
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeometry and electronic structure of Yb(III)[CH(SiMe3)2]3 from EPR and solid-state NMR augmented by computations†
Anton
Ashuiev
a,
Florian
Allouche
a,
Md. Ashraful
Islam
 b,
José P.
Carvalho
c,
Kevin J.
Sanders
b,
Matthew P.
Conley
d,
Daniel
Klose
a,
Giuseppe
Lapadula
a,
Michael
Wörle
a,
Dirk
Baabe
e,
Marc D.
Walter
e,
Andrew J.
Pell
*b,
Christophe
Copéret
*a,
Gunnar
Jeschke
*a,
Guido
Pintacuda
*b and
Richard A.
Andersen‡
f
b,
José P.
Carvalho
c,
Kevin J.
Sanders
b,
Matthew P.
Conley
d,
Daniel
Klose
a,
Giuseppe
Lapadula
a,
Michael
Wörle
a,
Dirk
Baabe
e,
Marc D.
Walter
e,
Andrew J.
Pell
*b,
Christophe
Copéret
*a,
Gunnar
Jeschke
*a,
Guido
Pintacuda
*b and
Richard A.
Andersen‡
f
aDepartment of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1-5, 8093 Zurich, Switzerland. E-mail: ccoperet@ethz.ch; gjeschke@ethz.ch
bUniversité de Lyon, Centre de RMN à Très Hauts Champs de Lyon (UMR 5082 - CNRS, ENS Lyon, Université Claude Bernard Lyon 1), F-69100 Villeurbanne, France. E-mail: andrew.pell@ens-lyon.fr; guido.pintacuda@ens-lyon.fr
cDepartment of Materials and Environmental Chemistry, Stockholm University, Svänte Arrhenius väg 16 C, 106 91 Stockholm, Sweden
dDepartment of Chemistry and Chemical Sciences, University of California Riverside, 501 Big Springs Road, Riverside, CA 92521, USA
eInstitut für Anorganische und Analytische Chemie, Technische Universität Braunschweig, Hagenring 30, 38106 Braunschweig, Germany
fDepartment of Chemistry, University of California, Berkeley, CA 94720, USA
First published on 20th February 2024
Abstract
Characterization of paramagnetic compounds, in particular regarding the detailed conformation and electronic structure, remains a challenge, and – still today it often relies solely on the use of X-ray crystallography, thus limiting the access to electronic structure information. This is particularly true for lanthanide elements that are often associated with peculiar structural and electronic features in relation to their partially filled f-shell. Here, we develop a methodology based on the combined use of state-of-the-art magnetic resonance spectroscopies (EPR and solid-state NMR) and computational approaches as well as magnetic susceptibility measurements to determine the electronic structure and geometry of a paramagnetic Yb(III) alkyl complex, Yb(III)[CH(SiMe3)2]3, a prototypical example, which contains notable structural features according to X-ray crystallography. Each of these techniques revealed specific information about the geometry and electronic structure of the complex. Taken together, both EPR and NMR, augmented by quantum chemical calculations, provide a detailed and complementary understanding of such paramagnetic compounds. In particular, the EPR and NMR signatures point to the presence of three-centre–two-electron Yb-γ-Me-β-Si secondary metal–ligand interactions in this otherwise tri-coordinate metal complex, similarly to its diamagnetic Lu analogues. The electronic structure of Yb(III) can be described as a single 4f13 configuration, while an unusually large crystal-field splitting results in a thermally isolated ground Kramers doublet. Furthermore, the computational data indicate that the Yb–carbon bond contains some π-character, reminiscent of the so-called α-H agostic interaction.
Introduction
Organometallic compounds, perhydrocarbyls in particular (MRn with M = metal; R = hydrocarbyl), often adopt unusual geometries or bonding patterns due to their low coordination numbers and the associated electronic consequences in metal–carbon bondings.1,2 These patterns continue to engage the broader community in understanding how preferences in geometric structure relate to the often complex electronic structure and reactivity in these compounds. As an example, among diamagnetic compounds, carbons with single bonds that exhibit significant π character to metals show specific deshielded nuclear magnetic resonance (NMR) chemical shifts and associated reactivity, indicating that alkyl groups are often better described as both σ- and π-donors to the metal (Fig. 1a).3,4 Reduced JC–H coupling constants for these carbons, and associated specific structural distortions, provide evidence for agostic interactions.5,6 Similar bonding peculiarities are also found in compounds with metal–carbon multiple bonds. The prototypical example is Ta(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHtBu)(CH2tBu)3, which contains a highly deshielded 13C signal and a very low JC–H coupling constant for the formally sp2 hybridized TaC carbon. These signatures are consistent with an α-CH interaction, better described as an additional π-donation from the α-carbon to the electron deficient d0 metal centre.3,4,7
CHtBu)(CH2tBu)3, which contains a highly deshielded 13C signal and a very low JC–H coupling constant for the formally sp2 hybridized TaC carbon. These signatures are consistent with an α-CH interaction, better described as an additional π-donation from the α-carbon to the electron deficient d0 metal centre.3,4,7
 | ||
| Fig. 1 (a) Typically occurred peculiarities in structure of organometallic compounds, in particular, Ln[CH(SiMe3)2]3. (b) Characterization approach, applied to paramagnetic organometallic lanthanide complexes. | ||
Peculiarities in geometric structure from X-ray diffraction analysis are also observed in monomeric lanthanide perhydrocarbyl complexes, Ln[CH(SiMe3)2]3 (Ln = Y,8 La,9 Ce,8 Sm,9 Lu10) that contain three short Ln⋯Cγ contacts between one Si–CH3 group of each of the three proximal –SiMe3 groups (Fig. 1a). The Ln⋯Cγ contacts result in lengthening of the Si–CH3 bonds close to the lanthanide by ∼0.04 Å with respect to the other Si–CH3 groups, indicating that these proximal Si–CH3 groups interact with the lanthanide. This interaction helps to satisfy the high coordination numbers required to isolate these f-block homoleptic complexes; a strategy that also facilitates isolation of Yb[C(SiMe3)3]2,11 Ln(AlMe4)3,12 and Ln[C(SiHMe2)3]2.13,14 Recent studies of Lu[CH(SiMe3)2]310 indicated that the Lu⋯Cγ interactions are not a result of γ-C–H agostic interactions (2-electron donation from a C–H bond to an electron deficient metal centre),6 but are rather due to a three-centre–two-electron (3c–2e) interaction between the Si–Me bond and Lu. In other words, the Lu⋯Cγ interaction is best described as a pseudo bridging Lu–Me–Si group (Fig. 1a), analogous to Al–Me–Al observed in dimeric AlMe315,16 or Ln(AlMe4)3.17 The critical data in that study10 were a combination of high quality X-ray diffraction results, variable temperature 1H and 13C NMR spectra in solution, as well as J-resolved 1H–13C and 29Si magic-angle spinning (MAS) solid-state NMR spectra. The experimental results were reliably reproduced using both wave-function based and density functional theory (DFT) models of Lu[CH(SiMe3)2]3, and natural bond orbital (NBO) analysis showed that the charges on Cα and Cγ are negative, while the charges on Siβ and Hγ are positive. This analysis was consistent with the proposed bridging Lu–Me–Si group to describe the Lu⋯Cγ interaction.
Extending this analysis using similar NMR methods to other Ln[CH(SiMe3)2]3 is however challenging, because most lanthanides contain a partially filled f-shell in their +3 oxidation state. The resulting paramagnetism perturbs the magnetic properties of the nearby nuclei, making data acquisition and interpretation complicated at best, and typically impractical in most instances. Similarly, while electron paramagnetic resonance (EPR) could offer a potential alternative for this class of compounds, the signals are typically not detectable at room temperature using routine continuous wave (CW) methods due to the short electronic spin relaxation times and large hyperfine couplings. However, both fields have seen impressive recent progress. In solid-state NMR, fast MAS and broadband adiabatic irradiation sequences allow for measurements on such samples18 and in EPR, new EPR pulsed techniques19 were developed for f-block metal complexes.20,21 In parallel, the recent advances in quantum chemical/DFT methods and dedicated formalisms22–30 now allow us to interpret NMR and EPR data in terms of the spatial and electronic structure.31–37
In this study, we showcase a methodology to assess the detailed electronic structure of a prototypical compound, namely Yb[CH(SiMe3)2]3, by combining state-of-the-art EPR, NMR and computation (Fig. 1b). We first synthesized and isolated a series of Yb[CH(SiMe3)2]x[O-2,6-tBu2-C6H3]3−x (x = 0, 1, 2, 3), and determined their structures by single-crystal X-ray diffraction at 100 K along with their basic magnetic properties by variable-temperature measurements of their magnetic susceptibilities. We next investigate the full geometric and electronic structure of Yb[CH(SiMe3)2]3 based on the most advanced EPR and solid-state NMR, supported by quantum chemical calculations. This approach enables a full characterization of the direct metal–carbon bonding as well as the nature of the secondary Yb⋯Cγ interaction at low and room temperature. We discuss how this interaction influences the structure and ligand dynamics in solution and in the solid state, and how the CH(SiMe3)2 ligand leads to an unusual behavior in the crystal field interactions of the Yb(III) ion.
Results and discussion
Synthesis and X-ray diffraction studies of Yb[CH(SiMe3)2]3
The syntheses of Yb[CH(SiMe3)2]3, Yb[CH(SiMe3)2]2[O-2,6-tBu2-C6H3], and Yb[CH(SiMe3)2][O-2,6-tBu2-C6H3]2 are shown in Fig. 2a (see ESI,† Section 1 for detailed synthetic procedures). Similar to the lutetium complexes isolated previously,10 the alkylation depends on the stoichiometry of the organolithium reagent. Key to successful isolation of these compounds is the use of sufficient volumes of pentane to dissolve the sparingly soluble polymeric Li[CH(SiMe3)2].38 Addition of Li[CH(SiMe3)2] as a slurry in smaller volumes of pentane results in poor yields and formation of mixtures of these compounds. All organoytterbium compounds shown in Fig. 2a are isolated as crystalline solids from concentrated pentane solutions. Yb[CH(SiMe3)2]3 is blue, Yb[CH(SiMe3)2]2[O-2,6-tBu2-C6H3] is brown, and Yb[CH(SiMe3)2][O-2,6-tBu2-C6H3]2 is red. The study below focuses on the structure and spectroscopic features of Yb[CH(SiMe3)2]3. X-ray structures of Yb[CH(SiMe3)2]2[O-2,6-tBu2-C6H3] and Yb[CH(SiMe3)2][O-2,6-tBu2-C6H3]2 are provided in ESI,† Section 2.2 (see Fig. S3–S6), and their NMR spectra are provided in ESI,† Section 7 (see Fig. S15–S20). | ||
| Fig. 2 (a) Synthesis scheme of Yb[CH(SiMe3)2]x[O-2,6-tBu2-C6H3]3−x (x = 1, 2, 3). (b) Structure of the Yb[CH(SiMe3)2]3 complex with displacement ellipsoids at 50% probability level. For clarity the hydrogen atoms and the solvent molecules CH2(SiMe3)2 are omitted. (c) Sketch of relevant bond lengths and angles of Yb[CH(SiMe3)2]3. The bond lengths are reported in Å, and the bond angles (in red) are reported in degrees (see also Table S2, ESI†). (d) Newman projection down the C(2)–Si(2) bond with bond lengths and angles (in red) obtained from the crystal structure of Yb[CH(SiMe3)2]3. (e) Structure of the Lu[CH(SiMe3)2]3 complex with displacement ellipsoids at 50% probability level. For clarity the hydrogen atoms and the solvent molecules CH2(SiMe3)2 are omitted. (f) Sketch of relevant bond lengths and angles of Lu[CH(SiMe3)2]3. The bond lengths are reported in Å, and the bond angles (in red) are reported in degrees (see also Table S2, ESI†) (f) Newman projection down the C(2)–Si(2) bond with bond lengths and angles (in red) obtained from the crystal structure of Lu[CH(SiMe3)2]3. For comparison panels (e)–(g) are reproduced from ref. 10 with permission from the American Chemical Society, copyright 2016 Conley et al.10 | ||
A view of the Yb[CH(SiMe3)2]3 complex is shown in Fig. 2b and Fig. S2 (ESI†). The relevant bond distances obtained from this structure are shown in Fig. 2c and d (see also Table S2, ESI†). For comparison the previously reported bond distances for Lu[CH(SiMe3)2]310 (Fig. 2e) are also shown in Fig. 2f and g (see also Table S2, ESI†). Yb[CH(SiMe3)2]3 crystallizes in the P31c space group and is isostructural with other isolated Ln[CH(SiMe3)2]3. Yb[CH(SiMe3)2]3 adopts a C3 symmetric structure with the ytterbium displaced from the plane defined by the three Yb–C bonds by 0.783(3) Å. The Yb–Cα (Yb–C1) distance is 2.324(3) Å, which is nearly the same as the Lu–Cα distance in Lu[CH(SiMe3)2]3 (2.319(3) Å) and the Sm–Cα distance in Sm[CH(SiMe3)2]3 (2.33(2) Å).
Yb[CH(SiMe3)2]3 contains short Yb⋯Cγ contacts at 2.963(3) Å, which is slightly longer than Lu⋯Cγ (2.936(2) Å) and Sm⋯Cγ (2.85(3) Å) distances in Lu[CH(SiMe3)2]3 and Sm[CH(SiMe3)2]3, respectively. The Neumann projection shown in Fig. 2d contains distances and angles for the proximal Si–CH3 group interacting with Yb. The C–H bond distances and angles are close to those expected for a tetrahedral sp3 hybridized carbon, again similar to results obtained for Lu[CH(SiMe3)2]3. These structural data infer that a similar 3c–2e Yb–Me–Si interaction is present in Yb[CH(SiMe3)2]3 under the conditions of low-temperature X-ray diffraction studies (i.e. 100 K).
Magnetic susceptibility studies
We performed magnetic susceptibility studies for Yb[CH(SiMe3)2]3, Yb[CH(SiMe3)2]2[O-2,6-tBu2-C6H3], Yb[CH(SiMe3)2][O-2,6-tBu2-C6H3]2, and Yb[O-2,6-tBu2-C6H3]3 (see ESI,† Section 6, Fig. S11–S14 and Table S11 for more details). The determined experimental values of the effective magnetic moment of all four complexes at 300 K are in the range between μeff (300 K) = 4.30 (χT = 2.31 cm3 K mol−1) − 4.53μB (χT = 2.57 cm3 K mol−1) and show only slight variations with the different substituents. Furthermore, these values are also in a good agreement with the (Hund-Landé) expectation value for the free Yb(III) ion, which is described by a single 2F7/2 term (4f13) with μeff (300 K) = 4.54μB (χT = 2.58 cm3 K mol−1), suggesting that all Yb complexes within the investigated series can be described by a single 4f13 configuration, similar to those of the free Yb(III) ion.EPR characterization of Yb[CH(SiMe3)2]3
We characterized the Yb(III) alkyl complex Yb[CH(SiMe3)2]3 using pulse EPR spectroscopy19 that, in combination with computational studies, enables the successful characterization of the spatial and electronic structure of paramagnetic complexes.20,37 The continuous-wave (CW) EPR spectrum of Yb[CH(SiMe3)2]3 (Fig. 3a, blue) was reasonably reproduced by simulation (Fig. 3a, red) by modeling the Yb ion as an electronic spin S = 1/2, with g tensor principal values of g⊥ = 0.7316 and g‖ = 7.5698. The spectrum also exhibits a well-resolved satellite structure, due to hyperfine coupling to the two spin-active isotopes 171Yb (spin I = 1/2 and natural abundance of 14.31%) and 173Yb (spin I = 5/2 and natural abundance of 16.13%) of the lanthanide. From spectral simulation (Fig. 3a, red), the 171,173Yb hyperfine tensors were determined to be A⊥(173Yb) = 221 MHz, A‖(173Yb) = 1650 MHz, and accordingly A⊥(171Yb) = 842 MHz, A‖(171Yb) = 6285 MHz. | ||
| Fig. 3 (a) CW EPR spectrum of a solid powder of Yb[CH(SiMe3)2]3, 10 K (blue) and simulation (red), together with an expansion of the low-field region. The black arrow marks the field position of the HYSCORE measurement. (b) Echo-detected EPR spectrum of Yb[CH(SiMe3)2]3. The field position for HYSCORE and EDNMR are marked with a black and red arrow, respectively. (c) HYSCORE spectrum of Yb[CH(SiMe3)2]3 (blue to yellow, see also Fig. S8, ESI†) and simulation (red) based on a single 1H hyperfine coupling Aiso(1H) = 3.8 ± 0.3 MHz. Anti-diagonal lines indicate the 1H and 29Si nuclear Zeeman frequencies. Spurious peaks at 21.5/2.0 MHz and 23.5/23.5 MHz are electronic artefacts. (d) 1H region of EDNMR spectrum of Yb[CH(SiMe3)2]3 (blue), together with simulation (red) of a single effective 1H coupling with Aeff(1H) = 13 ± 1 MHz (gray) and a 1H matrix peak (gray dashed) at a 1H nuclear Zeeman frequency of 39.4 MHz. | ||
A higher-resolution picture can be provided by pulsed EPR spectroscopy, which has evolved into a robust tool for the determination of geometry and electronic configuration of organometallic complexes of paramagnetic d- and f-block metals. In particular, 2D hyperfine sub-level correlation spectroscopy (HYSCORE)39 allows us to detect magnetically active nuclei (e.g.1H, 13C, 29Si etc.) that are coupled to paramagnetic centres by hyperfine interaction, due to close spatial proximity to these centres. The technique can separate the isotropic (primarily Fermi-contact) and anisotropic (primarily spin-dipolar) parts of the hyperfine interaction tensors that are linked to the electronic properties of the systems (e.g. metal–ligand spin density transfer), and bonding between the nuclei and the paramagnetic centre. The HYSCORE spectrum of Yb[CH(SiMe3)2]3 (Fig. 3c and Fig. S8, ESI†) reveals the presence of hyperfine coupling to protons in the CH(SiMe3)2 alkyl ligands. From the simulation of the HYSCORE spectrum a single 1H hyperfine tensor was determined to have Aiso(1H) = 3.8 ± 0.3 MHz, Tdip(1H) = 4.0 ± 0.5 MHz (Adip = [−4.0 ± 0.5; −4.0 ± 0.5; 8.0 ± 1.0] MHz), with the tensors principal axes frame (PAF) rotated by 45° ± 15° with respect to the z axis of the PAF of the g tensor. This interpretation of the HYSCORE data is consistent with the ELDOR-detected NMR (EDNMR)40 spectrum shown in Fig. 3d, from which we can extract a single splitting due to an effective hyperfine coupling to 1H with Aeff(1H) = 13 ± 1 MHz. Based on previous observations of similar isotropic and dipolar parts of hyperfine coupling tensors for the S = 1/2 Ti(III) alkyl complexes,37,41 the observed 1H hyperfine couplings could be assigned to the hydrogen atoms that are close in space to the Yb(III) centre, such as α-H atoms of CH(SiMe3)2 ligands. The positions of the α-H atoms within the structure of Yb[CH(SiMe3)2]3 were further refined using quantum chemical computations (vide infra). This finding indicates that the transferred spin density is significant only for the closest to the Yb(III) atoms, such as C1 and H1, while it is substantially lower for the rest of the ligands. The observed isotropic 1H hyperfine couplings that are even smaller than those observed for d1 metal alkyls37,41 suggest that, in contrast to the case of Cp3 Yb,20 there is no significant charge and spin density transfer to the ligands in Yb[CH(SiMe3)2]3. This is also consistent with the preferential presence of the unpaired electron in the 4f orbital rather than in the valence 5d orbital, which would result in more significant distribution of the spin density into the ligands of Yb(III). A difficulty of promoting 4f electrons of Yb(III) to the 5d orbital, which effectively prevents the formation of double YbC bonds, was previously discussed based on CASSCF calculations.42 Taking into account the results of magnetic susceptibility measurements (vide supra), we propose that the ground state of the Yb[CH(SiMe3)2]3 complex has a single electronic configuration of 4f13 (S = 1/2), close to the one of Yb(III) free ion.
NMR characterization of Yb[CH(SiMe3)2]3
Over the last twenty years, the development of instrumentation and spectroscopic methods in NMR allows us to target paramagnetic lanthanide complexes such as Yb[CH(SiMe3)2]3. Specifically, solid-state NMR with fast MAS and broadband adiabatic irradiation sequences18 can now be used to obtain data on microcrystalline powders, where the complex dynamics are simplified and only the relevant internal ligand motions are retained. Nonetheless, obtaining high-quality magnetic resonance data on these systems represents only part of the challenge, since the resulting spectra are often very difficult to interpret using standard assignment methods. In this respect, recent progress in wave-function based/DFT methods for the computation of NMR parameters of paramagnetic complexes has also been impressive. For NMR, there have been significant advances both in the development of the formalism of the paramagnetic shifts22–30 and the computational implementation.31–36 However, applying the developed methods to lanthanides has been a much more difficult matter.For the Yb[CH(SiMe3)2]3 complex, we are able to draw the conclusions regarding its electronic structure and on the positions of 1H nuclei closest to the metal centre (α-H atoms of [CH(SiMe3)2] ligands, H1) based on the EPR data and quantum chemistry calculations. However, the weak hyperfine interactions between the unpaired electron and the more distant nuclei prevented us from obtaining any high-resolution information about the rest of the molecule by EPR. This gap could, however, be filled by paramagnetic NMR.18 Note that the signals of H1 nuclei, most likely, cannot be observed by NMR, in particular, due to a severe reduction of sensitivity because the large paramagnetic relaxation enhancement expected for these nuclei in close proximity to Yb, or due to the fact that potentially large shifts push them out of the excitation window. Therefore, EPR and NMR spectroscopies could be seen as complementary techniques for the characterization of paramagnetic lanthanide organometallic compounds like Yb[CH(SiMe3)2]3, allowing to obtain the information from both internal and external coordination environment of the metal center.
The ligands of the complex are expected to exhibit substantial dynamics, which has the effect of averaging the signals. This was observed in the case of the Lu analogue, where variable temperature 1H solution NMR data were required to partially freeze out these motions and the individual 1H resonances were obtained by cooling the sample. However, this approach was not successful for the present Yb complex, where no such resolution was obtained over a temperature range between 285 and 182 K, and no detection was possible beyond that because of increased line-broadening (see ESI,† Section 3 and Fig. S7).
We therefore proceeded to examine the Yb[CH(SiMe3)2]3 complex via solid-state MAS NMR, which has the advantage of eliminating molecular rotational diffusion, so that only the internal ligand dynamics remain. Such spectra relate to the room-temperature crystal structure, thereby providing a direct bridge to the low-temperature XRD data.
One-dimensional solid-state MAS NMR spectra of 1H, 13C and 29Si are shown in Fig. 4a–c. Each of these spectra exhibits poorly resolved patterns, due to hyperfine interactions with the unpaired electron, which induce large shifts and shift anisotropies and extremely large inhomogeneous line-broadenings. We see shift anisotropies that are very large for all three nuclei, of the order of hundreds of ppm, in contrast to the Lu analogue, where the 1H and 13C diamagnetic chemical shift anisotropies (CSAs) were too small to be measured and for 29Si were of the order of tens of ppm. In particular, for the Lu analogue, 29Si CSAs were used to characterize the metal–ligand interaction. That approach is not at first sight obviously applicable to the paramagnetic Yb complex, since the interpretation of the shift anisotropy is more complex.
 | ||
Fig. 4 NMR spectra of a solid powder of Yb[CH(SiMe3)2]3, 300 K: (a) 1H MAS spectra at 30 kHz MAS, (b) 13C MAS spectra of at 14.286 kHz MAS and (c) 29Si MAS spectra at 30 kHz MAS, showing two distinct 29Si signals. The spinning sidebands are marked with asterisks (*). 2D aMAT spectrum: (d) 1H at 30 kHz, (e) 13C at 20 kHz and (f) 29Si at 20 kHz. The indirect dimension projections were deconvolved using asymmetric Gaussian functions to extract the isotropic shifts and quantify shift dispersion. (g)–(i) Extracted slices with isotropic shift corresponding to the maxima of the deconvolved projections and labelled with Roman numerals. Each slice was subsequently fitted considering a simple CSA model using the Haeberlen convention43 ( Δδ = Δδ = ![[small delta, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e125.gif) zz − δiso and η = (yy − xx)/Δδ, where δxx, δxx and are the components of the CSA tensor in the principal axes frame). See ESI,† Section 1.6 for more details. zz − δiso and η = (yy − xx)/Δδ, where δxx, δxx and are the components of the CSA tensor in the principal axes frame). See ESI,† Section 1.6 for more details. | ||
The first problem to be addressed is the lack of resolution of the individual signals in each of the three spectra, due to the overlapping spinning sideband manifolds of neighboring resonances. To solve this problem, we employed the 2D adiabatic magic-angle turning (aMAT) experiment,44 which correlates each spinning side-band manifold to its isotropic shift, thus removing the overlap, as seen in Fig. 4d–f for 1H, 13C and 29Si, respectively. In all three cases, the aMAT spectra are able to resolve two resonances with distinct isotropic shifts. For 29Si, the interpretation is relatively straightforward, with the two signals V and VI being due to the two inequivalent Si nuclei (specifically, one due to Si1 and the other to Si2). By extension, it is reasonable to tentatively interpret each 13C signal (III and IV) as being from the three methyl groups attached to each of the Si (C2/3/4 and C5/6/7), and each 1H signal (I and II) to the corresponding group of protons (H2/3/4 and H5/6/7).
For each of the signals I–VI, a slice extracted from the aMAT spectra was fitted to determine the parameters of the shift tensor (isotropic shift δ, shift anisotropy Δδ and asymmetry parameter η) (Fig. 4g–i). For each of the three nuclei, within each pair one signal (II, IV and V) possesses a larger isotropic shift and shift anisotropy compared to the second (I, III and VI). We can tentatively assign the former set of three signals to the same SiMe3 group, and the latter set of signals to the other SiMe3 group. This interpretation is supported by a transferred-echo double resonance (TEDOR) experiment45 (Fig. 5a), which displays a first unambiguous correlation between I and III, a second unambiguous correlation between II and IV, and no other correlations. These solid-state NMR spectra and our tentative interpretation indicate that each group of 1H, 13C and 29Si resonances represents a single SiMe3 group in which there are rotations about the C1–Si bond and about the Si–C bonds of each of the three methyls. The rate constants describing each rotation are larger than the spread of the individual chemical shifts of the exchanging sites. Note that the predicted shifts of H1 nuclei (see ESI,† Section 5.3 for the details) lie within a range between 230 and 200 ppm (Fig. 5b), and are not observed in any of the 1H NMR spectra (1D, aMAT, TEDOR), as expected.
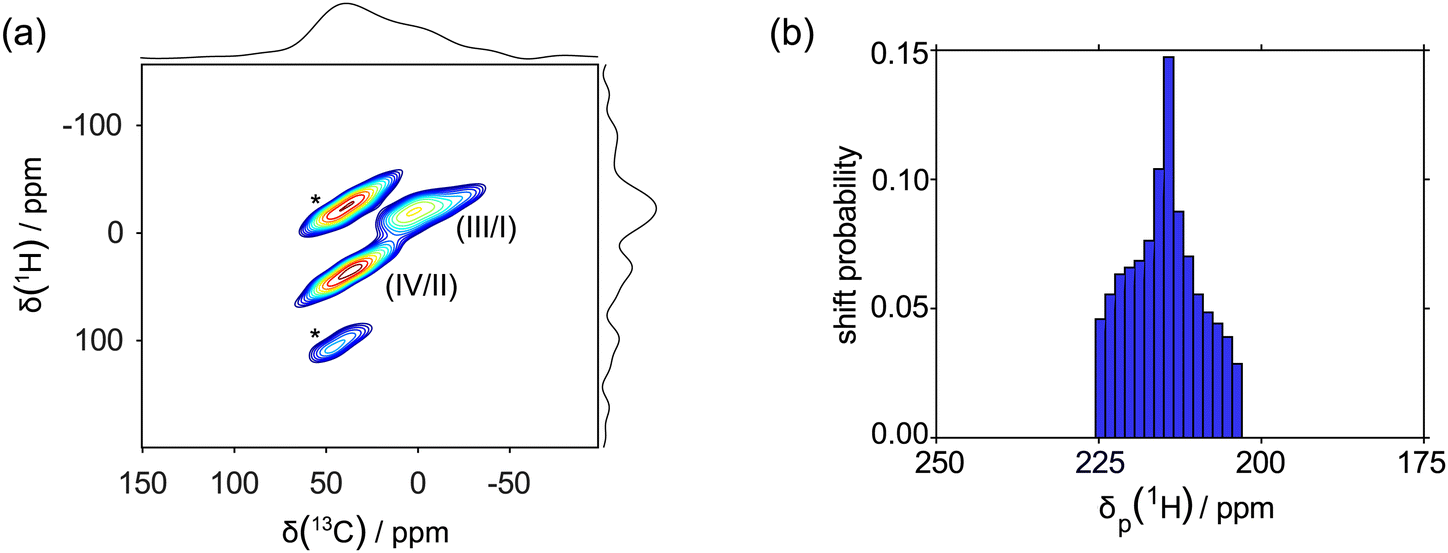 | ||
| Fig. 5 (a) 1H–13C correlation spectrum achieved by using the TEDOR experiment45 at 30 kHz MAS, yielding two distinct correlations of the resonances (III/I) and (IV/II), shown in Fig. 4, which were subsequently used to model the paramagnetic shifts in the following section. The spinning sidebands are marked with asterisks (*). (b) 1H NMR shift distributions considering solely the contributions arising from the presence of the unpaired electron and neglecting long-range PCS effects stemming from parametric centres in neighboring unit cells (δp). The shifts were calculated considering the Moon and Patchkovski,23 Vaara et al.24,25,28 and van den Heuvel and Soncini26,27 formalism using the g tensor obtained using multi-reference perturbation theory (SO-CASPT2) on the DFT optimized geometry (see Sections 1.8 and 5.3 of the ESI,† for details) and the experimental 1H hyperfine tensor. A distribution of relative orientations of the g and A tensors were considered using Zaremba, Conroy, Wolfsberg orientational averaging scheme with 1154 orientations.46–48 The relative orientation of the z axes of the g and A tensors was kept fixed at 45°, in order to match with the EPR results. | ||
Therefore, we suggest that the observed signals are consistent with the presence of the two distinct types of SiMe3 groups, associated with Si1 and Si2 nuclei. Note that the span (Ω = δ11 − δ33) of Si1 is Ω(Si1) = 676 ppm, and the span of Si2 is Ω(Si2) = 479 ppm. These values are significantly larger than those previously obtained for Lu[CH(SiMe3)2]3 (Ω(Si1) = 46.8 ppm), Ω(Si2 = 30.2 ppm),10 which is expected because that compound is diamagnetic. The pattern, observed for Lu[CH(SiMe3)2]3, was the strongest experimental evidence that, combined with the DFT interpretations, showed that Si1 was interacting with Lu through a Lu–Me–Si 3c–2e interaction.10 Although the Ω values for Yb[CH(SiMe3)2]3 are significantly larger, a similar pattern emerges; Si1 has a larger Ω(Si) than Si2, suggesting that Si1 is also interacting with Yb to form a 2c–2e Yb–Me–Si.
Computational studies of Yb[CH(SiMe3)2]3
| E 1 | E 2 | E 3 | E 4 | E 5 | E 6 | E 7 | |
|---|---|---|---|---|---|---|---|
| a In ORCA. | |||||||
| XRD | |||||||
| SF-CASSCF | 0 | 144 | 682 | 687 | 1031 | 1123 | 1203 |
| SF-CASPT2 | 0 | 205 | 823 | 928 | 1181 | 1392 | 1769 |
| SO-CASSCF | 0 | 525 | 889 | 1088 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 387 387 |
11007 |
11341 |
| SO-CASPT2 | 0 | 659 | 1043 | 1452 | 10420 |
11166 |
11660 |
| Optimized | |||||||
| SF-CASSCF | 0 | 168 | 694 | 694 | 1074 | 1083 | 1203 |
| SF-CASPT2 | 0 | 383 | 949 | 1002 | 1331 | 1393 | 1729 |
| SF-CASSCFa | 0 | 172 | 746 | 748 | 1161 | 1172 | 1297 |
| SO-CASSCF | 0 | 526 | 888 | 1070 | 10387 |
11006 |
11327 |
| SO-CASPT2 | 0 | 689 | 1068 | 1386 | 10428 |
11189 |
11613 |
| SO-CASSCFa | 0 | 565 | 959 | 1154 | 9214 | 9891 | 10241 |
The seven inner-core active 4f orbitals are shown in Fig. 6 along with the three energetically higher bonding type orbitals of the Yb–Ca bonds. Out of the seven active orbitals, the lowest one is non-bonding, followed by two sets of doubly-degenerate orbitals: one undergoes a slight π anti-bonding interaction with the Cα pz orbitals and the other one is non-bonding. The final two orbitals characterize a σ bonding interaction with the Cα hybrid orbitals. The seven 4f orbitals are very little admixed with the ligand orbitals, yet the ligand field splitting at the orbital level, or at the many-electron level, is relatively large compared to the case of O and N donor ligands (usually 200–300 cm−1). This is similar to the case of YbCp3, which has also been shown to exhibit a large ligand field splitting.20 The active 4f orbitals are mostly perturbed by electrostatic interactions, while the Yb valence 6s, 6p and 5d orbitals mostly participate in the bonding interactions with the Cα hybrid orbitals. As a result, population analysis of the ground spin-free CASSCF state (see Table 2) leads to significant electron occupations in the Yb(III) atomic valence and Rydberg orbitals.
 | ||
| Fig. 6 Surface plots of the three Yb–Cα bonding orbitals and the seven 4f active orbitals of the YbIII(CH(SiMe3)2)3 complex (separated by the vertical line) from the SF-CASSCF states. The canonical energies (in cm−1) are given in parentheses. The isosurface value is at ±0.02 a.u. The dominant compositions of the orbitals are also shown. | ||
| s Populations | p Populations | d Populations | f excessd | |
|---|---|---|---|---|
| a Basis set: ANO-RCC-TZVP(Yb, Si and Ca) + ANO-RCC-DZP (C,H) in MOLCAS. b Basis set: SARC-DKH-TZVP(Yb) and IGLO-III (rest) in ORCA. c Basis set: SARC-ZORA-TZVP(Yb) and TZVP (rest) in ORCA. d Excess of combined 4f + 5f orbital occupations than the formal 4f electron number. | ||||
| SF-CASSCF (M)a | 0.50(6s + 7s) | 1.52(6p + 7p) | 1.22(5d + 6d) | 0.02 |
| SF-CASSCF (M)b | 0.48(6s + 7s) | 0.76(6p + 7p) | 1.13(5d + 6d) | 0.14 |
| SF-CASSCF (L)b | 0.19(6s + 7s) | 0.60(6p + 7p) | 1.87(5d + 6d) | 0.52 |
| ZORA-DFT(PBE0) (M)c | 0.72(6s + 7s) | 1.06(6p + 7p) | 1.84(5d + 6d) | 0.18 |
| ZORA-DFT(PBE0) (L)c | 0.32(6s + 7s) | 0.77(6p + 7p) | 2.28(5d + 6d) | 0.60 |
At the spin-free (SF) CASSCF level, the 2F manifold of the central Yb(III) ion splits into three singlets, of relative energies of 0, 168 and 1203 cm−1, and two orbital doublets at 694 and ∼1074 cm−1, following the energy stabilization of the unpaired electron or hole in the trigonal ligand field. Dynamic correlation energy correction at the MS-CASPT2 method further increases the energy splitting by further stabilizing and destabilizing the bonding and anti-bonding type orbitals. At the spin–orbit (SO) level, the four Kramers doublets (KDs) of the ground J = 7/2 manifold are split by the axial ligand field in the order mJ = ±7/2 < ±5/2 < ±3/2 < ±1/2. The ligand field splitting is around 1400 cm−1, seven times larger than the room temperature thermal energy (200 cm−1). In fact, the first excited KD characterized by mJ = ±5/2 is around 690 cm−1 and is not meaningfully populated at room temperature. The irreducible spherical tensor ligand-field parameters extracted from the model spaces of the ground L (at SF) and J (at SO) manifolds are given in Table 3. The dominant ligand-field parameter is the second-rank axial component B02, but the components of orders three and six are also significant, as expected from the three-fold symmetry axis. The B02 component is related to the quadrupole moment of the ligand field and its negative value corresponds to an oblate coordinating sphere. The excited J = 5/2 manifold is energetically well separated by more than 10000 cm−1 in accordance with the SO coupling strength (ζ4f ≈ 2900 cm−1) of the Yb 4f electrons.52
| B 0 2 | B 2 2 | B 0 4 | B 2 4 | B 3 4 | B 4 4 | B 0 6 | B 2 6 | B 3 6 | B 4 6 | B 6 6 | S | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SF-CASSCF | −1839 | 11 | −24 | 3 | 317 | 3 | 43 | 2 | 185 | 3 | 216 | 487 |
| SF-CASPT2 | −2302 | 74 | −97 | 173 | 374 | 161 | −465 | 58 | 331 | 62 | 550 | 630 |
| SO-CASSCF | −1854 | 11 | −33 | 3 | 316 | 3 | 36 | 2 | 167 | 3 | 221 | 490 |
| SO-CASPT2 | −2319 | 75 | −104 | 168 | 368 | 164 | −454 | 51 | 296 | 55 | 580 | 634 |
Next, the SO-CASPT2 g factors, g⊥ = 0.72 and g‖ = 7.77 (see Table 4) are also in very good agreement with the experimental values and show a substantial improvement as compared to the SO-CASSCF values (g⊥ = 0.40, g‖ = 7.91, respectively) due to the better inclusion of the electron dynamical correlation. The anisotropic orbital and spin contributions to the magnetic moments as a result of the figand field and spin–orbit coupling admixing of the 4f orbitals are also shown. The magnetic moment of the ground KD is significantly anisotropic, as characterized by the prolate mJ = ±7/2 state stabilizing the unpaired electron in the bonding active 4fy(3x2−y2) and 4fx(x2−3y2) orbitals, as shown in Fig. 6. In addition, the EPR hyperfine coupling parameters to the central 171Yb (I = 1/2) nucleus were calculated from first principle methods using a finite nuclear model. The SO-CASSCF calculations are in good agreement with experiments as shown in Table 4. Quantitative evaluation of the hyperfine coupling value in the strong relativistic limit requires a forefront level of computational methods and is still in the developing phase. The Fermi contact (AFC), spin-dipolar (ASD) and orbital (Aorb) contributions to the hyperfine tensor in Table 4 indicate a dominant anisotropic nuclear spin – electron orbit coupling which leads to the large Yb(III) hyperfine A⊥ and A‖ components. The good agreement between calculated and experimental g tensor values and 171Yb hyperfine couplings indicates that our computational approach, based on DFT-optimized geometry and CAS methods, describes well the electronic structure of Yb(CH(SiMe3)2)3.
| g L x | g L y | g L z | g S x | g S y | g S z | g x | g y | g z | g iso | Δgax | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a SO-CASSCF in ORCA, experimental gx,y = 0.73, gz = 7.57 and A⊥ = 842 MHz, A‖ = 6285 MHz. | |||||||||||
| SO-CASSCF | 0.27 | 0.27 | 5.92 | 0.13 | 0.13 | 1.99 | 0.40 | 0.40 | 7.91 | 2.90 | 7.51 |
| SO-CASPT2 | 0.50 | 0.45 | 5.81 | 0.25 | 0.24 | 1.96 | 0.75 | 0.69 | 7.77 | 3.10 | 7.05 |
| SO-CASSCFa | 0.27 | 0.27 | 5.92 | 0.13 | 0.13 | 1.99 | 0.38 | 0.38 | 7.90 | 2.89 | 7.52 |
| A ⊥,SD | A ‖,SD | A ⊥,orb | A ‖,orb | A ⊥,FC | A ‖,FC | A ⊥ | A ‖ | |
|---|---|---|---|---|---|---|---|---|
| SO-CASSCFa | −24 | −748 | 287 | 6728 | 105 | 108 | 368 | 6088 |
 axis. The calculated isotropic part of the magnetic susceptibility χT at 300 K is 2.30 cm3 K mol−1 and is comparable to the experimental result of 2.57 cm3 K mol−1. The axial anisotropy of the molar magnetic susceptibility Δχax = χz − (χx + χy)/2 is very large in this complex and dominated by the anisotropic contribution from the ground KD (calculated to be ΔχKD1ax = 23.44 × 10−8 m3 mol−1), and is due to the large ligand field splitting and consequent non-homogeneous population in the 4f orbitals.
axis. The calculated isotropic part of the magnetic susceptibility χT at 300 K is 2.30 cm3 K mol−1 and is comparable to the experimental result of 2.57 cm3 K mol−1. The axial anisotropy of the molar magnetic susceptibility Δχax = χz − (χx + χy)/2 is very large in this complex and dominated by the anisotropic contribution from the ground KD (calculated to be ΔχKD1ax = 23.44 × 10−8 m3 mol−1), and is due to the large ligand field splitting and consequent non-homogeneous population in the 4f orbitals.
Indeed, the NBO analysis revealed a high degree of deviation of the natural hybrid orbitals (NHO) on α-C atoms from the Yb–C axis (θNHO–C–Yb = 17.8° for one [CH(SiMe3)2]3 ligand and 17.9° for the other two alkyl ligands). The value of such deviation can be used as a quantitative descriptor of the degree of the π character within the metal–carbon bonds (for a pure σ bond, no deviation would be expected).4 In fact, the degree of the π character within the Yb–C bonds is higher than the one observed for Ti(III), Ti(IV) and Zr(IV) neutral alkyl complexes, while being somewhat smaller than for the related cationic complexes.4,37
Furthermore, the NBO analysis revealed three low-lying natural orbitals, which have been attributed to the low-lying 5d orbitals, with their occupancies being slightly higher than 5% (see Table S11, ESI†). The spatial distribution of these orbitals (Fig. 7a) includes, in particular, three lobes of p-type on each of the α-C atoms of the[CH(SiMe3)2]3 ligands, all being in antiphase with the central d-type lobes (Fig. 7a, marked with arrows). These parts of the orbital can be viewed as π* orbitals of the Yb–C bonds,37 thus confirming the partial alkylidenic character of these bonds.3,4
 | ||
| Fig. 7 (a) Natural spin-α orbital, representing one 5d orbital of Yb[CH(SiMe3)2]3 (red for positive and blue for negative signs of the wavefunction, the same in all panels). The p-type parts of the orbital (two out of three) are marked with arrows. (b) Overlap of the same 5d orbital (wireframe contour) with natural spin-α orbital, corresponding to the bonding orbital of one of Si-Cγ bonds (solid contour). | ||
The obtained significant stabilization energies for these 5d orbitals (see Table S9, ESI†) imply that there are other secondary interactions within the structure of Yb[CH(SiMe3)2]3 that involve these 5d orbitals. One example of such an interaction is the three-centre–two-electron Yb–Me–Si interaction, previously observed for the Lu[CH(SiMe3)2]3 analogue.10 Indeed, the NBO analysis reveals the constructive overlap between these 5d orbitals and the bonding orbitals of the Si–Cγ bonds (Fig. 7b), thus showing that such interactions are also predicted for the Yb[CH(SiMe3)2]3 complex.
Next, to further probe the chemical bonding interactions, the DFT computed electron density was analysed by the quantum theory of atoms in molecules (QTAIM) method. The QTAIM approach is based on the topological analysis of the physically observable electron density, and is thus theoretically independent of the computational method. The bonding descriptors, namely the electron density (ρb), its Laplacian (∇2ρb), the potential energy density (Vb) and the kinetic energy density (Gb) at the so-called line critical point (LCP, i.e. the inflection point at the gradient path between two neighboring atoms) are of interest in this kind of analysis. Generally at the LCP, ρb > 0.20 a.u. with a negative Laplacian indicates a shared electron interaction between two atoms. Accompanied by |Vb|/Gb > 1 and Hb = Vb + Gb < 0, this typically indicates bound electrons at the LCP, often interpreted as a covalent interaction. For Yb[CH(SiMe3)2]3, the C–H bonds yield typical values of closed shell interactions with ρb > 0.25 and negative Laplacian, while the Si–C bonds give ρb ≈ 0.11 with a positive Laplacian, which is characteristic of an ionic interaction. By contrast, the values for both the Yb–Cα and Yb–Cγ (ρb = 0.078 a.u. and 0.016 a.u. respectively, with positive Laplacians, see Table 6) indicate donor–acceptor type bonding interactions.
| Bonds | ρ b | ∇2ρb |
|
H b |
|---|---|---|---|---|
| a On LuIII(CH(SiMe3)2)3 complex. | ||||
| Yb–Cα | 0.078 | 0.149 | 1.434 | −0.0286 |
| Yb–C(ag)γ | 0.016 | 0.058 | 0.865 | 0.0018 |

| Charges | q Yb | q Ca | q Sib |
|
|---|---|---|---|---|
| NBOa | 1.32 | −1.38 | 1.81 | −1.11 |
| QTAIM | 3.21 | −2.03 | 2.82 | −0.82 |

The NBO and QTAIM charges vary due to their different ways of partitioning the total electron density onto the individual atoms, but both indicate that a Yb–Cγ interaction is caused by the Coulomb attraction between the positively charged Yb and negatively charged Cγ atoms.
| δ = δorb + δFC + δPSO + δSD + δLR, | (1) |
 | (2) |
| Method | 29Si | 13C | 1H | ||||
|---|---|---|---|---|---|---|---|
| p | d | p | d | p | d | ||
| δ orb | −10.7 | −9.2 | 2.8 | 4.6 | 0.2 | 0.4 | |
| δ LR | 16.9 | 16.5 | 28.5 | 9.5 | 15.6 | 27.6 | |
| SO-CASPT2/CASSCF | δ SD | −67.4 | −9.6 | −4.4 | 0.5 | 7.4 | 2.1 |
| δ PSO | −201.0 | −29.2 | −15.9 | 1.7 | 21.4 | 6.6 | |
| δ FC | 3.0 | 7.6 | 2.4 | 0.5 | 0.2 | 0.0 | |
| δ total | −259.2 | −23.9 | 13.4 | 16.8 | 44.8 | 36.7 | |
| Δδ | −493.2 | −19.3 | −41.4 | −28.4 | 10.2 | 21.8 | |
| SO-CASPT2/PBE0 | δ SD | −61.2 | −10.2 | −1.1 | −0.4 | 6.3 | 1.9 |
| δ PSO | −78.4 | −10.3 | −8.6 | 1.0 | 7.4 | 2.4 | |
| δ FC | −24.4 | −77.4 | 67.7 | 14.7 | 3.0 | 0.4 | |
| δ total | −157.8 | −90.6 | 89.3 | 29.4 | 32.5 | 32.7 | |
| Δδ | −321.8 | −122.0 | −256.6 | −51.0 | 25.1 | 18.3 | |
| Exp | δ | −220 ± 20 | −30 ± 30 | 40 ± 20 | −20 ± 20 | 30 ± 20 | −20 ± 20 |
| Δδ | −542 ± 3 | 409 ± 3 | −183 ± 4 | 140[+30, −270] | 153 ± 1 | −56 ± 1 | |
On examining the computed shift tensors, we see that each nuclear species gives two distinct signals, with the shift and shift anisotropy of one species being consistently larger than those of the other species. There are clear quantitative discrepancies with the experimental values, but the general trends are reproduced. We can therefore assign the signals from the Me3Si(2) group, with the larger shifts and shift anisotropies, to the proximal (p) ligand, and the signals from Me3Si(1), with the smaller shifts and shift anisotropies, to the distal (d) ligand.
It is instructive to examine the breakdown of the shifts into the contributions given in eqn (1). The FC contribution is negligible in all cases, due to the very limited transfer of the unpaired spin density to these atoms over two, three or four bonds. The intra-molecular PCS is more significant, and is consistently larger for the proximal ligand than for the distal ligand, for all three nuclear species, which is a straightforward consequence of the former being in closer proximity to Yb than the latter. This conclusion is also true in the presence of rotational dynamics, where upon rotation about the C1–Si bond, the three methyls on Si2 are on average closer to the Yb ion than those on Si1. The same trend is seen for the more dominant PSO contribution, which for the same reasons is consistently larger by up to one order of magnitude for the proximal ligand. Finally, the long-range PCS is also dominant, due to the large magnetic anisotropy of Yb(III) in this complex. A comparison between the proximal and distal ligands reveals more comple trends than for the intra-molecular PCS, due to the packing of molecules in the crystal lattice.
Overall, the interpretation of the NMR data provides evidence that the same structural distortions, observed in the low-temperature XRD structure and associated with the presence of 3c–2e Yb–Me–Si interactions, persist at the higher temperature of the solid-state NMR measurements (ca. 300 K). Therefore, the Yb[CH(SiMe3)2]3 complex possesses the Yb⋯Cγ interaction, described as a pseudo bridging Yb–Me–Si group and previously observed10 for Lu[CH(SiMe3)2]3 (see Fig. 1), at both low- and room-temperature and irrespective of the presence of the incomplete electronic f-shell.
Conclusions
This study establishes that pulsed EPR and paramagnetic solid-state NMR spectroscopies are highly complementary and able, when coupled with the advanced computational methods, to provide detailed information about the geometry and electronic structure of paramagnetic compounds such as Yb[CH(SiMe3)2]3. Besides the specific geometry typically accessible by X-ray diffraction, this study provides direct spectroscopic evidences for the 3c–2e Yb–Me–Si interaction and for the presence of π-character in the Yb–C bond in this paramagnetic compound, paralleling what is observed for the diamagnetic Lu analog10 and so commonly among the corresponding d0 organometallics,3,4,7 often referred to as an α-H agostic interaction. In fact, the magnetic data for Yb[CH(SiMe3)2]3 indicates a f13 ground state configuration with no evidence for a multiconfigurational ground state that is often observed in lanthanide compounds,20,55–58 probably further explaining the observed similarities with the Lu isostructural compound. Yet, while magnetic susceptibility measurements indicate that the magnetic moment is consistent with that of a free Yb(III) ion, CAS-based calculations instead reveal an unusually large crystal-field splitting resulting in a ground KD that is almost completely thermally isolated even at room temperature and which results in a large magnetic anisotropy.Author contributions
A. A. and D. K. performed EPR measurements. F. A. and M. P. C. developed the synthetic procedures. J. P. C. and K. J. S. conducted NMR measurements. M. A. I., J. P. C. and A. A. performed quantum chemical calculations. M. W. and F. A. performed XRD studies. D. B. and M. D. W. conducted magnetic susceptibility measurements. M. D. W., A. J. P., C. C., G. J., G. P. and R. A. A. supervised the project. All authors participated in writing the manuscript. A. A., F. A., M. A. I., J. P. C., K. J. S. and M. P. C. declare equal contribution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. A. was supported by a SNF – ANR grant (Mr CAT 2-77275-15). J. P. C. was supported by the Swedish Research Council (2016-03441), M. A. I. by the Agence Nationale de la Recherche (ANR-21-CE29-0010-01) and by an installation grant of the ENS Lyon, and K. J. S. by the European Research Council (ERC-2015-CoG P-MEM-NMR). Calculations were performed thanks to an access grant to the Pôle Scientifique de Modélisation Numérique (PSMN) at the ENS Lyon. We acknowledge Laurent Maron (Université Paul Sabatier, Toulouse) for discussions at the early stage of this project. We acknowledge Maciej Korzynski (ETHZ) for a help with preparation of the manuscript. We acknowledge Elizaveta A. Suturina (University of Bath) and Aleksander Jaworski (Stockholm University) for the advices concerning quantum chemistry calculations in lanthanide complexes. We thank Martin Bröring (Institut für Anorganische und Analytische Chemie) at TU Braunschweig for providing access to the SQUID magnetometer.References
- J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Mill Valley, California, 2010 Search PubMed.
- G. Wilkinson, G. A. Stone and E. W. Abel, Comprehensive organometallic chemistry: The synthesis, reactions, and structures of organometallic compounds, Pergamon Press, 1982 Search PubMed.
- W. W. Lukens, M. R. Smith and R. A. Andersen, A π-donor spectrochemical series for X in (Me5C5)2TiX, and β-agostic interactions in X = Et and N (Me) Ph, J. Am. Chem. Soc., 1996, 118, 1719–1728 CrossRef CAS.
- C. P. Gordon, S. Shirase, K. Yamamoto, R. A. Andersen, O. Eisenstein and C. Copéret, NMR chemical shift analysis decodes olefin oligo- and polymerization activity of d0 group 4 metal complexes, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5867–5876 Search PubMed.
- M. Brookhart and M. L. H. Green, Carbon-hydrogen-transition metal bonds, J. Organomet. Chem., 1983, 250, 395–408 CrossRef CAS.
- M. Brookhart, M. L. Green and G. Parkin, Agostic interactions in transition metal compounds, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908 CrossRef CAS PubMed.
- C. P. Gordon, D. B. Culver, M. P. Conley, O. Eisenstein, R. A. Andersen and C. Copéret, π-Bond Character in Metal-Alkyl Compounds for C–H Activation: How, When, and Why?, J. Am. Chem. Soc., 2019, 141, 648–656 CrossRef CAS PubMed.
- A. G. Avent, C. F. Caro, P. B. Hitchcock, M. F. Lappert, Z. Li and X.-H. Wei, Synthetic and structural experiments on yttrium, cerium and magnesium trimethylsilylmethyls and their reaction products with nitriles; with a note on two cerium β-diketiminates, Dalton Trans., 2004, 1567–1577 RSC.
- P. B. Hitchcock, M. F. Lappert, R. G. Smith, R. A. Bartlett and P. P. Power, Synthesis and structural characterisation of the first neutral homoleptic lanthanide metal(III) alkyls: [LnR3][Ln = La or Sm, R = CH (SiMe3)2], J. Chem. Soc., Chem. Commun., 1988, 1007–1009 RSC.
- M. P. Conley, G. Lapadula, K. Sanders, D. Gajan, A. Lesage, I. Del Rosal, L. Maron, W. W. Lukens, C. Coperet and R. A. Andersen, The nature of secondary interactions at electrophilic metal sites of molecular and silica-supported organolutetium complexes from solid-state NMR spectroscopy, J. Am. Chem. Soc., 2016, 138, 3831–3843 CrossRef CAS PubMed.
- C. Eaborn, P. B. Hitchcock, K. Izod and J. D. Smith, A Monomeric Solvent-Free Bent Lanthanide Dialkyl and a Lanthanide Analog of a Grignard Reagent. Crystal Structures of Yb{C(SiMe3)3}2 and [Yb{C(SiMe3)3}I·OEt2]2, J. Am. Chem. Soc., 1994, 26, 12071–12072 CrossRef.
- W. J. Evans, R. Anwander and J. W. Ziller, Inclusion of Al2Me6 in the Crystalline Lattice of the Organometallic Complexes LnAl3Me12, Organometallics, 1995, 14, 1107–1109 CrossRef CAS.
- K. Yan, B. M. Upton, A. Ellern and A. D. Sadow, Lewis Acid-Mediated β-Hydride Abstraction Reactions of Divalent M(C(SiHMe2)3)2THF2 (M = Ca, Yb), J. Am. Chem. Soc., 2009, 131, 15110–15111 CrossRef CAS PubMed.
- K. Yan, A. V. Pawlikowski, C. Ebert and A. D. Sadow, A tris(alkyl)yttrium compound containing six β-agostic Si–H interactions, Chem. Commun., 2009, 656–658 RSC.
- H. C. Longuet-Higgins, The structure of some electron-deficient molecules, J. Chem. Soc., 1946, 139–143 RSC.
- M. Brookhart, M. L. H. Green and G. Parkin, Molecular structure and bonding in hexamethyldialuminum, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed.
- W. T. Klooster, R. S. Lu, R. Anwander, W. J. Evans, T. F. Koetzle and R. Bau, Neutron Diffraction Study of [Nd(AlMe4)3]0.5Al2Me6 at 100 K: The First Detailed Look at a Bridging Methyl Group with a Trigonal-Bipyramidal Carbon Atom, Angew. Chem., Int. Ed., 1998, 37, 1268–1270 CrossRef CAS PubMed.
- A. J. Pell, G. Pintacuda and C. P. Grey, Paramagnetic NMR in solution and the solid state, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 111, 1–271 CrossRef CAS PubMed.
- M. M. Roessler and E. Salvadori, Principles and applications of EPR spectroscopy in the chemical sciences, Chem. Soc. Rev., 2018, 47, 2534–2553 RSC.
- R. G. Denning, J. Harmer, J. C. Green and M. Irwin, Covalency in the 4f Shell of tris-Cyclopentadienyl Ytterbium (YbCp3) - A Spectroscopic Evaluation, J. Am. Chem. Soc., 2011, 133, 20644–20660 CrossRef CAS PubMed.
- A. Formanuik, A.-M. Ariciu, F. Ortu, R. Beekmeyer, A. Kerridge, F. Tuna, E. J. L. McInnes and D. P. Mills, Actinide covalency measured by pulsed electron paramagnetic resonance spectroscopy, Nat. Chem., 2016, 9, 578–583 CrossRef PubMed.
- R. J. Kurland and B. R. McGarvey, Isotropic NMR shifts in transition metal complexes: the calculation of the Fermi contact and pseudocontact terms, J. Magn. Reson., 1970, 2, 286–301 CAS.
- S. Moon and S. Patchkovskii, Calculation of NMR and EPR Parameters, John Wiley & Sons, Ltd, 2004, ch. 20, pp. 325–338 Search PubMed.
- T. O. Pennanen and J. Vaara, Density-functional calculations of relativistic spin–orbit effects on nuclear magnetic shielding in paramagnetic molecules, J. Chem. Phys., 2005, 123, 174102 CrossRef PubMed.
- T. O. Pennanen and J. Vaara, Nuclear magnetic resonance chemical shift in an arbitrary electronic spin state, Phys. Rev. Lett., 2008, 100, 133002 CrossRef PubMed.
- W. Van den Heuvel and A. Soncini, NMR chemical shift in an electronic state with arbitrary degeneracy, Phys. Rev. Lett., 2012, 109, 073001 CrossRef PubMed.
- W. Van den Heuvel and A. Soncini, NMR chemical shift as analytical derivative of the Helmholtz free energy, J. Chem. Phys., 2013, 138, 054113 CrossRef PubMed.
- J. Vaara, S. A. Rouf and J. Mares, Magnetic couplings in the chemical shift of paramagnetic NMR, J. Chem. Theory Comput., 2015, 11, 4840–4849 CrossRef CAS PubMed.
- G. Parigi, L. Benda, E. Ravera, M. Romanelli and C. Luchinat, Pseudocontact shifts and paramagnetic susceptibility in semiempirical and quantum chemistry theories, J. Chem. Phys., 2019, 150, 144101 CrossRef PubMed.
- L. Lang, E. Ravera, G. Parigi, C. Luchinat and F. Neese, Solution of a puzzle: high-level quantum-chemical treatment of pseudocontact chemical shifts confirms classic semiempirical theory, J. Phys. Chem. Lett., 2020, 11, 8735–8744 CrossRef CAS.
- S. A. Rouf, J. Mares and J. Vaara, 1H chemical shifts in paramagnetic Co(II) pyrazolylborate complexes: a first-principles study, J. Chem. Theory Comput., 2015, 11, 1683–1691 CrossRef CAS PubMed.
- R. Pigliapochi, A. J. Pell, I. D. Seymour, C. P. Grey, D. Ceresoli and M. Kaupp, DFT investigation of the effect of spin–orbit coupling on the NMR shifts in paramagnetic solids, Phys. Rev. B, 2017, 95, 054412 CrossRef.
- A. Mondal, M. W. Gaultois, A. J. Pell, M. Iannuzzi, C. P. Grey, J. Hutter and M. Kaupp, Large-scale computation of nuclear magnetic resonance shifts for paramagnetic solids using CP2K, J. Chem. Theory Comput., 2018, 14, 377–394 CrossRef CAS.
- A. Mondal and M. Kaupp, Computation of NMR shifts for paramagnetic solids including zero-field-splitting and beyond-DFT approaches. Application to LiMPO4 (M = Mn, Fe, Co, Ni) and MPO4 (M = Fe, Co), J. Phys. Chem. C, 2019, 123, 8387–8405 CrossRef CAS.
- A. Pyykkönen, R. Feher, F. H. Köhler and J. Vaara, Paramagnetic Pyrazolylborate Complexes Tp2M and Tp* 2M: 1H, 13C, 11B, and 14N NMR Spectra and First-Principles Studies of Chemical Shifts, Inorg. Chem., 2020, 59, 9294–9307 CrossRef PubMed.
- A. B. Andersen, A. Pyykkönen, H. J. A. Jensen, V. McKee, J. Vaara and U. G. Nielsen, Remarkable reversal of 13C-NMR assignment in d1, d2 compared to d8, d9 acetylacetonate complexes: analysis and explanation based on solid-state MAS NMR and computations, Phys. Chem. Chem. Phys., 2020, 22, 8048–8059 RSC.
- A. Ashuiev, F. Allouche, N. Wili, K. Searles, D. Klose, C. Copéret and G. Jeschke, Molecular and supported Ti(III)-alkyls: efficient ethylene polymerization driven by the π-character of metal-carbon bonds and back donation from a singly occupied molecular orbital, Chem. Sci., 2021, 12, 780–792 RSC.
- B. Tecle, A. F. M. Maqsudur Rahman and J. P. Oliver, X-ray crystal structure of trimethylsilylmethyllithium, J. Organomet. Chem., 1986, 317, 267–275 CrossRef CAS.
- P. Höfer, A. Grupp, H. Nebenführ and M. Mehring, Hyperfine sublevel correlation (HYSCORE) spectroscopy: a 2D ESR investigation of the squaric acid radical, Chem. Phys. Lett., 1986, 132, 279–282 CrossRef.
- P. Schosseler, T. Wacker and A. Schweiger, Pulsed ELDOR Detected NMR, Chem. Phys. Lett., 1994, 224, 319–324 CrossRef CAS.
- F. Allouche, D. Klose, C. P. Gordon, A. Ashuiev, M. Wörle, V. Kalendra, V. Mougel, C. Copéret and G. Jeschke, Low-Coordinated Titanium (III) Alkyl—Molecular and Surface—Complexes: Detailed Structure from Advanced EPR Spectroscopy, Angew. Chem., Int. Ed. Engl., 2018, 130, 14741–14745 CrossRef.
- B. O. Roos and P. Pyykkö, Bonding Trends in Molecular Compounds of Lanthanides: The Double-Bonded Carbene Cations LnCH2+ (Ln = Sc, Y, La–Lu), Chem. – Eur. J., 2010, 16, 270–275 CrossRef CAS PubMed.
- U. Haeberlen, Advances in magnetic resonance, Academic Press, 1976, vol. 1 Search PubMed.
- R. J. Clément, A. J. Pell, D. S. Middlemiss, F. C. Strobridge, J. K. Miller, M. S. Whittingham, L. Emsley, C. P. Grey and G. Pintacuda, Spin-transfer pathways in paramagnetic lithium transition-metal phosphates from combined broadband isotropic solid-state MAS NMR spectroscopy and DFT calculations, J. Am. Chem. Soc., 2012, 134, 17178–17185 CrossRef.
- G. Kervern, G. Pintacuda, Y. Zhang, E. Oldfield, C. Roukoss, E. Kuntz, E. Herdtweck, J.-M. Basset, S. Cadars and A. Lesage, Solid-state NMR of a paramagnetic DIAD-FeII catalyst: sensitivity, resolution enhancement, and structure-based assignments, J. Am. Chem. Soc., 2006, 128, 13545–13552 CrossRef CAS PubMed.
- S. K. Zaremba, Good lattice points, discrepancy, and numerical integration, Ann. Mat. Pura Appl., 1966, 73, 293–317 CrossRef.
- H. Conroy, Molecular Schrödinger equation. VIII. A new method for the evaluation of multidimensional integrals, J. Chem. Phys., 1967, 47, 5307–5318 CrossRef.
- V. B. Cheng, H. H. Suzukawa Jr and M. Wolfsberg, Investigations of a nonrandom numerical method for multidimensional integration, J. Chem. Phys., 1973, 59, 3992–3999 CrossRef CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The ORCA quantum chemistry program package, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- I. Fdez. Galván, M. Vacher, A. Alavi, C. Angeli, F. Aquilante, J. Autschbach, J. J. Bao, S. I. Bokarev, N. A. Bogdanov and R. K. Carlson, et al., OpenMolcas: from source code to insight, J. Chem. Theory Comput., 2019, 15, 5925–5964 CrossRef PubMed.
- F. Aquilante, J. Autschbach, A. Baiardi, S. Battaglia, V. A. Borin, L. F. Chibotaru, I. Conti, L. De Vico, M. Delcey and I. Fdez. Galván, et al., Modern quantum chemistry with [Open] Molcas, J. Chem. Phys., 2020, 152, 214117 CrossRef CAS PubMed.
- X. Zhou, M. F. Reid, M. D. Faucher and P. A. Tanner, Electronic spectra of Cs2NaYbF6 and crystal field analyses of YbX63-(X = F, Cl, Br), J. Phys. Chem. B, 2006, 110, 14939–14942 CrossRef CAS PubMed.
- F. Weinhold, C. R. Landis and E. D. Glendening, What is NBO analysis and how is it useful?, Int. Rev. Phys. Chem., 2016, 35, 399–440 Search PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, NBO 7.0: new vistas in localized and delocalized chemical bonding theory, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- R. L. Halbach, G. Nocton, J. I. Amaro-Estrada, L. Maron, C. H. Booth and R. A. Andersen, Understanding the Multiconfigurational Ground and Excited States in Lanthanide Tetrakis Bipyridine Complexes from Experimental and CASSCF Computational Studies, Inorg. Chem., 2019, 58, 12083–12098 CrossRef CAS PubMed.
- C. H. Booth, M. D. Walter, D. Kazhdan, Y.-J. Hu, W. W. Lukens, E. D. Bauer, L. Maron, O. Eisenstein and R. A. Andersen, Decamethylytterbocene complexes of bipyridines and diazabutadienes: multiconfigurational ground states and open-shell singlet formation, J. Am. Chem. Soc., 2009, 131, 6480–6491 CrossRef CAS PubMed.
- C. H. Booth, D. Kazhdan, E. L. Werkema, M. D. Walter, W. W. Lukens, E. D. Bauer, Y.-J. Hu, L. Maron, O. Eisenstein and M. Head-Gordon, Intermediate-valence tautomerism in decamethylytterbocene complexes of methyl-substituted bipyridines, J. Am. Chem. Soc., 2010, 132, 17537–17549 CrossRef CAS PubMed.
- M. D. Walter, P. T. Matsunaga, C. J. Burns, L. Maron and R. A. Andersen, Synthesis and Reactions of [Cp2*Yb]2(μ-Me) and [Cp2*Yb]2(μ-Me)(Me) and Related Yb2(II,III) and Yb2(III,III) Compounds, Organometallics, 2017, 36, 4564–4578 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2237830–2237832. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cp00281d |
| ‡ Richard A. Andersen passed away on June 16, 2019 while the early draft of this manuscript was being written; this manuscript is dedicated to his memory. |
| This journal is © the Owner Societies 2024 |