
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of particle size on the optical and electronic properties of hydrogenated silicon nanoparticles†
Eimear
Madden
 and
Martijn A.
Zwijnenburg
*
and
Martijn A.
Zwijnenburg
*
Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: m.zwijnenburg@ucl.ac.uk
First published on 21st March 2024
Abstract
We use a combination of many-body perturbation theory and time-dependent density functional theory to study the optical and electronic properties of hydrogen terminated silicon nanoparticles. We predict that the lowest excited states of these silicon nanoparticles are excitonic in character and that the corresponding excitons are completely delocalised over the volume of the particle. The size of the excitons is predicted to increase proportionally with the particle size. Conversely, we predict that the fundamental gap, the optical gap, and the exciton binding energy increase with decreasing particle size. The exciton binding energy is predicted to counter-act the variation in the fundamental gap and hence to reduce the variation of the optical gap with particle size. The variation in the exciton binding energy itself is probably caused by a reduction in the dielectric screening with decreasing particle size. The intensity of the excited state corresponding to the optical gap and other low energy excitations are predicted to increase with decreasing particle size. We explain this increase in terms of the ‘band structure’ becoming smeared out in reciprocal space with decreasing particle size, increasing the ‘overlap’ between the occupied and unoccupied quasiparticle states and thus, the oscillator strength. Fourier transforms of the lowest excitons show that they inherit the periodicity of the frontier quasiparticle states. This, combined with the delocalisation of the exciton and the large exciton binding energy, means that the excitons in silicon nanoparticles combine aspects of Wannier–Mott, delocalisation and effect of periodicity of the underlying structure, and Frenkel, large exciton binding energy, excitons.
Introduction
Silicon nanoparticles (SiNPs) are both interesting from a theoretical and practical perspective. From a theoretical perspective they are perhaps the simplest concrete example of semiconductor nanodots, often referred to as quantum dots, as well as the most amenable to computational studies because of the simple structure and composition. From a practical perspective, SiNPs have been prepared both embedded in a matrix and as colloidal particles, and find use in a range of applications including light emitting diodes, lasers, solar-cells, data storage and bioimaging and sensing.1–3Because of the covalent character of the bonding in silicon, undercoordinated silicon atoms on the surface of the SiNPs need to be terminated by capping groups. These capping groups can, for example, be hydrogen atoms, hydroxyls or organic groups. Experimentally, the exact nature of the capping group depends on the synthesis conditions. In the case of organic groups, they are generally specifically introduced during synthesis, for example, so that the synthesised SiNPs have a special affinity to a biomolecule the SiNP will be used to sense.2 Computationally, mostly hydrogen terminated SiNPs, the most general of SiNPs, are studied under the implicit assumption that the perturbation induced by capping agents other than hydrogen on the optical and electronic properties of the silicon core of the SiNPs are small and that hydrogen terminated SiNPs are thus good models of SiNPs terminated with other capping agents. Additionally, SiNPs can take different shapes, corresponding to different cuts from the bulk silicon structure,4 the most common studied of which are spherical and octahedral SiNPs.
The key optical and electronic properties of SiNPs include the fundamental gap, the optical gap and the exciton binding energy (see Fig. 1). The fundamental gap is the energy required to excite a non-interacting electron–hole pair. The optical gap is the energy required to instead excite an interacting electron–hole pair, often referred to as an exciton, which is bound by the mutual Coulombic interaction between electron and hole. For particles with a singlet electronic ground state like the SiNPs this optical gap corresponds to the lowest energy singlet excitation. The exciton binding energy, finally, is a measure of how much the exciton is stabilised relative to a non-interacting electron–hole pair, and the difference between the fundamental and optical gap (see Fig. 1). Analytical models for the electronic and optical properties of semiconductor nanoparticles based on the effective mass approximation (EMA),5 or using particle in a sphere wavefunctions as basis,6 predict that the fundamental gap of such particles varies as 1/r2 with the particle size r and that the exciton binding energy, or at least its Coulombic part, varies as 1/r. The optical gap, as the fundamental gap minus the exciton binding energy, is predicted by the same models to vary as 1/rn, where n varies with particle size but always lies somewhere between 2 and 1. Analytical models, however, always include necessary approximations. Numerical quantum chemical calculations that make less assumptions yield n values that deviate from the values predicted by the analytical models.7–10 For example, in the case of the fundamental gap such calculations predict n values ranging from 0.7 to 1.5 instead of 2.
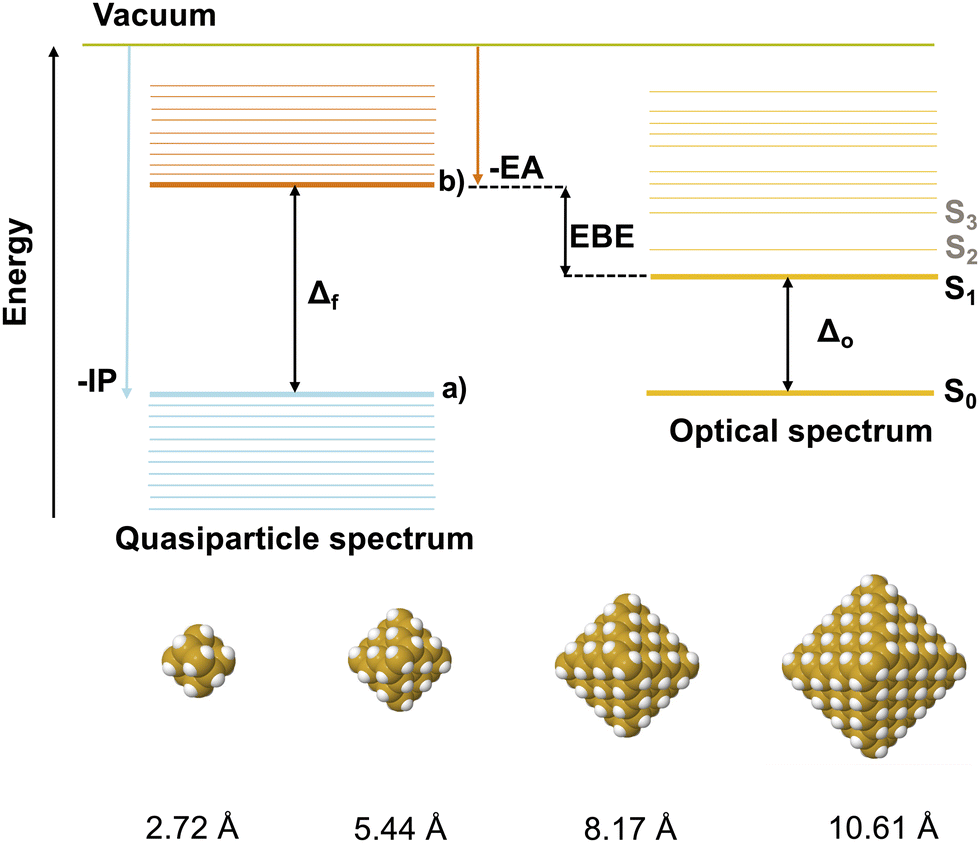 | ||
| Fig. 1 Top: Schematic illustrating the quasiparticle and optical spectra and the definitions of the ionisation potential (IP), electron affinity (EA), fundamental (Δf) and optical (Δo) gaps, and the exciton binding energy (EBE), the difference between the latter two. Here (a) labels the highest occupied quasiparticle state, (b) the lowest unoccupied quasiparticle state, S0 the electronic ground state and S1 the first electronic excited state. Bottom: DFT optimised structures and radii of the four smallest hydrogen terminated SiNPs, Si10H16, Si35H36 and Si84H64 and Si165H100. | ||
Changes in the SiNP size impact not only the fundamental and optical gap, and the exciton binding energy. Various other electronic and optical properties of the SiNPs also change with particle size. For example, with the optical gap, the whole UV-Vis absorption spectrum of SiNPs shifts to the blue with decreasing particle size. The photoluminescence maxima of SiNPs have also been predicted to shift with decreasing particle size to larger energies/shorter wavelengths.11–15 Similarly, the associated Stokes shift, the difference in energy between the optical gap and the photoluminescence maxima, is predicted to increases with decreasing particle size.11–15 Finally, the oscillator strength of the excitations near the optical gap was predicted to increase with decreasing particle size, which was linked in the literature to a transition from an indirect to a direct ‘band gap’ upon nanostructuring of the silicon.16,17
Because of the fundamental interest in SiNPs as model systems for semiconductor quantum dots, as well as the technological applications of SiNPs, hydrogenated silicon nanoparticles have been studied using a wide range of theoretical approaches, including tight-binding models,8,16,18–20 empirical pseudopotential theory,9,21–23 (tight-binding) density functional theory (DFT),7,13,17,24–27 (tight-binding) time-dependent DFT (TDDFT),11,12,14,15,28–36 quantum Monte Carlo methods,35,37 and many-body perturbation theory in the form of GW and GW in combination with solving the Bethe Salpter Equation (GW-BSE).10,31,38–41 However, few of these studies consider the combination of the fundamental gap, optical gap and exciton binding energy, how each of them changes with the particle size, and how these changes are interrelated in one integrated study. Similarly, the exact nature of the delocalisation of the exciton and the frontier quasiparticle states, the states corresponding to the ionisation potential and electron affinity (Fig. 1), are not extensively explored.
Here we revisit the optical and electronic properties of hydrogen terminated SiNPs by performing GW-BSE and time-dependent DFT calculation on octahedral SiNPS containing 10 to 455 silicon atoms, see Fig. 1 and Fig. S1 (ESI†). We specifically focus on the (de)localisation of the electronic and optically excited state over the particles in both real and reciprocal space, and how the particle's fundamental gap, optical gap and exciton binding energy change with particle size. Finally, we also consider the effect of particle size on the predicted intensities of the excitations near the optical gap and predict the particles’ UV-Vis absorption spectra.
Methodology
Octahedral SiNPs, see Fig. 1 and Fig. S1 (ESI†), were cut using the nanocut code,42 from the bulk silicon structure, as taken from the Materials Project.43 The geometry of these SiNPs was subsequently optimised using DFT calculations, employing the B3LYP hybrid density functional,44–46 in combination with the D3 dispersion correction by Grimme and co-workers,47 with Becke–Johnson damping, and the def2-SVP or def2-TZVP basis sets.48 The SiNPs were optimised with their symmetry fixed to their highest possible point group symmetry (Td), as well as for the D2 subgroup and in the absence of any enforced symmetry (C1). Harmonic frequency calculations, where tractable, were conducted to ensure that the optimised structures correspond minima with only positive and no negative frequencies.Subsequently, the quasiparticle spectrum and specifically the highest occupied and lowest unoccupied quasiparticle states, the ionisation potential and electron affinity, of the DFT optimised particles were predicted by different GW variants: single-shot G0W0, eigenvalue-only self-consistent GW (evGW) and quasiparticle self-consistent GW (qsGW) calculations.49–52 These GW calculations utilised the B3LYP orbitals as starting points and again used the def2-SVP or def2-TZVP basis sets. The results of the various GW calculations were then used as inputs for solving the Bethe–Salpeter equation to obtain vertical excitation energies, oscillator strength values, static polarizabilities and ultimately the nanoparticles’ optical gap values.53 For G0W0 the predicted properties will show a dependency on the functional used in the underlying DFT calculation. evGW and qsGW reduce and practically eliminate, respectively, this starting-point dependency by iterating the eigenvalues or the underlying ground state, respectively, until self-consistency is achieved.51 Moreover, in the case of finite-sized systems, the results of the combination of either evGW and qsGW and solving the Bethe–Salpeter equation agrees well with coupled-cluster benchmarks, as explicitly shown for singlet excitation energies for organic molecules,50 and yield excitation energies there that are clearly superior to G0W0-BSE.
Additionally, time-dependent density functional theory (TDDFT) calculations were carried out on the DFT optimised SiNPs, again using the same B3LYP functional and def2-SVP or def2-TZVP basis sets to predict the particle's absorption spectrum. These TDDFT calculations used the Tamm–Dancoff approximation to avoid TDDFT stability issues.54
Throughout all these calculations, version 7.5 of the Turbomole code was employed in combination with a tight integration grid (m5), stringent SCF convergence criteria (denconv set at 1 × 10−7) and the RI-J approximation.55–57 Additionally, the RI-K approximation was applied in all GW and BSE calculations, and in the G0W0 and evGW calculations we exploited the RIGW algorithms implemented in Turbomole,52 where only the highest occupied and lowest unoccupied quasiparticle states are explicitly calculated and the remainder of the Kohn–Sham spectrum is shifted accordingly. The latter approximation reduces the scaling of the calculations from N6 to N4, allowing for the study of much larger nanoparticles, but is not used for the qsGW calculations. The use of symmetry in the GW and BSE calculations in Turbomole is limited to Abelian point groups, therefore the GW and BSE calculations on SiNPs use the Abelian D2 or C1 sub-groups of the non-Abelian Td point group instead of the full Td symmetry of the particles. Finally, where plotting spectra or commenting on the strength of particular excitations we always use oscillator strength values calculated within the velocity gauge.
The character of the excited states predicted by the BSE and TDDFT was analysed in terms of the most prominent natural transition orbitals (NTOs),58 and using the excited state analysis software TheoDORE,59 in terms of the exciton size, charge-transfer character, and contributions of different atoms. Because TheoDORE is limited to analysing the results of calculation performed in C1, the latter analysis was limited in the case of BSE to particles for which we were able to perform BSE calculations in C1.
The effective static dielectric constants of the particles were estimated based on their static polarizability calculated from evGW-BSE. Firstly, the static evGW-BSE polarizability values α were converted into polarizability volume α′ values using:
| α′ = α/(4pε0) | (1) |
| requiv. = ((3/(4π))Vm)1/3 | (2) |
| εr = (requiv.3 + 2α′)/(requiv.3 − α′) | (3) |
The bandlike nature of the frontier orbitals and NTOs was analysed by performing a discrete Fourier transform on the orbitals or NTOs. This Fourier transform was performed using an inhouse Python code based around routines from the numpy library and with a cube file of the orbitals/NTOS as input.61 The Fourier transform was plotted as a function of kx and ky defined as:
| kx/y = 2π/lx/y | (4) |
The different properties of the SiNPs are plotted versus the radius of the silicon core of the SiNPs. The radius is calculated from the average edge length of the silicon cores (L), assuming the particles are ideal octahedra, as the distance between the centroid and the vertices:
| R = (1/2)21/2L | (5) |
Finally, TDDFT optical absorption spectra were generated by representing all excitations in a given window above the lowest excited state by Gaussian functions with a width of 0.05 eV, followed by a point-by-point summation over these Gaussian functions for an equally spaced grid of points in that window. The size of the window depends on the size of the specific SiNP, as with increasing size of the SiNPs the spectrum of excited states becomes denser making it computationally harder to calculate all excitations in a window, as with the basis-set size.
Results and discussion
Fundamental gap
The top panel of Fig. 2 shows the fundamental gap and the energies of the highest occupied and lowest unoccupied quasiparticle states relative to vacuum as obtained from evGW and qsGW, as well as their DFT Kohn–Sham counterparts, for the different octahedral SiNPs studied. As expected, based on the literature,7–10 the fundamental gap values are predicted to decrease with increasing particle size while the energy of the highest occupied and lowest unoccupied quasiparticle states become less and more negative, respectively. | ||
| Fig. 2 Top: Quasiparticle spectrum for each SiNP showing the four highest occupied and four lowest unoccupied Kohn–Sham orbitals/quasiparticle states obtained from DFT, evGW and qsGW (the latter two where tractable). evGW fundamental gap values are also presented for each SiNP with the DFT Kohn–Sham gaps given in between parentheses. Bottom left: Plot of the optical gap values predicted using TDDFT, evGW-BSE and qsGW-BSE versus particle radius, as well fits to the evGW-BSE/def2-SVP and TDDFT/def2-SVP optical gaps where a is the optical gap of bulk silicon (1.1 eV), r is the SiNP radius, and b and n are fitting constants (see Table S10 for fitted values, ESI†), and data and fits for the experimental data of Wolkin et al.,63 and Furukawa et al.64 Bottom right: Plot of the exciton binding energy as a function of particle radius, as calculated from the fundamental gap and optical gap values predicted by evGW and evGW-BSE and DFT and TDDFT, respectively (open symbols for Si10H16 calculated using the lowest bright excited state). All results obtained using D2 symmetry, B3LYP and the def2-SVP basis-set, except if otherwise indicated. | ||
The results in the top panel of Fig. 2 are obtained using the def2-SVP basis-set. However, Tables S1–S6 (ESI†) show that values calculated with the larger def2-TZVP basis-set, calculations which, in the case of evGW and qsGW, are only computationally tractable for the smaller SiNPs, are very similar, and importantly show the same trend. The same holds for the results of qsGW calculations when compared to their evGW counterparts, validating the use of the RIGW algorithm which combined with the evGW scheme allows us to study much larger particles than possible otherwise. The DFT Kohn–Sham gap is considerably smaller than its evGW counterpart, while the energy of the highest occupied and lowest unoccupied Kohn–Sham states is less negative and more negative, respectively, than the corresponding evGW quasiparticle states. The fact that the DFT Kohn–Sham gap is smaller than the fundamental gap predicted by evGW, as well as the fact that DFT predicts a more negative energy of lowest unoccupied Kohn–Sham states than the evGW-EA, is expected because for pure density functionals, the unoccupied orbitals feel the same field of N − 1 electrons as the occupied orbitals instead of the correct N electrons.62 Introduction of exact exchange corrects this issue to a degree but not completely. However, importantly the DFT Kohn–Sham and evGW results display the same trend and the difference appears to reduce with increasing particle size.
For all method/basis-set combinations the fundamental gap is found, as expected from the analytical models discussed in the introduction and previous calculations,7–10 to vary as approximately one over the particle-size to some power n (see Fig. S2, ESI†). To exemplify this trend and to compare with the scaling of the fundamental gap previously reported in the literature and discussed in the introduction, we fit the fundamental gap to:
| Δf = a + b/rn | (6) |
Finally, the highest occupied quasiparticle state for all octahedral SiNPs belongs to the T2 irrep and is hence triply degenerate. The lowest unoccupied quasiparticle state is non-degenerate and, in all cases, belongs to the A1 irrep. We do not observe a switch of the character of the highest occupied quasiparticle state for the smaller particles from T2 to T1 as seen in some early calculations on spherical cuts.9 The Kohn–Sham orbitals underlying the highest occupied and lowest unoccupied quasiparticle states for the three smallest SiNPs, Si10H16, Si35H36 and Si84H64 are shown in Fig. 3, where in the case of the triply degenerate highest occupied Kohn–Sham orbital, only one out of three is shown.
 | ||
| Fig. 3 Highest occupied (top) and lowest unoccupied (bottom) molecular orbitals of the three smallest SiNPs, Si10H16 (left), Si35H36 (centre) and Si84H64 (right), where the phase of the orbitals is shown as red and blue, respectively. The highest occupied molecular orbital is triply degenerate and only one of the three degenerate orbitals is shown per particle. All results obtained using evGW, D2 symmetry, B3LYP and the def2-SVP basis-set. | ||
Optical gap
The bottom left panel of Fig. 2 shows the optical gap, the lowest energy singlet-to-singlet excitation, of the different octahedral SiNPs as a function of particle-size as calculated with TDDFT, evGW-BSE and qsGW-BSE. For all SiNPs other than Si10H16, the optical gap corresponds to a dipole-allowed excitation, while for Si10H16 the first dipole-allowed excitation lies higher in energy. Because of the Td symmetry of the octahedral SiNPs, the dipole-allowed excitations have T2 character (or B1/2/3 character and non-zero oscillator strength in the case of the qs/evGW-BSE calculation in D2). The optical gap for Si10H16 in contrast has A2 (A in D2) character. It should be added here that the fact that the optical gap of Si10H16 is non-bright and has A2 (A) character is not due to a change of the character of the highest occupied and lowest unoccupied quasiparticle states, which as discussed above, have T2 and A1 character for all octahedral SiNPs studied. Instead, the change in character of the optical gap stems from an A2 (A) excitation from the second highest occupied quasiparticle state to the second lowest unoccupied quasiparticle state, having T1 and T2 character, respectively, coming down in energy below the lowest T2 excitation from the highest occupied to the lowest unoccupied quasiparticle state. Fig. S3 in the ESI,† shows the equivalent of Fig. 2 but then including for Si10H16 the lowest bright dipole-allowed excitation. Finally, for the larger octahedral SiNPs, Si84H64 and larger, the lowest dipole-allowed T2 excitation is effectively degenerate with the lowest non-dipole-allowed T1 excitation.Similar to the fundamental gap, the bottom left panel of Fig. 2, as well Tables S7 and S8 (ESI†), show that def2-SVP and def2-TZVP predicted values agree reasonably well, with the def2-TZVP results shifted down relative to their def2-SVP counterparts, as do the results of BSE calculations on top of evGW and qsGW calculations. Moreover, in contrast to the fundamental gap, the evGW-BSE/qsGW-BSE and TDDFT predictions of the optical gap agree well, something that is not generally the case for inorganic nanoparticles, or at least without tuning the amount of exact exchange used in the TDDFT calculations.65,66 The optical gap of Si35H36 predicted by evGW-BSE/qsGW-BSE here agrees well with the onset of the BSE predicted spectra of Si35H36 by Rocca et al.67 Similarly, the TDDFT predicted optical gap value for Si35H36 is comparable to that predicted by Lethhonen and co-workers,14,15 and Wang and co-workers,12 both of which used B3LYP, and slightly smaller than that reported by Foerster and Besley,33 in line with the fact that the latter used CAM-B3YLP which is known to result in a blue-shift of the predicted optical absorption spectra relative to B3LYP.
Wolkin et al. reported the experimental photoluminescence energies (PLEs) of silicon nanoparticles of a range of sizes embedded in a porous silicon matrix generated through electrochemical and photo assisted stain etching, where the synthesized size range overlaps with that studied here by TDDFT/def2-SVP and evGW-BSE/def2-SVP.63 Similarly, Furukawa et al. reported the optical gap of hydrogenated silicon nanoparticles prepared by reactive etching with a similar size to that studied here.64 In both cases, the experimental values are ∼0.4–0.5 eV lower than the optical gaps predicted using TDDFT/def2-SVP and evGW-BSE/def2-SVP, (see Fig. 2 bottom left), but display a similar trend to the predicted optical gaps. In the Discussion section below, we discuss in detail how this discrepancy between the experimentally measured and predicted optical gap values is likely due to issues with the experimental data rather than the predictions.
For all method/basis-set combinations the optical gap is, like the fundamental gap, found to vary as approximately one over the particle-size to some power n. We fit the optical gap to a similar equation as the fundamental gap:
| Δo = a + b/rn | (7) |
Exciton binding energy
The exciton binding energy values, shown in the bottom right panel of Fig. 2, obtained from the difference in the fundamental and optical gap values predicted with evGW/evGW-BSE or DFT/TDDFT, decrease in both cases with increasing particle size. The absolute values predicted by DFT/TDDFT are slightly larger than those predicted by evGW/evGW-BSE but both agree that the lowest excited state in 1–2 nm SiNPs is stabilised by hundreds of meV relative to a free electron and hole, instead of 10–20 meV in the bulk.68,69 This not only confirms that the lowest excited state in such particles is excitonic in character, but also that the dielectric screening of charges in such particles is less efficient than in the bulk and decreases with decreasing particle size.To provide a more qualitative picture, we predicted the static dielectric constant of the of the four smallest SiNPs: Si10H16, Si35H36, Si84H64, Si165H100 by calculating their static polarizability using evGW-BSE and using that as an input to obtain the predicted static dielectric constants. As can be seen from Fig. 4 the predicted static dielectric constants of the SiNPs are smaller than that of bulk silicon (11.4–12.0 experimentally,70,71 11.5–12.7 G0W0/G0W0 + BSE)72 but increase with particle size. The predicted values are similar and follow a similar trend with particle size to that predicted by Wang and Zunger using their empirical pseudopotential approach and those predicted by Delerue and co-workers using a tight-binding method.73,74
 | ||
| Fig. 4 The static dielectric constant of the four smallest SiNPs Si10H16, Si35H36, Si84H64, Si165H100 as a function of particle size. The static polarizability was predicted using evGW-BSE, with D2 symmetry and B3-LYP, and used to obtain the static dielectric constant according to eqn (1)–(3). | ||
Overall, our predicted static dielectric constants and those from the literature, support the suggestion above that the larger exciton binding energy of the small SiNPs is the result of the reduced dielectric screening of the exciton in such particles, and the decrease in exciton binding energy with particle size is due to an increase in the static dielectric constant of the SiNPs tending towards the bulk value.
Finally, like the fundamental and optical gap, and as expected from the analytical models discussed in the introduction, the exciton binding energy varies as approximately one over the particle-size to some power n. To exemplify this trend, we fit the optical gap to a similar equation as the fundamental and optical gap:
| EBE = a + b/rn | (8) |
Character of the lowest excited state
As already mentioned above, the lowest excited state in octahedral SiNPs is excitonic in character. Moreover, as, as discussed above, the highest occupied and lowest unoccupied quasiparticle states (and highest occupied and lowest unoccupied Kohn–Sham states) have T2 and A1 character respectively. As a result, the lowest optically allowed excited state, which corresponds to the lowest T2 state in both the evGW-BSE and TDDFT calculations, has significant ‘HOMO’ to ‘LUMO’ character.Using TheoDORE it was possible to calculate the exciton radius, defined as the root-mean-square separation between the instantaneous electron and hole position,75 and the atoms over which the excited electron and hole are (de)localised for the lowest excited state as calculated with TDDFT and evGW-BSE. As TheoDORE is currently limited to analysing the results of C1 calculations, this limited us to an analysis of the smaller SiNPs, for which it was computationally tractable to run TDDFT and evGW-BSE calculations in C1, i.e., Si10H16–Si165H100 for TDDFT and Si10H16 and Si35H36 in the case of evGW-BSE. This analysis shows that the excited electron and hole are delocalised over the silicon atoms of the core with little or no contribution of the terminating hydrogens. The TheoDORE analysis also showed that the exciton radius was similar for TDDFT and evGW-BSE and increases proportionally with the SiNP radius (Fig. 5), as would be expected for an excited state delocalised over the whole nanocrystal volume. Visualisation of the hole and electron components of the leading natural transition orbitals for the lowest evGW-BSE excited state for the different particles, as shown in Fig. 5 (and Fig. S5 and S6 for their TDDFT counterparts, ESI†), yields the same picture.
 | ||
| Fig. 5 Left: Plot of the exciton size for the lowest excited state (filled symbols) and lowest optically allowed excited state when that is not the lowest excited state (open symbols) as a function of the SiNP radius, as obtained with evGW-BSE and TDDFT. Right: The leading NTOs of the three smallest SiNPs, Si10H16, Si35H36 and Si84H64, calculated via evGW-BSE. (top hole orbitals, bottom particle (excited electron) orbitals, the red and blue signifies the phases of the NTOs). All results obtained using C1 symmetry, B3LYP and def2-SVP basis-set. As the calculation were performed in C1 instead of the Td point group, the triply degenerate T2 excited state is described as a triplet of degenerate C1 excited states, the NTOs of only one of those degenerate states is shown here for each particle. | ||
Oscillator strengths
Bulk silicon has an indirect bandgap and as a result displays negligible absorption of light at the indirect band gap. Light absorption only starts a couple of tenths of eV above this gap. As discussed in the introduction, it has been predicted in the literature based on tight-binding and DFT calculations that the ‘bandgap’, or more accurately, the fundamental gap, of SiNPs is more direct-like and that nanostructuring of the silicon induces a transition from an indirect to a direct gap material. The predicted oscillator strengths of the lowest excited-state of the SiNPs as calculated with evGW-BSE and TDDFT indeed are found to decrease with particle size, see Fig. 6, in line with similar predictions from previous tight-binding,16 and DFT calculations.7 This observation does not change when we considered in the case of TDDFT not just the lowest excited-state, but all excited-states within 0.2 or 0.5 eV from the optical gap, to account for the fact that with increasing particle size the spectrum inherently gets denser. We should note, however, that even for the smallest SiNPs considered, the predicted oscillator strength values are rather low; one or two orders of magnitude lower than those predicted for the localised excitations in similar sized oxide and chalcogenide nanoparticles.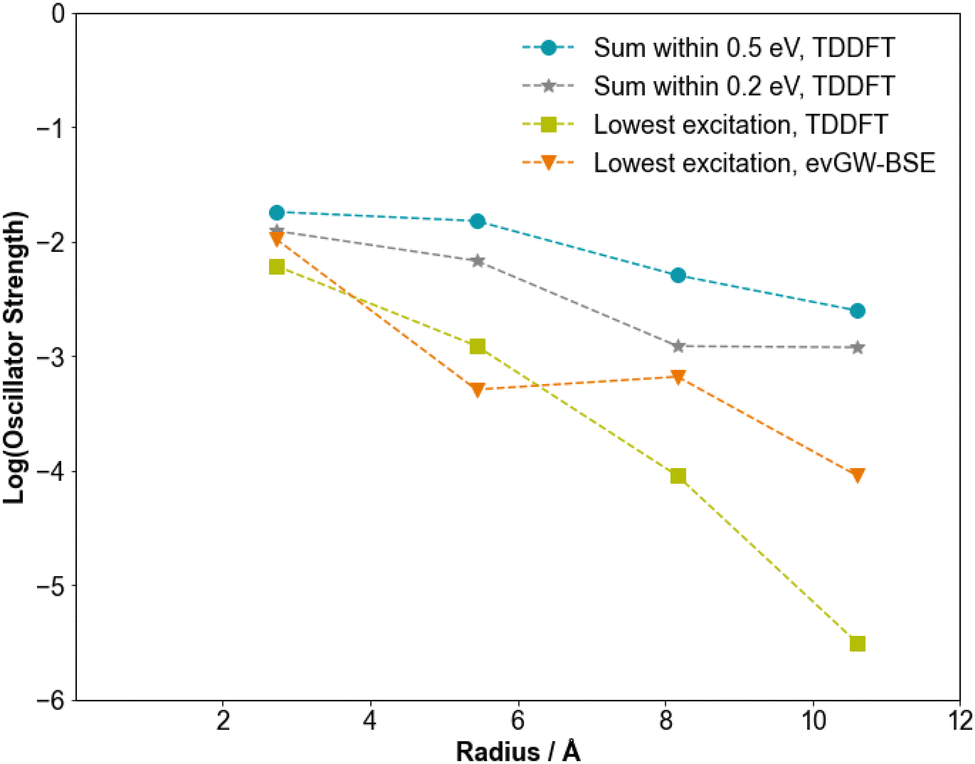 | ||
| Fig. 6 Log of the predicted oscillator strengths of various excitations of SiNPs. The blue circles represent the sum of the oscillator strengths for the excitation energies which lie within 0.5 eV of the lowest excitation for each SiNP, calculated using TDDFT. The grey stars represent the sum of the oscillator strengths for the excitations which lie within 0.2 eV of the lowest excitation for each SiNP, calculated using TDDFT. The green squares represent the oscillator strength of the lowest excitation for each SiNP calculated using TDDFT. The orange triangles represent the oscillator strength of the lowest excitation for each SiNP calculated using evGW-BSE. All calculations used the B3LYP functional and the def2-SVP basis-set. | ||
Kocevski and co-workers have analysed the changing nature of the gap of SiNPs with particle size in terms of the Fourier transform of the frontier orbitals of spherical SiNPs.17 While bands strictly only exist under periodic boundary conditions and the wavevector k is only a proper quantum number in that limit, one can study signatures of ‘periodicity’ in the spatial nature of the frontier orbitals, and by extension the corresponding quasiparticle states, by considering the discrete Fourier transform of the orbitals as a function of kx, ky and kz. Fig. 7 shows the absolute values of the Fourier transforms of the three components of the Kohn–Sham HOMO and the LUMO as a function of kx and ky with the information along the z-direction projected on the xy plane by summing up the contributions along the z-axis for each xy value. The wavevectors are expressed as fractions of 2π/a0, where a0 is the lattice parameter of the primitive cell of bulk silicon (taken to be 3.89 Å). Just like Kocevski and co-workers we observe that with increasing particle size the spots in the plot become sharper, suggesting the orbitals become more band-like. Similarly, we also observe, just like them, that for the largest particles, the pattern in the spots becomes clearly different for the HOMO and LUMO in line with the valence band maximum and conduction band maximum for the bulk lying at different k values.
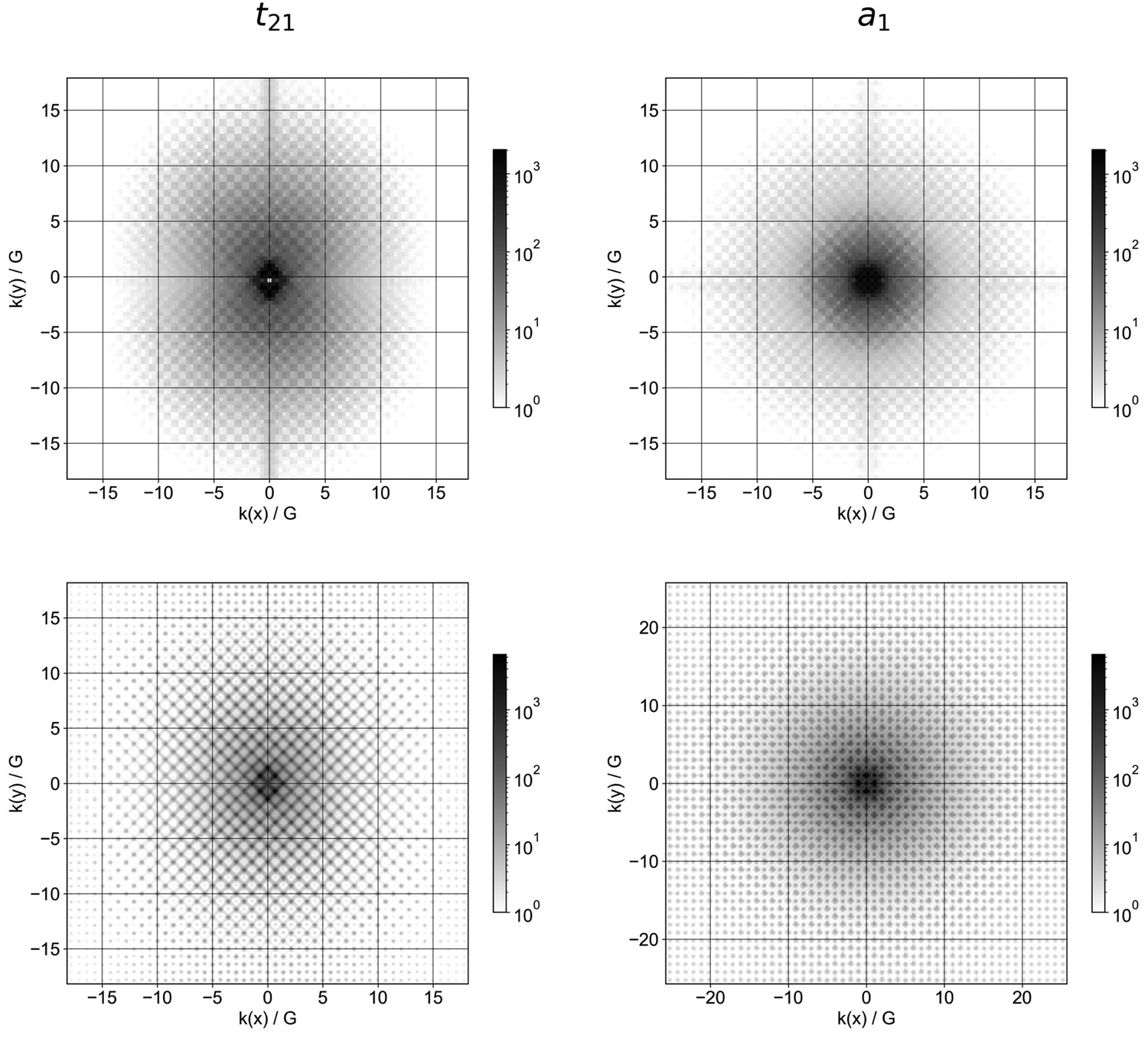 | ||
| Fig. 7 Fourier transforms of one of three degenerate HOMO orbitals (left) and the LUMO (right) of the smallest SiNP, Si10H16 (top) and the largest SiNP, Si455H195 (bottom), calculated using DFT and Td symmetry. The Fourier transforms of the other two HOMO orbitals can be found in the ESI† (Fig. S7). | ||
Interestingly, as can be seen from Fig. 7 and 8, the NTOs of the lowest excited state appear to display similar regular spot patterns in their Fourier transform as their frontier orbital equivalents. The NTOs, and thus the lowest energy exciton, appear to inherit the periodicity of the frontier orbitals, and thus the periodicity of the underlying atomic structure of the SiNP, as well as the differences in ‘localisation’ in reciprocal space between them.
 | ||
| Fig. 8 Fourier transforms of the hole (left) and electron (right) component of the leading NTO of Si165H100, calculated using evGW-BSE and D2 symmetry. As the calculation were performed in D2 instead of the Td point group, the triply degenerate T2 excited state is described as a triplet of degenerate B1/2/3 excited states, the Fourier transforms of the leading NTOs of only one of those states are shown here. The others can be found in the ESI† (Fig. S8). | ||
Optical absorption spectra
Finally, we predicted the optical absorption spectra for the four smallest SiNPs using TDDFT. The predicted optical absorption spectra in Fig. 9 follow the trends observed above for the absorption onset. The spectra exhibit a progressive blueshift with decreasing SiNP particle size in line with the increase in the optical gap with decreasing SiNP size, discussed above. Similarly, the intensity of the peaks reduces with increasing SiNP size in line with the trend observed above in Fig. 6. Lastly, in line with what we hypothesised above, as the size of the SiNP increases, the spectra become denser, the gaps between individual excitations decreases, which is evident from the clear broadening of the peaks in the spectra for the larger SiNPs. | ||
| Fig. 9 Predicted optical absorption spectra of the four smallest SiNPs; Si10H16, Si35H36, Si84H64 and Si165H100, as calculated using TDDFT, Td symmetry, the B3LYP functional and a Gaussian broadening of 0.05 eV. The blue lines represent the TDDFT spectra calculated with the def2-TZVP basis set, and the green lines the TDDFT spectra calculated with the def2-SVP basis set. Calculations with the def2-TZVP basis set were limited to the smaller SiNPs because of the scaling of the cost of the calculations with system size. | ||
Comparing the optical absorption spectra obtained with def2-SVP and def2-TZVP basis-sets for Si10H16 and Si35H36 we observe a small red-shift of the def2-TZVP results compared to their def2-SVP counterparts, in line with what was observed for the optical gap above. More interestingly perhaps, we also observe that the def2-TZVP predicted oscillator strength values are consistently larger than their def2-SVP counterparts, though importantly follow a similar trend.
Discussion
The difference between the predicted optical gap values and the experimentally measured values can in part be explained by the need to use the def2-SVP basis-set to be able to study the larger particles and the fact that calculations using this basis-set overestimate, as discussed above, the optical gap, relative to calculations with larger basis-sets. Also, the calculations ignore vibronic effects, inclusion of which likely would also reduce the predicted optical gap. However, the major source of the discrepancy might be the interpretation of the experimental values. While the PLE values of Wolkin et al.63 have often been used as a proxy for the optical gap of SiNPs, such a comparison strictly requires the Stokes shift to be negligible. This might be a fair approximation in the bulk limit, but TDDFT/def2-SVP calculations in which the excited-state is relaxed (see Table S12, ESI†) and previous work on excited-state relaxation in SiNPs11–15 suggests it is not for the SiNPs in the size range studied. The PLE data thus provides more of an experimental lower limit to the optical gap values. In contrast, Furukawa et al. use a Tauc analysis to extract the optical gap of the particles directly.64 However, the suitability of a Tauc analysis to extract the optical gap of nanoparticles, which, for example, display vibrational broadening, is not known, something with which the low oscillator strength associated with the optical gap also will not help. These issues with the interpretation of the experimental values are compounded by additional uncertainty for both datasets in the measured particle size, often obtained indirectly, as well as the effects of polydispersity, and the exact chemical composition. We suspect that such issues with the optical gap data are also inherited by the fitted n values. Additionally, in the case of the data by Furakawa et al., the small size-range data available makes extrapolation and thus fitting a n value difficult, even in the absence of other issues.As mentioned in the Results section, evGW-BSE, qsGW-BSE and TDDFT all predict very similar trends for the variation of the optical and fundamental gaps with particle size, where the optical and fundamental gap increase with decreasing particle size in 1/rn fashion, with similar n values for all methods and basis-sets. This 1/rn trend is, as discussed in the introduction, the expected trend from analytical models of the optical and fundamental gap of semiconductor particles, and is the trend seen in the experimental data for the optical gap (proxies) and previous calculations. However, the here predicted n values are smaller than those obtained from experiment and most of the previous calculations which report a n value or report sufficient data for us to fit a n value for. In the case of experiments, this difference in n is, as discussed above, probably related to issues with the interpretation of the experimental data. In the case of the comparison with the previous calculations, the calculations that give the larger n values use much more approximate methods than those used here, while the most recent calculation for both the optical and fundamental gaps give n values very similar to those obtained here.
As discussed above, based on the ev/qsGW-BSE predicted exciton binding energies, the excitation corresponding to the optical gap for all SiNPs studied here is clearly excitonic in nature with exciton binding energies one to two orders of magnitude larger than that for bulk silicon. The predicted exciton binding energies, >0.5 eV, lie in the range expected for Frenkel excitons. However, the fact that the exciton radii are similar to the SiNP radii and that the natural transition orbitals appear fully delocalised over the volume of the particles is more suggestive of a Wannier–Mott exciton. As is the fact that the Fourier transform of the natural transition orbitals show a similar spot pattern as the corresponding quasiparticle states, and thus likely display the same periodicity. The predicted large exciton binding energies thus seem completely driven by the poor dielectric screening by the vacuum outside the particles, in line with the fact that the reduction of the exciton binding energy goes hand in hand with an increase of the particle's predicted dielectric constant. The excitons in the SiNPs thus both combine Frenkel and Wannier–Mott like character.
Interestingly, the scaling of the predicted fundamental gap, optical gap and exciton binding energy with particle size is very similar in terms of the exponent n. This does not mean, however, that they vary by the same amount, as the b prefactors are significantly different. The variation in the evGW fundamental gap, our best estimate of the fundamental gap, with particle size, is approximately twice as large as the variation in the evGW-BSE optical gap. The exciton binding thus counter-acts the variation in the fundamental gap with particle size. This is expected based on the analytical models, discussed in the introduction, though such models would also predict that exciton binding energy, or at least its Coulombic component, would have a different scaling (a fundamentally different n value) than the fundamental gap, which is at odds with our predictions here. The similar scaling of the fundamental gap, optical gap and exciton binding energy with particle size also implies that these properties, by necessity, scale approximately linearly with each other. An example of that can be seen in Fig. S4 (ESI†) for the scaling of the evGW-BSE exciton binding energy with the evGW-BSE predicted fundamental gap.
In the literature the effect of nanostructuring on the intensity of the absorption of light near the optical gap is often discussed in terms of the ‘band gap’ changing from indirect to direct. However, our results here and the previous work of Kocevski and co-workers suggest that reducing the size of the particles does not so much change the ‘band gap’ from indirect to direct but rather that the ‘band structure’ becomes more smeared out, in line with the fact that k is not a good quantum number for a finite-size system. The reduction in oscillator strength with increasing particle size that we observe here, and which was previously observed by others,7,16 is probably driven simply by a reduction in the ‘overlap’ between the orbitals from which the electron gets excited and the orbital the electrons gets excited into. The ‘overlap’ in such a scenario then would be largest in the case of the smeared out ‘band structure’ for the small particles and reduces in size when with increasing particle size the orbitals, just like the valence band maximum and conduction band minimum for infinite crystals, become more sharply localised at different points in k-space.
Conclusions
The lowest excited state of hydrogen terminated silicon nanoparticles is predicted to be excitonic in character and the corresponding exciton is predicted to be completely delocalised over the volume of the particle. The size of the exciton increases proportionally with the particle radius. Conversely, the optical gap, the energy of the lowest optically excited state, increases when decreasing the particle radius in a 1/rn fashion. The fundamental gap, the energy required to generate a non-interacting electron–hole pair, which is the equivalent of the bandgap for an infinite solid, increases in a similar 1/rn fashion upon decreasing the particle size with a similar n as for the optical gap. However, even though the optical and fundamental gap are predicted to have a similar scaling with particle size, the change in the fundamental gap with particle size is much larger than for the optical gap. The exciton binding energy, which itself also changes with particle size, counter-acts the variation in the fundamental gap with particle size and reduces the variation of the optical gap. The variation in the exciton binding energy itself is, based on a prediction of the static dielectric constant of the particles, probably the result of a reduction in the dielectric screening with decreasing particle size.The intensity of the excited state corresponding to the optical gap, and the excitations lying close to it in energy, are predicted to clearly increase with decreasing particle size. This increase in intensity upon reduction of the particle size has been explained in the literature as being due to a transition from an indirect to a direct ‘band gap’. However, Fourier transforms of the orbitals corresponding to the frontier quasiparticle states suggest that the origin more likely is the fact that the ‘band structure’ becomes smeared out with decreasing particle size, in line with the fact that the wavevector k is not a good quantum number for a finite-size system. This smearing increases the ‘overlap’ between the occupied and unoccupied states, and consequently also increases the oscillator strengths.
Fourier transforms of the natural transition orbitals that describe the excited states show that the lowest exciton inherits the periodicity of the frontier quasiparticle states. This combined with the delocalisation of the exciton over the volume of the particle and the large exciton binding energy means that the lowest exciton in these silicon nanoparticles combine aspects of Wannier–Mott (delocalisation and effect of periodicity of the underlying structure), and Frenkel (large exciton binding energy) excitons.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
EM acknowledges the UK Engineering and Physical Sciences Research Council (EPSRC) for a DTP studentship (EP/T517793/1). The authors thank Dr Katarina Brlec, Dr Christof Holzer, Dr Seán Kavanagh, Prof. Johannes Lischner, Mr Adair Nicolson, Dr Felix Plasser, Dr Miguel Rivera and Dr Joe Willis for useful discussion.References
- K. Dohnalová, T. Gregorkiewicz and K. Kůsová, Silicon quantum dots: surface matters, J. Phys.: Condens. Matter, 2014, 26, 173201 CrossRef PubMed.
- C. M. Gonzalez and J. G. C. Veinot, Silicon nanocrystals for the development of sensing platforms, J. Mater. Chem. C, 2016, 4, 4836–4846 RSC.
- D. Beri, Silicon quantum dots: surface matter, what next?, Mater. Adv., 2023, 4, 3380–3398 RSC.
- H. F. Wilson, L. McKenzie-Sell and A. S. Barnard, Shape dependence of the band gaps in luminescent silicon quantum dots, J. Mater. Chem. C, 2014, 2, 9451–9456 RSC.
- L. Brus, Electronic Wave Functions In Semiconductor Clusters: Experiment and Theory, J. Phys. Chem., 1986, 90, 2555–2560 CrossRef CAS.
- L. E. Brus, Electron-electron and electron-hole interactions in small semiconductor crystallites: the size dependence of the lowest excited electronic state, J. Chem. Phys., 1984, 80, 4403–4409 CrossRef CAS.
- B. Delley and E. F. Steigmeier, Quantum confinement in Si nanocrystals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 1397 CrossRef CAS PubMed.
- J. P. Proot, C. Delerue and G. Allan, Electronic structure and optical properties of silicon crystallites: application to porous silicon, Appl. Phys. Lett., 1992, 61, 1948–1950 CrossRef CAS.
- F. A. Reboredo, A. Franceschetti and A. Zunger, Dark excitons due to direct Coulomb interactions in silicon quantum dots, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 13073–13086 CrossRef CAS.
- S. Wippermann, M. Vörös, D. Rocca, A. Gali, G. Zimanyi and G. Galli, High-pressure core structures of Si nanoparticles for solar energy conversion, Phys. Rev. Lett., 2013, 110, 046804 CrossRef CAS PubMed.
- X. Wang, R. Q. Zhang, S. T. Lee, T. A. Niehaus and T. Frauenheim, Unusual size dependence of the optical emission gap in small hydrogenated silicon nanoparticles, Appl. Phys. Lett., 2007, 90, 123116 CrossRef.
- X. Wang, R. Q. Zhang, T. A. Niehaus, T. Frauenheim and S. T. Lee, Hydrogenated silicon nanoparticles relaxed in excited states, J. Phys. Chem. C, 2007, 111, 12588–12593 CrossRef CAS.
- A. Puzder, A. J. Williamson, J. C. Grossman and G. Galli, Computational studies of the optical emission of silicon nanocrystals, J. Am. Chem. Soc., 2003, 125, 2786–2791 CrossRef CAS PubMed.
- O. Lehtonen and D. Sundholm, Computational studies of semiconductor quantum dots, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085424 CrossRef.
- O. Lehtonen, D. Sundholm and T. Vänskä, Density-functional studies of excited states of silicon nanoclusters, Phys. Chem. Chem. Phys., 2008, 10, 4535–4550 RSC.
- F. Trani, G. Cantele, D. Ninno and G. Iadonisi, Tight-binding calculation of the optical absorption cross section of spherical and ellipsoidal silicon nanocrystals, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 075423 CrossRef.
- V. Kocevski, O. Eriksson and J. Rusz, Transition between direct and indirect band gap in silicon nanocrystals, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 245401 CrossRef.
- S. Yuan Ren, Quantum confinement of edge states in Si crystallites, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 4665 CrossRef.
- K. Leung and K. B. Whaley, Electron-hole interactions in silicon nanocrystals, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 7455 CrossRef CAS.
- M.-F. Ng and R. Q. Zhang, Dimensionality Dependence of Optical Properties and Quantum Confinement Effects of Hydrogenated Silicon Nanostructures, J. Phys. Chem. B, 2006, 110, 21528–21535 CrossRef CAS PubMed.
- B. G. Lee, J. W. Luo, N. R. Neale, M. C. Beard, D. Hiller, M. Zacharias, P. Stradins and A. Zunger, Quasi-Direct Optical Transitions in Silicon Nanocrystals with Intensity Exceeding the Bulk, Nano Lett., 2016, 16, 1583–1589 CrossRef CAS PubMed.
- J. W. Luo, S. S. Li, I. Sychugov, F. Pevere, J. Linnros and A. Zunger, Absence of redshift in the direct bandgap of silicon nanocrystals with reduced size, Nat. Nanotechnol., 2017, 12, 930–932 CrossRef CAS PubMed.
- L. W. Wang and A. Zunger, Electronic Structure Pseudopotential Calculations of Large (.apprx.1000 Atoms) Si Quantum Dots, J. Phys. Chem., 1994, 98, 2158–2165 CrossRef CAS.
- E. W. Draeger, J. C. Grossman, A. J. Williamson and G. Galli, J. Chem. Phys., 2004, 120, 10807–10814 CrossRef CAS PubMed.
- G. Cantele, E. Degoli, E. Luppi, R. Magri, D. Ninno, O. Bisi, S. Ossicini and G. Iadonisi, Electronic, structural and optical properties of hydrogenated silicon nanocrystals: the role of the excited states, Phys. Status Solidi C, 2005, 2, 3263–3267 CrossRef CAS.
- N. Zonias, P. Lagoudakis and C.-K. Skylaris, J. Phys.: Condens. Matter, 2010, 22, 025303 CrossRef PubMed.
- E. Degoli, G. Cantele, E. Luppi, R. Magri, D. Ninno, O. Bisi and S. Ossicini, Optical properties of passivated silicon nanoclusters: the role of synthesis, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 155411 CrossRef.
- I. Vasiliev, S. Öǧüt and J. R. Chelikowsky, Ab initio absorption spectra and optical gaps in nanocrystalline silicon, Phys. Rev. Lett., 2001, 86, 1813–1816 CrossRef CAS PubMed.
- A. Gali, M. Vörös, D. Rocca, G. T. Zimanyi and G. Galli, Nano Lett., 2009, 9, 3780–3785 CrossRef CAS PubMed.
- R. Q. Zhang, A. de Sarkar, T. A. Niehaus and T. Frauenheim, Phys. Status Solidi B, 2012, 249, 401–412 CrossRef CAS.
- D. Rocca, M. Vörös, A. Gali and G. Galli, Ab initio optoelectronic properties of silicon nanoparticles: excitation energies, sum rules, and Tamm-Dancoff approximation, J. Chem. Theory Comput., 2014, 10, 3290–3298 CrossRef CAS PubMed.
- A. Nurbawono, S. Liu and C. Zhang, Modeling optical properties of silicon clusters by first principles: from a few atoms to large nanocrystals, J. Chem. Phys., 2015, 142, 154705 CrossRef PubMed.
- A. Foerster and N. A. Besley, Quantum Chemical Characterization and Design of Quantum Dots for Sensing Applications, J. Phys. Chem. A, 2022, 126, 2899–2908 CrossRef CAS PubMed.
- W. M. I. Hassan, M. P. Anantram, R. Nekovei, M. M. Khader and A. Verma, Tailoring optical absorption in silicon nanostructures from UV to visible light: a TDDFT study, Sol. Energy, 2016, 126, 44–52 CrossRef CAS.
- R. Derian, K. Tokár, B. Somogyi, Á. Gali and I. Štich, Optical Gaps in Pristine and Heavily Doped Silicon Nanocrystals: DFT versus Quantum Monte Carlo Benchmarks, J. Chem. Theory Comput., 2017, 13, 6061–6067 CrossRef CAS PubMed.
- S. Botti, H.-C. Weissker and M. A. L. Marques, Ab initio electronic gaps of Ge nanodots: the role of self-energy effects, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 155440 CrossRef.
- A. J. Williamson, J. C. Grossman, R. Q. Hood, A. Puzder and G. Galli, Quantum Monte Carlo Calculations of Nanostructure Optical Gaps: Application to Silicon Quantum Dots, Phys. Rev. Lett., 2002, 89, 196803 CrossRef PubMed.
- E. Luppi, F. Iori, R. Magri, O. Pulci, S. Ossicini, E. Degoli and V. Olevano, Excitons in silicon nanocrystallites: the nature of luminescence, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 033303 CrossRef.
- Y. Cho, S. J. Bintrim and T. C. Berkelbach, Simplified GW/BSE Approach for Charged and Neutral Excitation Energies of Large Molecules and Nanomaterials, J. Chem. Theory Comput., 2022, 18, 3438–3446 CrossRef CAS PubMed.
- N. L. Matsko, Study of volume and surface plasmons in small silicon–hydrogen nanoclusters using the GW method, Phys. Chem. Chem. Phys., 2018, 20, 24933–24939 RSC.
- M. G. Zauchner, A. Horsfield and J. Lischner, Accelerating GW calculations through machine-learned dielectric matrices, npj Comput. Mater., 2023, 9, 184 CrossRef.
- B. Aradi, Nanocut, https://github.com/aradi/nanocut.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, Commentary: The Materials Project: a materials genome approach to accelerating materials innovation, APL Mater., 2013, 1, 011002 CrossRef.
- A. D. Becke, Density–functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- M. J. van Setten, F. Weigend and F. Evers, The GW-Method for Quantum Chemistry Applications: Theory and Implementation, J. Chem. Theory Comput., 2013, 9, 232–246 CrossRef CAS PubMed.
- X. Gui, C. Holzer and W. Klopper, Accuracy Assessment of GW Starting Points for Calculating Molecular Excitation Energies Using the Bethe-Salpeter Formalism, J. Chem. Theory Comput., 2018, 14, 2127–2136 CrossRef CAS PubMed.
- D. Golze, M. Dvorak and P. Rinke, The GW compendium: a practical guide to theoretical photoemission spectroscopy, Front. Chem., 2019, 7, 377-1–377-66, DOI:10.3389/fchem.2019.00377.
- C. Holzer and W. Klopper, Ionized, electron-attached, and excited states of molecular systems with spin–orbit coupling: two-component GW and Bethe-Salpeter implementations, J. Chem. Phys., 2019, 150, 204116 CrossRef PubMed.
- K. Krause and W. Klopper, Implementation of the Bethe−Salpeter equation in the TURBOMOLE program, J. Comput. Chem., 2017, 38, 383–388 CrossRef CAS PubMed.
- J. C. Taylor, Tamm-Dancoff Method, Phys. Rev., 1954, 95, 1313–1317 CrossRef.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Turbomole, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, TURBOMOLE: modular program suite for ab initio quantum-chemical and condensed-matter simulations, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed.
- Y. J. Franzke, C. Holzer, J. H. Andersen, T. Begušić, F. Bruder, S. Coriani, F. Della Sala, E. Fabiano, D. A. Fedotov, S. Fürst, S. Gillhuber, R. Grotjahn, M. Kaupp, M. Kehry, M. Krstić, F. Mack, S. Majumdar, B. D. Nguyen, S. M. Parker, F. Pauly, A. Pausch, E. Perlt, G. S. Phun, A. Rajabi, D. Rappoport, B. Samal, T. Schrader, M. Sharma, E. Tapavicza, R. S. Treß, V. Voora, A. Wodyński, J. M. Yu, B. Zerulla, F. Furche, C. Hättig, M. Sierka, D. P. Tew and F. Weigend, TURBOMOLE: Today and Tomorrow, J. Chem. Theory Comput., 2023, 19, 6859–6890 CrossRef CAS PubMed.
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- F. Plasser, TheoDORE: a toolbox for a detailed and automated analysis of electronic excited state computations, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS PubMed.
- J. B. Maglic and R. Lavendomme, MoloVol: an easy-to-use program for analyzing cavities, volumes and surface areas of chemical structures, J. Appl. Crystalogr., 2022, 55, 1033–1044 CrossRef CAS PubMed.
- E. Madden, GitHub, 2023 DOI:10.5281/zenodo.10377882.
- E. J. Baerends, O. V. Gritsenko and R. van Meer, The Kohn–Sham gap, the fundamental gap and the optical gap: the physical meaning of occupied and virtual Kohn–Sham orbital energies, Phys. Chem. Chem. Phys., 2013, 15, 16408 RSC.
- M. V. Wolkin, J. Jorne, P. M. Fauchet, G. Allan and C. Delerue, Electronic States and Luminescence in Porous Silicon Quantum Dots: The Role of Oxygen, Phys. Rev. Lett., 1999, 82, 197–200 CrossRef CAS.
- S. Furukawa and T. Miyasato, Quantum size effects on the optical band gap of microcrystalline Si-H, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 5726–5729 CrossRef CAS PubMed.
- M. C. C. Wobbe, A. Kerridge and M. A. Zwijnenburg, Optical excitation of MgO nanoparticles; a computational perspective, Phys. Chem. Chem. Phys., 2014, 16, 22052–22061 RSC.
- M. A. Zwijnenburg, The effect of particle size on the optical and electronic properties of magnesium oxide nanoparticles, Phys. Chem. Chem. Phys., 2021, 23, 21579–21590 RSC.
- D. Rocca, D. Lu and G. Galli, Ab initio calculations of optical absorption spectra: solution of the Bethe-Salpeter equation within density matrix perturbation theorJ, Chem. Phys., 2010, 133, 164109 Search PubMed.
- K. L. Shaklee and R. E. Nahory, Valley-orbital splitting of free excitons? The absorption edge of Si, Phys. Rev. Lett., 1970, 24, 942–945 CrossRef CAS.
- M. A. Green, Improved value for the silicon free exciton binding energy, AIP Adv., 2013, 3, 112104 CrossRef.
- R. A. Faulkner, Higher Donor Excited States for Prolate-Spheroid Conduction Bands: A Reevaluation of Silicon and Germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1969, 184, 713–721 CrossRef CAS.
- J. A. Van Vechten, Quantum Dielectric Theory of Electronegativity in Covalent Systems. I. Electronic Dielectric Constant, Phys. Rev., 1969, 182, 891–905 CrossRef CAS.
- F. Zheng, J. Tao and A. M. Rappe, Frequency-dependent dielectric function of semiconductors with application to physisorption, Phys. Rev. B, 2017, 95, 03520 Search PubMed.
- L.-W. Wang and A. Zunger, Electronic Structure Pseudopotential Calculations of Large (.apprx.1000 Atoms) Si Quantum Dots, Phys. Rev. Lett., 1994, 73, 1039 CrossRef CAS PubMed.
- C. Delerue, G. Allan and M. Lannoo, Concept of dielectric constant for nanosized systems, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 115411 CrossRef.
- S. A. Bäppler, F. Plasser, M. Wormit and A. Dreuw, Exciton analysis of many-body wave functions: bridging the gap between the quasiparticle and molecular orbital pictures, Phys. Rev. A: At., Mol., Opt. Phys., 2014, 90, 052521 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: DFT optimised structures of all relevant particles, tables of G0W0(-BSE), evGW(-BSE) and qsGW(-BSE) results and DFT predicted photoluminescence energies and Stokes shifts, and plots of the fundamental gap versus particle size, optical gap versus particle size using the lowest bright excited state for Si10H16 and of the natural transition orbitals obtained with TDDFT and additional Fourier transforms can be found in the supporting information. See DOI: https://doi.org/10.1039/d4cp00119b |
| This journal is © the Owner Societies 2024 |