, 945 CrossRef CAS PubMed.
- K. Rurack and M. Spieles, Anal. Chem., 2011, 83, 1232 CrossRef CAS PubMed.
- G. Chapman, M. Henary and G. Patonay, Anal. Chem. Insights, 2011, 6, 29 CAS.
- I. Ibrahim, H. N. Lim, O. K. Abou-Zied, N. M. Huang, P. Estrela and A. Pandikumar, J. Phys. Chem. C, 2016, 120, 22202 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, Revision E.1, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- R. Ghosh and D. K. Palit, J. Phys. Chem. A, 2015, 119, 111128 Search PubMed.
- A. H. Ismail, M. F. Khyasudeen, J. Husband, A. R. Ibrahim, Y. Baqi and O. K. Abou-Zied, J. Lumin., 2020, 223, 117261 CrossRef CAS.
- F. Santoro, A. Lami and M. Olivucci, Theor. Chem. Acc., 2007, 117, 1061 Search PubMed.
- W. A. Lees and A. Burawoy, Tetrahedron, 1963, 19, 419 CrossRef CAS.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, 3rd edn, 2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00289j |
| This journal is © the Owner Societies 2024 |