
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical properties of superatomic Li3O clusters from a density functional theory perspective: formation of chloride and adsorption behavior on graphynes†
Xiao
Wang
a,
Meng
Zhang
 *a and
Wei
Cao
*b
*a and
Wei
Cao
*b
aSchool of Physics, East China University of Science and Technology, Shanghai 200237, China. E-mail: mzhang@ecust.edu.cn
bNano and Molecular Systems Research Unit, University of Oulu, FIN-90014, Finland. E-mail: wei.cao@oulu.fi
First published on 19th March 2024
Abstract
Superatomic clusters have received a lot of attention due to their ability to mimic the electronic configurations of individual atoms. Despite numerous studies of these clusters, their ability to mimic the chemical properties of individual atoms is still unclear. This also applies for Li3O/Li3O+ clusters which simulate the Na atom and its ion, but their capabilities to form a salt or be adsorbed on surfaces remain unexplored. In this work, a density functional theory investigation was performed to study the chemical formation and adsorption behavior of the superatomic Li3O cluster. The results show that Li3O mimics the chemical properties of the sodium element to form Li3O chloride and be adsorbed on graphdiyne and γ-graphyne with similar binding energy as the sodium adsorbate cases. Beyond the isolated cluster individuals, superatoms are demonstrated as elements from the 3D periodic table to construct compounds and attach onto solid surfaces.
1. Introduction
One of the breakthroughs in cluster research was the discovery of superatoms. With appropriate size and composition, these specific clusters have similar electronic structures to those of atoms in the periodic table. If superatoms are placed into the same positions as their master atoms, a three-dimensional (3D) periodic table can be constructed1 where superatoms are piled up above their masters. Detected in gaseous or free form, the superatoms are labeled by more conspicuous peaks compared to those from neighbouring isomers. The experimental observations imply more abundance and better stability of the superatomic clusters compared with their peers.2,3 As a result, they are considered as building blocks for new synthetic materials. Additionally, in molecules composed of superatomic clusters, the (sub)cluster structures may be kept and enrich potential functionalities originating from component clusters. Hence, molecules formed by superatoms are considered to own superior functionalities over molecules directly formed by atoms.4–8Over the past twenty years, many theoretical and experimental studies have been conducted to create superatoms and explore their structures and properties. So far, a variety of superatoms, including superalkalis,9–11 superalkaline-earth-metal atoms,12 superhalogens,13–15 multivalent superatoms,16,17 and magnetic superatoms18–20 have been designed and identified using different electron-counting rules such as the jellium rule, octet rule and Wade–Mingos rule etc. These research studies further interpretate the concept of a superatom and provide a variety of structural building blocks for cluster-assembled materials (CAMs). For example, C60 can form an FCC solid through van der Waals force at room temperature and pressure.21–23 An alkali-like superatom TaSi16 can combine with C60, resulting in a superatomic complex with high thermal and chemical robustness.24,25 Other CAMs, such as ZnO-based CAMs, Al-based CAMs and zintl cluster-based CAMs, have also been synthesized and investigated.26–31 In fact, clusters can be assembled on substrates as well as in confined spaces. A proper substrate is the key to host clusters and preserves their superatomic identities. For instance, weak interactions between the FeCa8 superatom and the h-BN or graphene can maintain the structure and characteristics of the superatomic cluster. On the contrary, the strong interaction between the FeCa8 and a calcium substrate leads to a destruction of the magnetic properties of the cluster.32 Therefore, it is important to understand the interactions between superatomic clusters and their ligands or hosting matrices, and the interactions’ impacts on the structures and properties of bonded or adsorbed systems.
Among the studied superatoms, the structure and properties of superalkali atom Li3O in its neutral or charged form have been systematically investigated.33–36 In our previous work,37 the predicted Li3O+ has been identified in the gas form. Its formation process has been proposed and the possibility of application as a building unit for energy storage has been uncovered. It is further noticed that Li-rich antiperovskite (LiRAP) conductors Li3OA (A = halogen or superhalogens) with lithium superionic conductivity have been created and considered to have great promise for solid-state electrolytes.38–40 Recent first-principles studies of Li3O functionalized graphyne, graphdiyne and h-BN sheets indicated that superalkali clusters can inhibit metal agglomeration. Thanks to facilitated charge transfers, the cluster decorated sheets are found to be suitable for hydrogen storage.41,42 Despite the above advances, the superatomic cluster's bonding schemes with elements and interactions with matrices are far less understood compared to knowledge of the structural and electronic configurations of individual clusters. The chemistry of superatomic clusters (including the Li3O) remains elusive.
In this work, we carried out a density functional theory study to investigate the physical and chemical properties exhibited by Li3O clusters when interacting with ligands or substrates. At the free form level, the Li3O cluster can mimic a single Na atom in electronic configuration. It also mimics the bonding or interaction schemes of the Na when bonding with a halogen atom or being adsorbed on two-dimensional graphynes. A chemical bond is formed between the superatomic Li3O cluster and chlorine, and the adsorption energies of Li3O onto the graphynes are almost the same as the case using Na adsorbate. From the above results, it is hoped that this work will provide a theoretical reference for further application of these clusters in compound formations and surface interactions.
2. Computational methods
All computations in this work were performed through density functional theory (DFT) calculations implemented in the Cambridge Serial Total Energy Package (CASTEP) package.43 They are detailed in the geometric optimization and electronic structure of the pristine Li3O superatom, the cluster's interaction with halogen elements, and adsorption on the graphdiyne (GDY) and γ-graphyne (γ-GY). The exchange–correlation was treated with the generalized gradient approximation (GGA) in the form of the Perdew–Burke–Ernzerhof (PBE) functional together with the employment of OTFG ultra soft pseudopotentials.44 Koelling–Harmon was applied as a relativistic effect treatment and spin polarization was chosen during the geometric optimization. A value of 10−6 eV per atom was set for the total energy SCF convergence threshold, and 450 eV as the cut-off energy to expand the plane waves included in the basis set. The quality of the Monkhorst–Pack k-points was specified on 3 × 3 × 1 for geometry optimization and 6 × 6 × 1 k-point grid for the calculation of density of states. Our calculations were carried out using 3 × 3 graphynes supercells with periodic boundary conditions in the basal plane to simulate infinite planar sheets. A vacuum region of 20 Å was applied in the z-direction to ensure negligible interaction between adjacent layers.The binding/adsorption energies of the Na atom or Li3O cluster interacted with other Cl/F or GDY/γ-GY systems were computed as follows.
| E = Etotal − [E(X) + E(Y)] | (1) |
3. Results and discussion
3.1 Characteristics of the Li3O cluster
Following geometry optimization, the most stable Li3O cluster owns a highly symmetric D3h triangle structure with the oxygen atom in the center as displayed in Fig. 1(a). The bond length between the lithium and oxygen atoms is 1.702 Å, in line with the reported value from Yokoyama et al.45 An Li3O cluster has nine valence electrons coming from Li and O sites: each Li atom contributes one valence electron, and the O atom contributes six valence electrons. | ||
| Fig. 1 The lowest energy structures of (a) the superatomic Li3O cluster, (b) (Li3O)Cl, and (c) (Li3O)F halides. Lithium, oxygen, chlorine and fluorine atoms are represented in violet, red, green and blue, respectively. | ||
The molecular orbital (MO) characteristics of the superatom Li3O are illustrated in Fig. 2 and the atomic orbitals (AOs) of the sodium atom are also depicted for comparison. It is shown that the electronic structure of Li3O can be described as 1S21P62S1, which bears resemblance to that of an individual sodium atom. Obviously, it is apt to lose one electron to achieve a closed shell electron configuration of 1S21P6, thus mimicking the chemical behavior of the alkali atoms. We further studied the electronic structure of the cluster in detail. The lowest valence MO of Li3O exhibits 1S character, followed by two degenerate 1Pz orbitals. The degenerate 1Px and 1Py orbitals are pushed 0.37 eV higher in energy owing to the planar structure of the Li3O cluster. This is slightly different from the sodium atom case where three degenerate p orbitals exist. These occupied MOs of Li3O are mainly composed of the AOs of the central atom O. Then, the following highest occupied molecular orbital (HOMO) of the cluster has a half-filled 2S superorbital character. Three 2P orbitals are the next higher energy orbitals. Similar to the case in 1P orbitals, 2P orbitals degenerate into two groups of one (Pz) and two (Px and Py) orbitals, as induced by the two-dimensional planar structure of the cluster. It should be noted that in Fig. 2, the electron cloud pervades the entire cluster. A shape difference stays between the cluster's 2S MO and the Na's 3s AO. The former appears flattened, while the latter is spherical. This discrepancy arises from the planar geometry of the Li3O. Therein, the delocalized electron distribution from each atom is confined around the 2D plane. An energy gap (Egap) of 0.631 eV was found between the highest occupied molecular orbital and the lowest unoccupied molecular orbital (HOMO–LUMO). Although the Egap value of the Li3O cluster is smaller than that of the Na atom (1.633 eV), its chemical stability is approximate to that of the Na atom, which will be discussed in the following section.
 | ||
| Fig. 2 Relative energy levels and orbitals of the Li3O cluster (left) and Na atom (right). EAO represents the empty atomic orbital. | ||
3.2 Chemistry of Li3O
In order to examine the similarity between the Li3O cluster and the Na atom, the chemistry of the Li3O cluster is discussed by considering the interaction of the Li3O cluster with halogen atoms and adsorption on graphynes.After careful optimization, the triangle Li3O cluster interacts with a Cl atom through two Li atoms through an edge connection. The binding energy (BE) of Li3O to Cl is −5.718 eV. The bond length (distance between Li and Cl atom) is 2.268 Å, very close to the value of 2.373 Å in the NaCl. Table 1 shows that the natural charge on the Cl is - 0.613 |e|, indicating that almost one electron transfers from the Li3O cluster to the Cl atom to form an ionic bond. Each Li atom of Li3O contributes an average charge of 0.494 |e|, sharing with O and Li atoms. The electrostatic interaction between Li3O+ and Cl− causes a slight variation in the average bond length of Li–O, which is elongated by 0.04 Å compared to the bare Li3O. All these observations indicate that the compound (Li3O)Cl is actually a ‘‘binary salt’’ consisting of Li3O+ and Cl−. The same ionic interaction is also observed in (Li3O)F which has a similar structure of (Li3O)Cl as shown in Fig. 1(c). The BE is −6.275 eV and the distance of the Li and F atom is 1.78 Å. The Li3O subunit carries 0.682 |e| net charge in (Li3O)F. Each Li atom transfers 0.516 |e| to O and F atoms. These characteristic data are also very close to those of NaF molecules. Given the above, it can be seen that Li3O is capable of combining with Cl or F atoms to form stable ionic compounds analogous to the sodium salts.
| System | Binding energies (eV) | Bond length | Charge (|e|) | |
|---|---|---|---|---|
| Li3O/Na | Cl/F | |||
| (Li3O)Cl | −5.718 | 2.268 | 0.613 | −0.613 |
| NaCl | −4.319 | 2.373 | 0.713 | −0.713 |
| (Li3O)F | −6.275 | 1.78 | 0.682 | −0.682 |
| NaF | −5.251 | 1.94 | 0.74 | −0.74 |
We also constructed the 3Li–O–Cl clusters with all atoms randomly distributed and without a predetermined geometric structure for Li3O. Extensive two dimensional (2D) and three-dimensional (3D) structures were constructed to determine the lowest-energy geometries for the 3Li–O–Cl clusters. The details of the metastable structures are depicted in Fig. S1 in the ESI.† During the geometric optimization process, the O atom and the three Li atoms spontaneously tend to assemble into a superatomic cluster structure before interacting with the Cl atom. In other words, the introduction of the highly reactive chlorine with strong chemical properties does not impact the formation of the Li3O superatomic cluster in its free state. Hence, the superatom Li3O is highly stable and can be assembled easily, consistent with our previous experimental findings based on time-of-flight mass spectrometry.37
Next, the molecular orbitals of (Li3O)Cl and the corresponding NaCl are investigated and shown in Fig. 3. Since the main intention here is to provide a comparison of the orbital shapes between two kinds of compounds, the orbitals in Fig. 3 are arranged in the order of energy without strictly adhering to their exact energy positions. In NaCl, the Na 3s orbital combines with Cl 3pz to form molecular orbitals labeled as 4σ and 4σ* in Fig. 3 (left). They are named as HOMO−2 and LUMO−1. The 4σ orbital is weakly bonded and mostly Cl-like in character. The antibond 4σ* orbital is centered almost entirely on the Na atom. HOMO−1 comes from Cl 3px and 3py which could be labeled as 2π orbitals and displayed as HOMO−1. The electronic configuration of (Li3O)Cl is analogous to that of NaCl. It is obvious that the superatomic S and P orbitals behavior is similar to the s and p AOs of the Na atom. The σ, σ* and π orbitals of (Li3O)Cl are also analogous to the σ, σ* and π orbitals of NaCl, respectively. Two molecular HOMOs are similar to σ* orbitals indicating that the two molecules have some chemical similarities. This finding is in line with previous studies of the halogen-like superatom Al13 and chlorine atom, superatom B6 and Ni atom. Although there are some variations in orbital order that occur in the case of bonding with the Cl atom, the similarity between the Li3O and Na is still exhibited.
 | ||
| Fig. 3 Molecular orbitals of the NaCl (left) and (Li3O)Cl (right). | ||
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CC–), while the linkage between two neighboring benzene rings is the –CC– carbon link in γ-GY.
C–CC–), while the linkage between two neighboring benzene rings is the –CC– carbon link in γ-GY.
 | ||
| Fig. 4 The optimized structures of (a) GDY and (b) γ-GY. | ||
The geometrical structures of Li3O cluster adsorption on GDY and γ-GY are systematically studied. First, all possible adsorption sites are considered, including the hollow sites above the center of the carbon rings, the top site right above a carbon atom and the bridge site above the carbon bonds. After careful structural optimization, the most stable configurations of the Li3O decorated GDY and γ-GY are obtained and displayed in Fig. 5. In the complex of Li3O@GDY, the Li3O unit prefers to stay above the center of the triangle pore formed by the –CC–CC– linkages. An adsorption energy of −3.70 eV is obtained and listed in Table 2, which is a little lower than the previous study.42 The shape of the Li3O cluster changes from a triangle to an umbrella where the plane formed by three Li atoms is parallel to the plane of GDY. The Li–O bond length increases to 1.71 Å after absorption. Meanwhile, the bond angles of Li–O–Li reduced to 113° from 120° in the pristine cluster. Li3O also prefers to bind on the hollow site of the C12-ring due to it having the largest binding energy of −3.56 eV in the Li3O@γ-GY case. The bond length of Li–O is 1.72 Å and the Li–O–Li angle changes to 107°. Additionally, the binding energy between Li3O and GDY is larger than that between Li3O and γ-GY. This is because each Li atom of the Li3O unit binds with four sp hybridized carbon atoms in the Li3O@GDY configuration. Due to the chemical activity of the triple carbon bonds of the GDY, the top site of the acetylenic ring is often the most stable adsorption site for single metal atom adsorption. However, according to the previous works,50–53 the atoms would automatically migrate to the corner when the transition metal (TM) or noble metal atoms were located at the center of the acetylenic ring. While alkali metals such as Li, Na and K would be anchored at the triangle center of GDY.54 These are in line with the adsorption of Na on the GDY and γ-GY, as depicted in Fig. 5(c) and (d). In addition, from Table 2, the adsorption energy of the Na atom on GDY or γ-GY is almost the same as the corresponding one of Li3O on GDY or γ-GY.
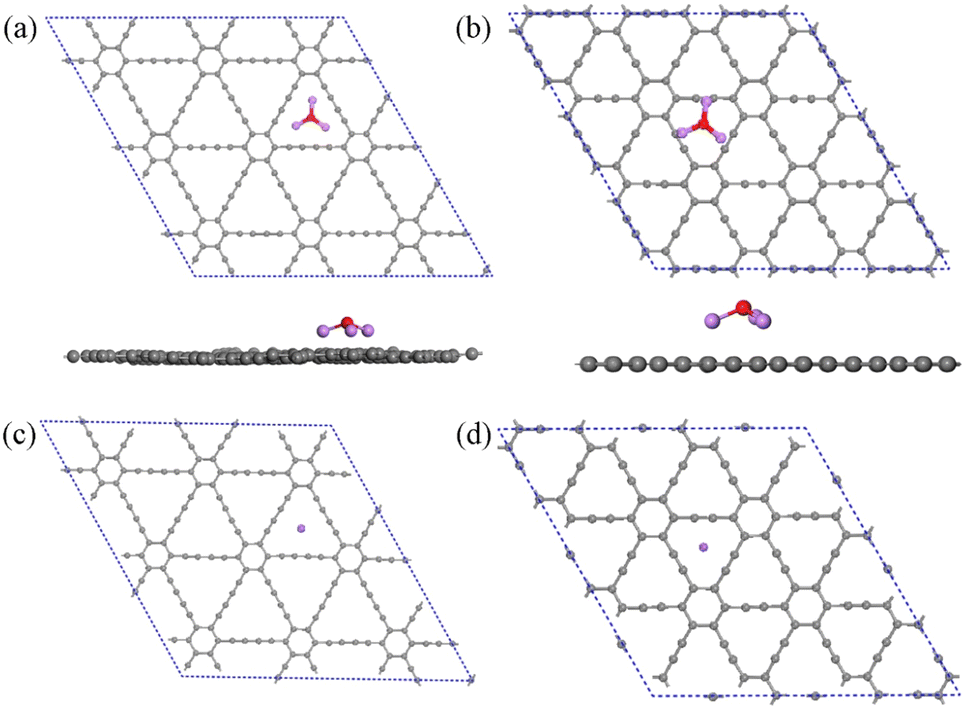 | ||
| Fig. 5 The optimized structures of Li3O adsorption on (a) GDY and (b) γ-GY. The optimized structures of Na atom adsorption on (c) GDY and (d) γ-GY. | ||
| System | Adsorption energies (eV) | Charge (|e|) | |||
|---|---|---|---|---|---|
| Li atoms | O atom | Li3O/Na | Carbon sheet | ||
| Li3O@GDY | −3.70 | 1.35 | −0.84 | 0.51 | −0.51 |
| Na@GDY | −3.71 | 0.84 | −0.84 | ||
| Li3O@GY | −3.56 | 1.50 | −0.87 | 0.63 | −0.63 |
| Na@GY | −3.18 | 0.80 | −0.80 | ||
To better understand and compare the binding mechanism of these complexes, we studied the electronic structures of Na@GDY and Li3O@GDY as typical cases. The Na atom donates a single valence electron in the 3s orbital to the LUMO of GDY. Consequentially, a singly occupied molecular orbital (SOMO) is formed in Na@GDY. It is composed of p orbitals perpendicular to the GDY plane as shown in Fig. 6. Li3O@GDY has a similar intramolecular charge transfer mechanism. Due to the low ionization potential, the Li3O cluster donates its electrons to the acetylenic linkages of the GDY sheets and these electrons occupy the empty pz orbitals of the carbon frame. It is worth noting that Fig. 6 shows inconsistent electron clouds around the adsorbed Na on Na@GDY and Li3O on Li3O@GDY. While the Na atom does not exhibit obvious electron density in the center of the triangle, the adsorbed superatom behaves differently. The electron clouds are centered next to the oxygen and keep a similar P-type distribution as in the free cluster (Fig. 2) or in ionic bonds (Fig. 3). However, almost no electron cloud could be detected on the Li, denoting the charge transfer from the Li to neighboring atoms. A zoomed-in image of the adsorbed structure in Fig. 6(c) further denotes slight increase of the electron densities on the C atoms next to the Li atoms. This proves that the charge transfer happens from Li3O to the GDY after absorption. In other words, the inner shell electronic structure is kept within the cluster to keep the superatomic feature, but the valence electron from 2S is involved in charge transfer in the adsorption.
 | ||
| Fig. 6 SOMO orbital diagram of the (a) Na@GDY, (b) Li3O@GDY and (c) zoomed-in region next to Li3O on (b). | ||
Furthermore, the bandgap energy of the adsorbed system is investigated. The pristine GDY is a semiconductor with a bandgap of 0.483 eV. However, both Na@GDY and Li3O@GDY exhibit metal behavior after decorations of the Na or Li3O. The bandgap energy decreases due to the redistribution of electrons following intramolecular charge transfers. Similar phenomena have also been reported in TM adsorbed GDY/GY due to electron transfers between the TM adatoms and the GDY/GY.50,51
The partial density of states (PDOS) of the Na@GDY and Li3O@GDY are further investigated and illustrated in Fig. 7. It can be observed that the PDOS shapes of both systems are quite similar. The s and p orbitals of Na atoms, as well as s orbitals of Li in the Li3O clusters, exhibit pronounced hybridization with C-2p orbitals of the GDY system around 2–6 eV. This suggests that both Na atoms and Li3O clusters can interact with GDY stably. The Li3O cluster adsorbed on GDY is comparable to that of a single Na atom adsorbed on GDY. However, in Li3O@GDY, besides the significant hybridization between the C-2p orbitals of GDY and the s orbitals of Li in the 2.5–6 eV range, the PDOS of Li below the Fermi level overlaps with O-2p. The overlaps indicate strong bonding between Li and O and denote high stability of the Li3O superatom after absorption. This is different from the case in Na@GDY where no overlap is observed at a similar energy range.
 | ||
| Fig. 7 The PDOS of the (a) Na@GDY and (b) Li3O@GDY. The Fermi level is denoted by a vertical dashed line. | ||
4. Conclusion
To summarize, the superatom identity of the Li3O cluster has been systemically studied using DFT in this work. A Li3O cluster can easily lose one electron to achieve a closed shell electron configuration of 1S21P6. It bears a great resemblance to the Na atom in the electronic configurations and chemical properties to form chloride and adsorption on graphynes. Li3O is able to form ionic bonds with chlorine or fluorine element and the molecular orbitals of (Li3O)Cl are analogous to those in the sodium salts. We also compared adsorptions of the Li3O cluster and Na atom on graphdiyne and γ-graphyne. Similar to the Na atom, the Li3O cluster preferentially interacts with the large triangular hole in extensively delocalized π-conjugated GDY and γ-GY. Interestingly, the adsorption energies are found to be the same for the Li3O@GDY and Na@GDY, and very close in the Li3O@GY and Na@GY cases. Meanwhile, the bandgaps of GDY and GY after adsorbing either superatom Li3O or Na atom reduce to zero due to the intramolecular charge transfer. The effects of doping Li3O into the surface of GDY on the electronic structures were further investigated. Electrons transfer from the Li3O cluster to the GDY sheets and occupy its empty pz orbitals, similar to an electron from Na to the GDY. In conclusion, Li3O can combine with halogen elements and interact with graphynes as a superatom counterpart of the sodium element.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 101002219), East China University of Science and Technology, China, and Strategic Research Council within the Research Council of Finland (decision 358422) JustH2Transit.References
- P. Jena, Beyond the periodic table of elements: the role of superatoms, J. Phys. Chem. Lett., 2013, 4, 1432–1442 CrossRef CAS PubMed.
- P. Jena and Q. Sun, Super atomic clusters: Design rules and potential for building blocks of materials, Chem. Rev., 2018, 118, 5755–5870 CrossRef CAS PubMed.
- E. A. Doud, A. Voevodin, T. J. Hochuli, A. M. Champsaur, C. Nuckolls and X. Roy, Superatoms in materials science, Nat. Rev. Mater., 2020, 5, 371–387 CrossRef.
- S. Khanna and P. Jena, Assembling crystals from clusters, Phys. Rev. Lett., 1992, 69, 1664–1667 CrossRef CAS PubMed.
- S. Khanna and P. Jena, Atomic clusters: Building blocks for a class of solids, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13705–13716 CrossRef CAS PubMed.
- S. Giri, S. Behera and P. Jena, Superalkalis and superhalogens as building blocks of supersalts, J. Phys. Chem. A, 2014, 118, 638–645 CrossRef CAS PubMed.
- M. Qian, A. C. Reber, A. Ugrinov, N. K. Chaki, S. Mandal, H. M. Saavedra, S. N. Khanna, A. Sen and P. S. Weiss, Cluster assembled materials: toward nanomaterials with precise control over properties, ACS Nano, 2010, 4, 235–240 CrossRef CAS PubMed.
- A. C. Reber, S. N. Khanna and A. W. Castleman, Superatom compounds, clusters, and assemblies: Ultra alkali motifs and architectures, J. Am. Chem. Soc., 2007, 129, 10189–10194 CrossRef CAS PubMed.
- G. L. Gutsev and A. Boldyrev, DVM Xα calculations on the electronic structure of “superalkali” cations, Chem. Phys. Lett., 1982, 92, 262–266 CrossRef CAS.
- E. Rehm, A. I. Boldyrev and P. R. Schleyer, Ab initio study of superalkalis. First ionization potentials and thermodynamic stability, Inorg. Chem., 1992, 31, 4834–4842 CrossRef CAS.
- J. Tong, Y. Li, D. Wu, Z. R. Li and X. R. Huang, Low ionization potentials of binuclear superalkali B2Li11, J. Chem. Phys., 2009, 131, 164307 CrossRef PubMed.
- D. Bergeron, P. Roach, A. Castleman, N. Jones and S. Khanna, Al cluster superatoms as halogens in polyhalides and as alkaline earths in iodide salts, Science, 2005, 307, 231–235 CrossRef CAS PubMed.
- L. P. Ding, P. Shao, C. Lu, F. H. Zhang and L. Y. Wang, Ironbased magnetic superhalogens with pseudohalogens as ligands: An unbiased structure search, Sci. Rep., 2017, 7, 45149 CrossRef PubMed.
- G. Gutsev and A. Boldyrev, DVM-Xα calculations on the ionization potentials of MXk+1− complex anions and the electron affinities of MXk+1 “superhalogens”, Chem. Phys., 1981, 56, 277–283 CrossRef CAS.
- X. Zhao, W. Liu, J. Wang, C. Li and G. Yuan, Theoretical study of ‘Mixed’ ligands superhalogens: Cl-M-NO3 (M= Li, Na, K), Chem. Phys. Lett., 2016, 658, 197–202 CrossRef CAS.
- J. U. Reveles, S. Khanna, P. Roach and A. Castleman, Multiple valence superatoms, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18405–18410 CrossRef CAS PubMed.
- L. J. Li, F. X. Pan, F. Y. Li, Z. F. Chen and Z. M. Sun, Synthesis, characterization and electronic properties of an endohedral plumbaspherene [Au@Pb12]3−, Inorg. Chem. Front., 2017, 4, 1393–1396 RSC.
- K. Pradhan, J. U. Reveles, P. Sen and S. Khanna, Enhanced magnetic moments of alkali metal coated Sc clusters: New magnetic superatoms, J. Chem. Phys., 2010, 132, 124302 CrossRef PubMed.
- V. Chauhan, V. M. Medel, J. U. Reveles, S. N. Khanna and P. Sen, Shell magnetism in transition metal doped calcium superatom, Chem. Phys. Lett., 2012, 528, 39–43 CrossRef CAS.
- X. Zhang, Y. Wang, H. Wang, A. Lim, G. Gantefoer, K. H. Bowen, J. U. Reveles and S. N. Khanna, On the existence of designer magnetic superatoms, J. Am. Chem. Soc., 2013, 135, 4856–4861 CrossRef CAS PubMed.
- A. M. Rao, P. Zhou, K. A. Wang, G. T. Hager, J. M. Holden, Y. Wang, W. T. Lee, X. X. Bi, P. C. Eklund, D. S. Cornett, M. A. Duncan and I. J. Amster, Photoinduced polymerization of solid C60 films, Science, 1993, 259, 955–957 CrossRef CAS.
- S. Jalali-Asadabadi, E. Ghasemikhah, T. Ouahrani, B. Nourozi, M. Bayat-Bayatani, S. Javanbakht, H. A. R. Aliabad, I. Ahmad, J. Nematollahi and M. Yazdani-Kachoei, Electronic structure of crystalline buckyballs: fcc-C60, J. Electron. Mater., 2016, 45, 339–348 CrossRef CAS.
- E. L. Shirley and S. G. Louie, Electron excitations in solid C60: Energy gap, band dispersions, and effects of orientational disorder, Phys. Rev. Lett., 1993, 71, 133–136 CrossRef CAS PubMed.
- M. Nakaya, T. Iwasa, H. Tsunoyama, T. Eguchi and A. Nakajima, Heterodimerization via the covalent bonding of Ta@Si16 nanoclusters and C60 molecules, J. Phys. Chem. C, 2015, 119, 10962–10968 CrossRef CAS.
- T. Ohta, M. Shibuta, H. Tsunoyama, T. Eguchi and A. Nakajima, Charge transfer complexation of Ta-encapsulating Ta@Si16 superatom with C60, J. Phys. Chem. C, 2016, 120, 15265–15271 CrossRef CAS.
- A. Dmytruk, I. Dmitruk, I. Blonskyy, R. Belosludov, Y. Kawazoe and A. Kasuya, ZnO clusters: Laser ablation production and time-of-flight mass spectroscopic study, Microelectron. J., 2009, 40, 218–220 CrossRef CAS.
- C. Wang, S. Xu, L. Ye, W. Lei and Y. Cui, Theoretical investigation of ZnO and its doping clusters, J. Mol. Model., 2011, 17, 1075–1080 CrossRef CAS PubMed.
- Z. Z. Zhu and J. T. Zhao, Electronic structures and geometry of the Al12Si cluster solid, Acta Phys. Sin., 1999, 8, 356–360 CAS.
- X. G. Gong, Structure and stability of cluster-assembled solid Al12C(Si): A first-principles study, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1091–1094 CrossRef CAS.
- M. Qian, A. C. Reber, A. Ugrinov, N. K. Chaki, S. Mandal, H. M. Saavedra, S. N. Khanna, A. Sen and P. S. Weiss, Cluster assembled materials: Toward nanomaterials with precise control over properties, ACS Nano, 2010, 4, 235–240 CrossRef CAS PubMed.
- S. Mandal, A. C. Reber, M. Qian, P. S. Weiss, S. N. Khanna and A. Sen, Controlling the band gap energy of cluster-assembled materials, Acc. Chem. Res., 2013, 46, 2385–2395 CrossRef CAS PubMed.
- A. Singh and P. Sen, Finding the right substrate support for magnetic superatom assembly from density functional calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 035438 CrossRef.
- S. Chen, H. L. Xu, L. Zhao and Z. M. Su, Superalkali atoms bonding to the phenalenyl radical: structures, intermolecular interaction and nonlinear optical properties, J. Mol. Model., 2015, 21, 209 CrossRef PubMed.
- S. Chen, H. L. Xu, L. Zhao and Z. M. Su, Superatoms (Li3O and BeF3) induce phenalenyl radical π-dimer: fascinating interlayer charge-transfer and large NLO responses, Dalton Trans., 2014, 43, 12657–12662 RSC.
- S. Zein and J. V. Ortiz, Interpretation of the photoelectron spectra of superalkali species: Li3O and Li3O−, J. Chem. Phys., 2011, 135, 164307 CrossRef CAS PubMed.
- M. Gutowski and J. Simons, Anionic and neutral states of Li3O, J. Phys. Chem., 1994, 98, 8326–8330 CrossRef CAS.
- H. Pauna, X. Y. Shi, M. Huttula, E. Kokkonen, T. H. Li, Y. H. Luo, J. Lappalainen, M. Zhang and W. Cao, Evolution of lithium clusters to superatomic Li3O, Appl. Phys. Lett., 2017, 111, 103901 CrossRef.
- Y. Zhao and L. L. Daemen, Superionic conductivity in lithium-rich anti-perovskites, J. Am. Chem. Soc., 2012, 134, 15042–15047 CrossRef CAS PubMed.
- Y. Zhang, Y. Zhao and C. Chen, Ab initio study of the stabilities of and mechanism of superionic transport in lithium-rich antiperovskites, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 134303 CrossRef.
- H. Fang, S. Wang, J. Liu, Q. Sun and P. Jena, Superhalogen-based lithium superionic conductors, J. Mater. Chem. A, 2017, 5, 13373–13381 RSC.
- A. You, Y. Liu, J. Xiao, X. Yue, H. Z. Huang and J. G. Song, Computational exploration of graphyne and graphdiyne decorated with OLi3 as potential hydrogen storage candidates, Int. J. Hydrogen Energy, 2023, 48, 26314–26327 CrossRef CAS.
- N. N. Zhang, Y. T. Shi, J. W. Li, Y. J. Zhang, J. H. Guo, Z. G. Fu and P. Zhang, Computational exploration of graphyne and graphdiyne decorated with OLi3 as potential hydrogen storage candidates, Appl. Phys. Lett., 2023, 123, 083902 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. Probert, K. Refson and M. C. Payne, First principles methods using CASTEP, Z. Kristallogr. - Cryst. Mater., 2005, 220, 567–570 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- K. Yokoyam, H. Tanaka and H. Kudo, Structure of hyperlithiated Li3O and evidence for electronomers, J. Phys. Chem. A, 2001, 105, 4312–4315 CrossRef.
- X. Wang, Y. H. Luo, T. Yan, W. Cao and M. Zhang, Strain enhanced lithium adsorption and diffusion on silicene, Phys. Chem. Chem. Phys., 2017, 19, 6563–6568 RSC.
- J. J. He, S. Y. Ma, P. Zhou, C. X. Zhang, C. Y. He and L. Z. Sun, Magnetic properties of single transition-metal atom absorbed graphdiyne and graphyne sheet, J. Phys. Chem. C, 2012, 116, 26313–26321 CrossRef CAS.
- D. W. Ma, T. X. Li, Q. G. Wang, G. Yang, C. Z. He, B. Y. Ma and Z. S. Lu, Graphyne as a promising substrate for the noble-metal single-atom catalysts, Carbon, 2015, 95, 756–765 CrossRef CAS.
- J. Kim, S. Kang, J. Lim and W. Y. Kim, Study of Li adsorption on graphdiyne using hybrid DFT calculations, ACS Appl. Mater. Interfaces, 2019, 11, 2677–2683 CrossRef CAS PubMed.
- T. W. He, S. K. Matta, G. Will and A. J. Du, Transition-metal single atoms anchored on graphdiyne as high-efficiency electrocatalysts for water splitting and oxygen reduction, Small Methods, 2019, 1800419 CrossRef.
- X. Chen, P. F. Gao, L. Guo, Y. N. Wen, Y. Zhang and S. L. Zhang, Two-dimensional ferromagnetism and spin filtering in Cr and Mn-doped graphdiyne, J. Phys. Chem. Solids, 2017, 105, 61–65 CrossRef CAS.
- X. Liu, M. Xu, Y. Han and C. G. Meng, Adsorption, diffusion and aggregation of Ir atoms on graphdiyne: a first-principles investigation, Phys. Chem. Chem. Phys., 2020, 22, 25841–25847 RSC.
- G. L. Xu, R. Wang, Y. C. Ding, Z. S. Lu, D. W. Ma and Z. X. Yang, First-principles study on the single Ir atom embedded graphdiyne: an efficient catalyst for CO oxidation, J. Phys. Chem. C, 2018, 122, 23481–23492 Search PubMed.
- X. J. Li and S. K. Li, Investigations of electronic and nonlinear optical properties of single alkali metal adsorbed graphene, graphyne and graphdiyne systems by first-principles calculations, J. Mater. Chem. C, 2019, 7, 1630–1640 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05478k |
| This journal is © the Owner Societies 2024 |