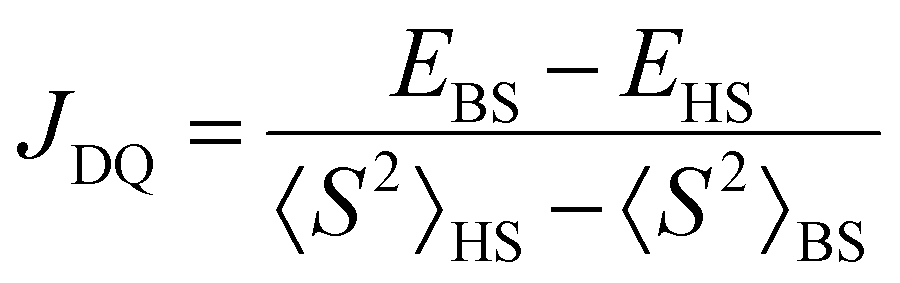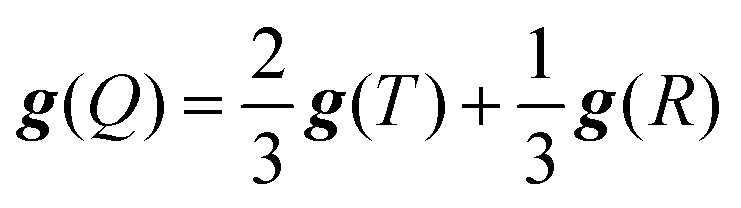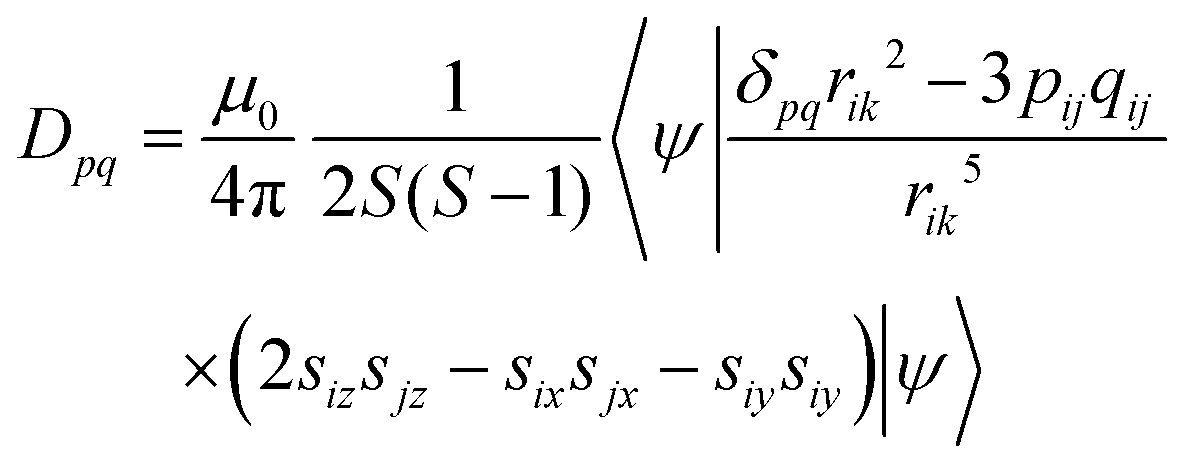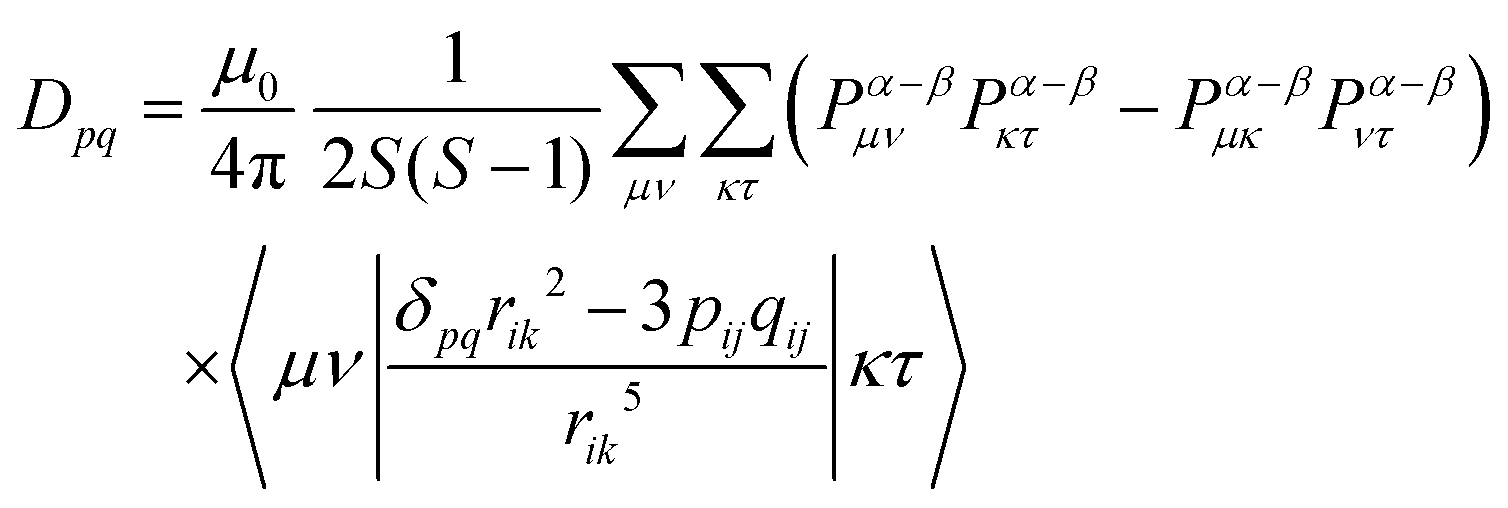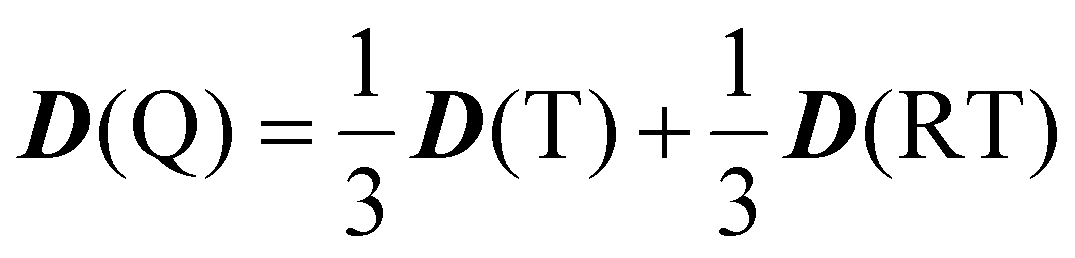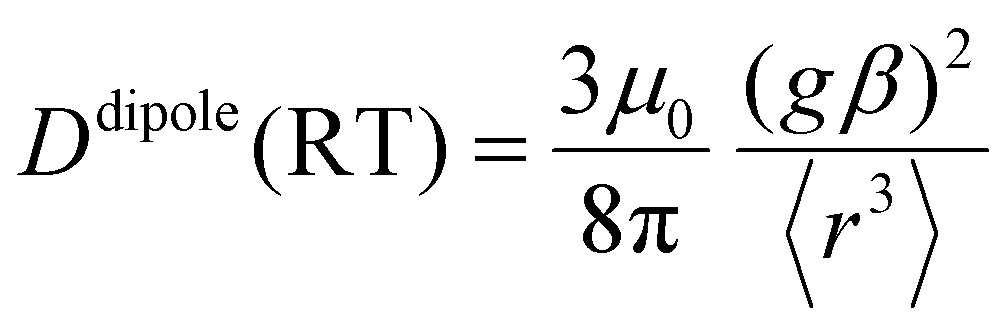Theoretical investigation of multi-spin excited states of anthracene radical-linked π-conjugated spin systems by computational chemistry†
Received
30th December 2023
, Accepted 12th February 2024
First published on 14th February 2024
Abstract
Multi-spin excited states of chromophore radical-linked π-conjugated spin systems are investigated by molecular orbital calculations based on density functional theory (DFT). The investigated systems consist of an anthracene photosensitive unit leading to a triplet-excited-state (S = 1), π-conjugated linker to propagate spin exchange-coupling, and stable organic radical with a doublet-ground-state (S = 1/2). The intramolecular exchange coupling (JDQ), g value, and fine-structure interaction of their excited states depended on the π-conjugation network (π-topology), type of radical, and molecular structure of the π-linker (length and dihedral angle). The exchange interaction was dependent on the π-topology and the type of radical species. A decrease in the dihedral angle between the anthracene moiety and phenyl linker in the photo-excited state led to larger exchange coupling. With an increase in the π-linker length (r), the magnitude of the exchange coupling gradually decreased in the photoexcited states according to JDQ = JEx0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−βr), similar to the ground-state exchange. The g values of the quartet (Q) state depended only on the radical type (independent of the linker). Conversely, the fine-structure interaction of the Q state was independent of the radical type and depended on both the linker length and the dihedral angle.
exp(−βr), similar to the ground-state exchange. The g values of the quartet (Q) state depended only on the radical type (independent of the linker). Conversely, the fine-structure interaction of the Q state was independent of the radical type and depended on both the linker length and the dihedral angle.
Introduction
Multi-spin excited states of photoexcited chromophore-radical linked systems are of current interest in chemistry and materials science.1–3 Strong exchange coupling between the excited-triplet-state (S = 1) of the chromophore and doublet-state (S = 1/2) of the radical substituent achieves a robust spin alignment, acting as an optical switch in the molecular spin state.1 On the other hand, the weak exchange interaction between them leads to electron spin hyperpolarisation by a variety of mechanisms4–7 such as radical–triplet pair mechanisms (RTPMs), electron spin polarization transfer (ESPT) or reverse quartet mechanisms (RQM). These polarization transfer mechanisms involve not only exchange interactions but also magnetic parameters such as fine-structure interactions and g values.8–12 The exchange interaction also causes enhancement/acceleration of the intersystem crossing (EISC) of the chromophore. EISC is suggested by three possible mechanisms:1 electron spin exchange (strictly speaking, exchange integral) between a SOMO on the radical and the HOMO or LUMO on the singlet excited chromophore (this mechanism is usual EISC),13,14 spin–orbit interaction borrowing the radical moiety,15 and ISC via higher-energy state.16,17 Several applications of EISC have been reported, such as preventing the decomposition of light-unstable materials18–20 and upconversion by triplet–triplet annihilation21 and information transfer.22,23 Understanding the dependence of molecular structure (e.g., type of radical substituent, π-topology and linker length) on the exchange interaction and magnetic parameters is important to exploit the photoexcited chromophore-radical linked systems in molecular-based future technology such as molecular spintronics devices or information science applications.
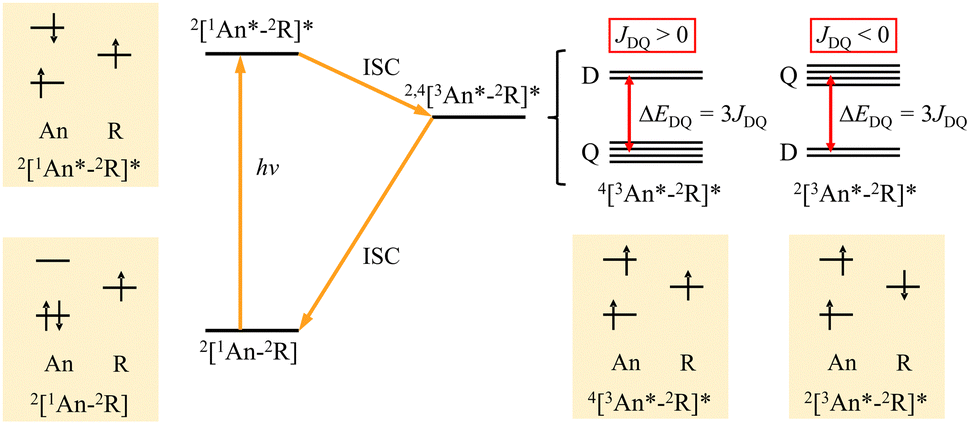
We have reported high-spin photoexcited states of anthracene radical-linked π-conjugated spin systems and demonstrated the control of the spin states utilizing their π-topology.15,24–27 The energy relaxation process with spin conversion in the multi-spin excited states and the schematic picture of electron configuration in each state are shown in Fig. 1. The energy gap between the lowest quartet photo-excited state (Q) and the lowest doublet photo-excited state (D) depends on the magnitude of the exchange coupling constant (JDQ) between the triplet-state (S = 1) of the excited chromophere and the doublet-state (S = 1/2) of the radical substituent, and the order of the energy level changes according to the sign of JDQ. Several studies to clarify the magnitude and sign of JDQ have been reported,12,28,29 but in experiments, the magnitude of JDQ that can be measured is limited. In addition, although the estimation of JDQ between the triplet excited state and radical spin has been carried out using the broken symmetry density functional theory (DFT) or complete active space self-consistent field (CASSCF) methods,30–33 there are still limited comprehensive investigations for the molecular structure of the coupling moiety (π-linker) together with structural evaluation of exchange interactions, fine-structure interaction and g values. In this paper, we report the DFT calculations of electron spin interactions in the photo-excited anthracene–radical π-conjugated systems to understand how the magnetic parameters depend on them.
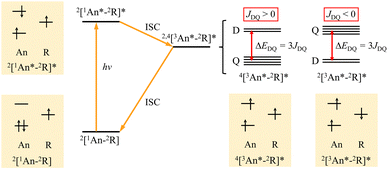 |
| | Fig. 1 Excited-state processes and schematic picture of the electron configuration of anthracene–radical linked system. | |
Methods
All calculations were carried out using an ORCA 5.0.3 program package.34 Molecular structure visualisation and spin density mapping were performed using Avogadro.35 The D state (pseudo-doublet excited state) used for the estimation of JDQ was calculated for the optimized geometry of the Q state by the broken-symmetry DFT. The JDQ between the triplet excited state of the anthracene moiety (3An*) and doublet radical moiety (2R) was expressed using the Heisenberg–Dirac–van Vleck (HDvV) spin Hamiltonian,where ST and SR are the spin operators of the triplet excited state of the anthracene moiety (chromophore) and the doublet state of the radical substituent, respectively. In this notation, a negative J value corresponds to an antiferromagnetic interaction, whereas a positive one is related to a ferromagnetic interaction. Since the D state obtained by the broken-symmetry DFT calculation has spin contamination from higher-energy spin states, the calculated energy is overestimated. For the Hamiltonian (1), JDQ can be related to the energy difference (ΔEDQ) between D and Q as JDQ = 3ΔEDQ when the spin contamination is negligible. In the DFT calculations, an approximate spin projection is used to remove the effect of spin contamination. Thus, JDQ can be estimated by Yamaguchi equation (eqn (2)),36| | 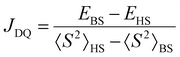 | (2) |
where 〈S2〉HS and 〈S2〉BS are the expected values of S2 for the high spin (HS) and broken symmetry (BS) states obtained in the DFT calculations, respectively.
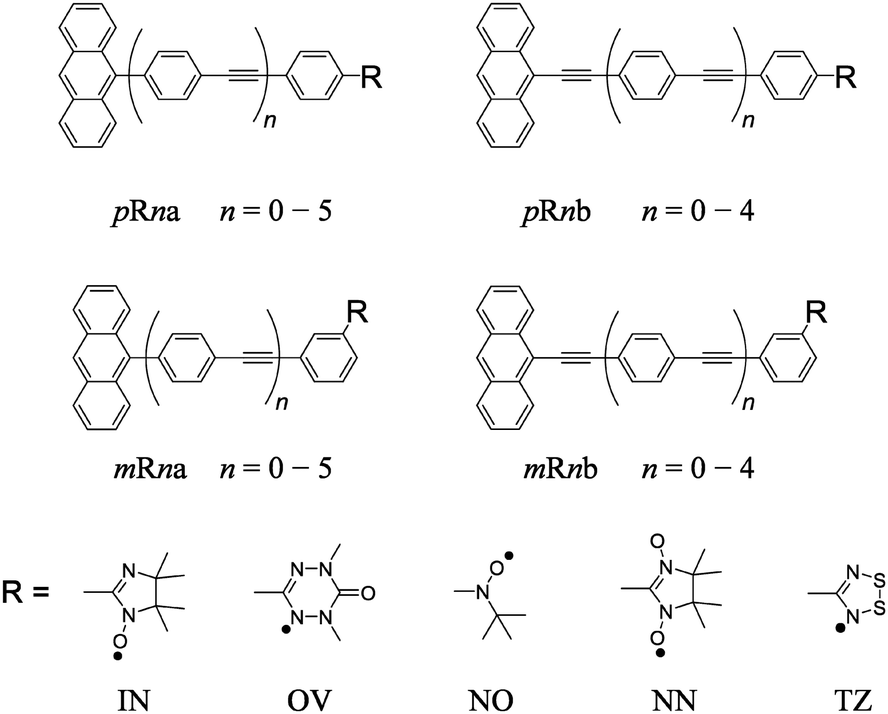
In this work, we have treated a series of anthracene coupled to stable radicals with π-conjugated linker (π-linker) shown in Fig. 2. Here, two types of π-topology are connected to the anthracene excited chromophore at meta (m) or para (p) position of the phenyl group in the linker by the radical moiety. Five kinds of radical substituents (IN: imino nitroxide, OV: 1,5-dimethyl-6-oxoverdazyl, NO: tert-butyl nitroxide, NN: nitronyl nitroxide, and TZ: 1,2,3,5-dithiadiazolyl) and a series of oligophenylene ethynylene linkers with different lengths (n means the number of phenyl-ethynyl repeating units) are investigated. We denote their molecules as p(m)Rna(b), where p(m), R, n and a(b) mean the type of π-topology, kind of radical species, number of phenyl-ethynyl repeating units in the π-linkers, and type of π-linker, respectively.
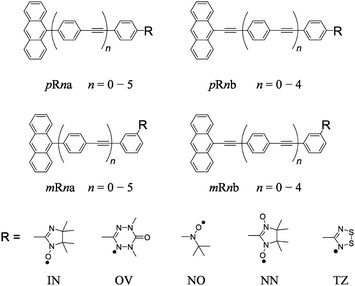 |
| | Fig. 2 Molecular structure of anthracene π-conjugated stable radical used in this work. | |
Geometry optimizations of doublet ground state (D0) and Q state were performed by DFT using unrestricted-B3LYP/6-31G(d,p) basis sets. The fine-structure interaction and g tensors of Q state and D state using broken symmetry DFT were calculated for the optimized geometry. The spin–spin term (DSS) of the fine-structure tensor was estimated using the unrestricted natural orbital (UNO). A full table of the results of the calculations is shown in the ESI.†
Results and discussion
The functional and basis-set dependencies were checked for pOV0a. In pOV0a, the sign and magnitude of JDQ (broken-symmetry method), g value, and zero-field splitting parameters (D and E) were almost independent of each functional, such as B3LYP, CAM-B3LYP, B3PW91 and O3LYP, and basis sets such as 6-31G(d,p), 6-311G(2d,2p), def2-SVP and def2-TZVP (Table S1, ESI†). The calculated JDQ values by the CASSCF method became smaller than those by the broken-symmetry method according to the increase in the active space and by adding perturbations. However, the magnitudes were still in the same order. It is reported that the calculated JDQ values of minimal CASSCF with multi-reference perturbation theory methods underestimate the exchange interactions by 60–80%.33 For organic diradicals in the ground state, the exchange interaction (J) is known to be overestimated by DFT methods that include large amounts of Hartree–Fock exact exchange, such as the B3LYP method, but agrees to some extent with experimental values by the application of appropriate scaling factors.37 When the writing of this paper was completed, a preliminarily experimental result for pOV1a was obtained, which suggested probably a fairly smaller J value than that of CASSCF.38 This may be a problem peculiar to multi-spin systems. However, there are still many unanswered issues, such as the cause of the issue, generality, solvent matrix effects and so forth. Therefore, in this work, the calculation and discussions will proceed using the traditional and widely used calculation methods. Although the absolute value of the exchange interaction may be inaccurate, the discussion focuses on the trend for the π-linker structure, which is more reliable. The broken-symmetry method using B3LYP/6-31G(d,p) is a popular method with low computational cost. Therefore, in this work, we chose this method to clarify the trend of JDQ in terms of molecular structure in further discussions.
It is well established both experimentally and theoretically that the spin polarization effect plays an important role in the spin alignment in the ground state and the photo-excited state of π-conjugated spin systems.1,3,15,39Fig. 3a shows that the spin alignment is qualitatively understood by taking the spin polarization effect depicted by the arrows into account (the arrow direction indicates the sign of the spin density and the alternating sign of the spin density between the adjacent carbon atoms is expected from the spin polarization effect). The lowest excited states were detected by the time-resolved ESR measurements,40 but it was difficult experimentally to detect higher energy spin states with different spin multiplicities, except when they were in energy proximity.41–43 Theoretical calculations can approach the electronic states that are not accessible experimentally, and therefore, making it possible to estimate the intra-molecular exchange interaction in the π-conjugated multi-spin photoexcited systems.
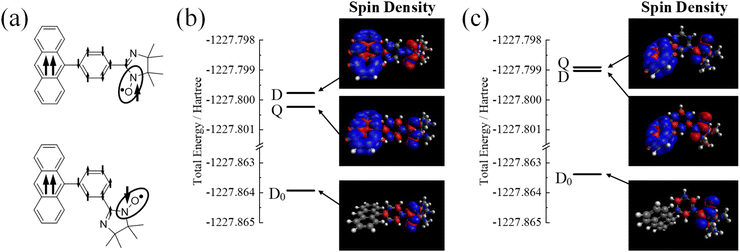 |
| | Fig. 3 Spin alignment and photo-excited states of pIN0a and mIN0a. (a) Schematic picture of the spin alignment in the lowest photo-excited state of pIN0a and mIN0a. (b) Total energy and spin density mapping of pIN0a. (c) Total energy and spin density mapping of mIN0a. | |
Fig. 3b and c show the calculated total energy and spin densities of pIN0a and mIN0a, respectively. The lowest excited states of pIN0a and mIN0a were Q and D states, respectively, which agreed with the results of the TRESR experiments.15 In the lowest excited states of pIN0a and mIN0a, the spin density distribution in the phenyl group jointing the triplet anthracene to the radical moiety is consistent with the qualitative picture in Fig. 3a. The JDQ of pOV0a calculated from eqn (2) was close to the experimental result29 and the previous theoretical calculation.30,31 The dihedral angles (ϕ) between the anthracene moiety and the adjacent phenyl group were 73.1° and 58.6° in the ground and excited states of pIN0a, respectively. These values were also close to those of the previous theoretical studies.30,31
Radical species and π-topology dependencies of exchange interactions and dihedral angles
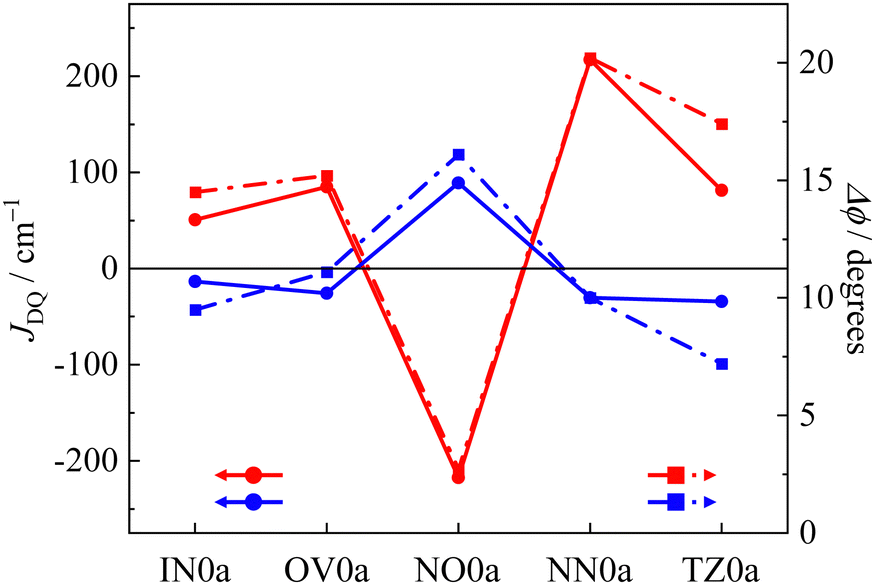
Since the rate of intersystem crossing (ISC) depends on the potential energy surfaces between the initial and final states, the molecular structure in the excited state is an important factor related to the rate.44,45 Here, we selected the subtraction of the dihedral angle between the ground and excited states (Δϕ) as the index of the spin (unpaired electron) delocalization in the photo-excited states, which are characteristics in the photo-excited states with singly occupied “HOMO” and “LUMO”. We compared the exchange interaction between the radical moiety and the chromophore to their Δϕ in the excited state. Fig. 4 shows the radical species dependencies of the JDQ value and Δϕ of p(m)R0a. The dihedral angle (ϕ) between the anthracene and phenyl moieties depicted in Fig. 5a is indicated on the right axis of Fig. 4. The p(m)IN0a, p(m)OV0a, p(m)NN0a, and p(m)TZ0a gave ferromagnetic interactions in the para-joint type (p) and antiferromagnetic interactions in the meta-joint type (m). Only p(m)NO0a showed the opposite trend: antiferromagnetic in the para-joint type (p) and ferromagnetic in the meta-joint type (m). This result is due to the direct bonding of the NO group with large spin density to the π-linker.27 On the other hand, the radical species dependence in the magnitude of JDQ is regardless of their π-topology; that is, |JDQ| is larger in the para-joint type than in the meta-joint type. The magnitude of the effective exchange interaction in the ground state of the π-conjugated multi-spin systems is proportional to the spin densities (ρAi and ρBj) between the connecting sites i and j, when the two moieties (A and B) are weakly interacting (corresponding to the disjoint case in the category of Borden and Davidson46), that is, Jeff = JijρAiρBj/4SASB.39,47,48 Thus, if the phenyl group is regarded as a spin-coupler, the meta position has a small spin density, resulting in a small exchange interaction. A similar trend was observed in the JDQ values of the excited states.
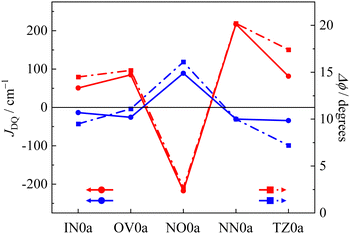 |
| | Fig. 4
J
DQ
vs. Δϕ plot. Closed circles and closed squares are JDQ values and Δϕ values, respectively (red: para joint, blue: meta joint). | |
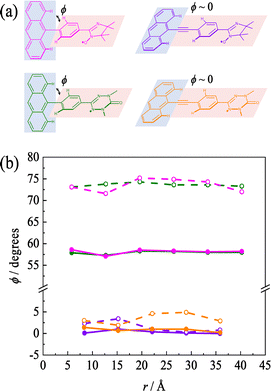 |
| | Fig. 5 (a) Molecular structure of pIN0a (pink), pOV0a (green), pIN0b (orange) and pOV0b (violet). (b) Dihedral angles (ϕ) in a series of pINna (pink), pOVna (green), pINnb (orange) and pOVnb (violet) with the different length oligophenylene ethynylene linkers; ground state (open circle) and excited state (closed circle). | |
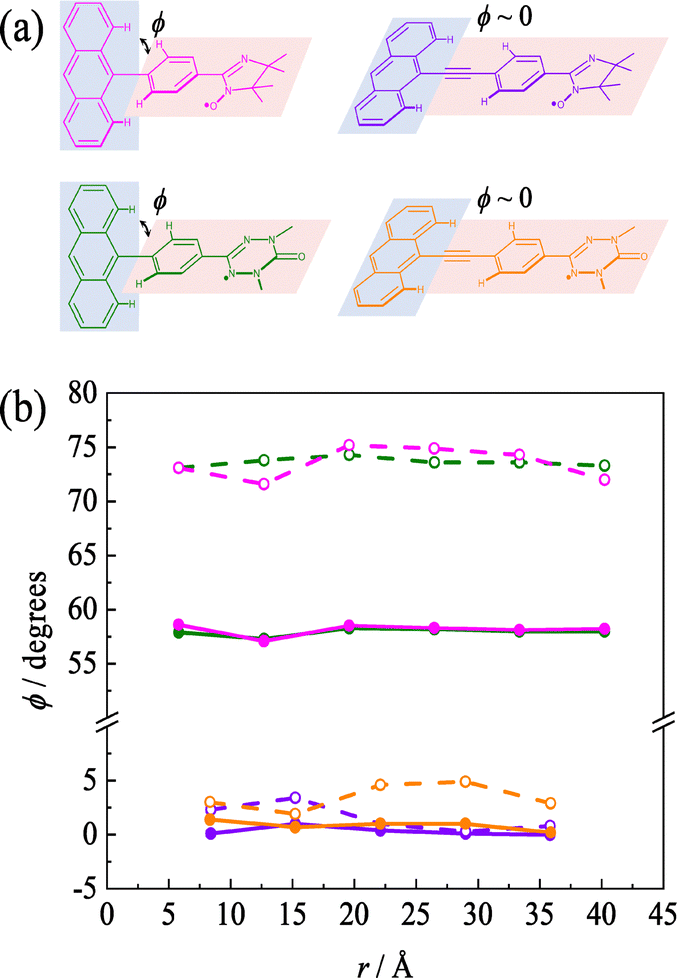
All pR0a series has a twisted conformation of the phenyl linker about ϕ ∼ 75° for the anthracene chromophore in the ground state due to the steric hindrance between the anthracene moiety and the attached phenyl group, which is independent of radical substituents and π-topology. In the previous theoretical studies for pOV0a and pIN0a, the ϕ in the Q state is reported to be ∼56°,30 which is much flatter than that of the ground state and also smaller than 67° of the T1 state of phenyl-anthracene (the details as shown Fig. S1, ESI†). In the present calculations, the structural optimization of the Q state in the para-joint type, except for the tert-butyl nitroxide radical (NO) species, gave almost the same tendency as in the previous report.30,31 Thus, attachment of the radical substituents leads to a decrease of ϕ in the Q states compared to the T1 state of phenyl-anthracene. The ϕ in the Q states of pNN0a and pTZ0a was much flatter than that of the D0 state. The decrease in ϕ leads to more propagation through π-conjugation, in which the magnetic interaction becomes larger. Thus, a decreasing ϕ in the excited state leads to increasing |JDQ|. Therefore, the pNN0a with the largest exchange interaction in the para joint type also has the largest change in dihedral angle, that is, the largest Δϕ. In contrast, the optimized structures of the Q state in the meta-joint type, except the tert-butyl nitroxide radical (NO) species, gave almost the same dihedral angle as that of the T1 state of the phenyl-anthracene, which indicates a small amount of electronic and magnetic stabilization between the anthracene and phenyl moiety. For NO0a with a large spin density directly bonded to the π-linker, the signs of JDQ and Δϕ in the Q state are opposite to those of other radical species. These calculations show that in all molecules calculated here, the subtraction of the dihedral angle (Δϕ) between the ground and excited states correlates well with the JDQ values. These findings mean that in the a type molecules, Δϕ works as an excellent index of the excess spin (unpaired electron) delocalization characteristics in the photo-excited states.
Linker dependence of exchange interactions and dihedral angles
Since the dihedral angle increases by the steric hindrance between the anthracene moiety and the attached phenyl group, the insertion of an ethynyl group between them decreases the steric hindrance significantly, as shown in Fig. 5a. Thus, the b type molecules have smaller dihedral angle than the a type molecules. The changes in the structure and the magnetic parameters were calculated for both types when the number of phenyl–ethynyl repeating units, that is, the linker length, was varied in the π-linker. The dihedral angles (ϕ) between the anthracene moiety and the phenyl group vs. linker length plot is shown in Fig. 5b. Here, the linker length (r) was defined as the distance between the carbon atoms at the 9-position of anthracene and the carbon atoms of OV or IN at the joint position to the π-linker. Similar to the previous section, the ϕs in the excited state and ground state for the pINna and pOVna series (n: the number of phenyl–ethynyl repeating units) were around 57° and 73°, respectively, independent of the linker length. In contrast, the ϕs for the pINnb and pOVnb series were close to zero in both the excited and ground states, leading to Δϕ ∼ 0. This trend was also observed for other radical species and meta-joint molecules.
There is no systematic study of the exchange interaction in the π-conjugated multi-spin organic systems in the photo-excited state. Therefore, although the exchange interactions between excited triplet-state chromophores and stable radicals may have similar characteristics to those of the ground state spin systems, it is worth examining the behaviour. In the ground-state biradical systems, the exchange interaction through π-conjugation between radical substituents was studied experimentally and computationally.49–54 In this ground-state biradical system, the exchange interaction is reported to decay exponentially as follows:49
| | | J = J0exp(−βr) | (3) |
where
β and
r were the decay constant and the linker length, respectively. The exchange interaction can be determined experimentally by analysing the shape of the ESR spectra when the magnitude of the exchange interaction is comparable to the hyperfine coupling constant.
50,51 It was also estimated by the broken symmetry DFT calculations. In these results, the decay constant due to π-bonding was independent of the type of radical substituent and varied significantly with the structure of the linker.
52–55 In addition, the conductance of a single molecular π-conjugated wire decreased exponentially with increasing number of the repeating units of the π-linker.
56–58 The linker species dependence of the decay constant
β of the conductance was in good agreement with that of the exchange interaction in the case of the neutral radicals. The exchange interaction between charge-separated radical pairs
via oligo-phenylene groups was also investigated using magnetic field effects on triplet yields of photoinduced electron transfer reactions.
59 The distance dependence of the rate constant in the charge separation and the recombination processes was also exponential decay, and their decay constants (
β) were consistent with those of the exchange interaction between photoinduced radical pairs.
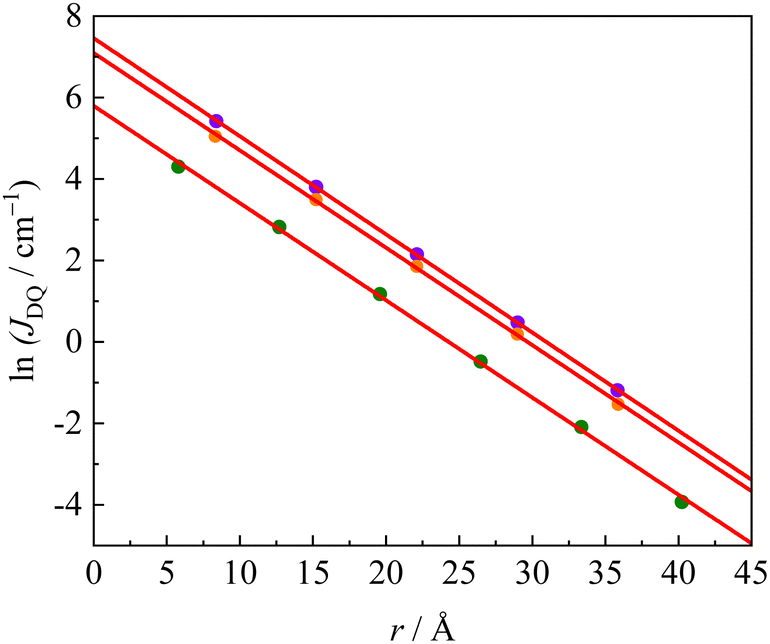
60–62 In this work, we investigated the decay of the exchange interaction in the multi-spin photoexcited systems for the repetition number of the phenyl–ethynyl group in the π-linker, which connects the anthracene triplet-state chromophore to the radical substituents. A plot of ln|
JDQ|
vs. the linker length is shown in
Fig. 6. Since in some molecules of the
INnb series, the broken symmetry DFT calculation converged to the D
0 state, not the D state, they are not shown in
Fig. 6. The slope in the plot of ln|
JDQ| to the linker length corresponds to the decay constant,
β. The
β values of
pOVna,
pOVnb, and
pINnb series were determined to be 0.239, 0.241 and 0.239 Å
−1, respectively. The
β values in the equation,
JDQ =
JEx0exp(−
βr), determined in this study, showed similar characteristics to the ground-state bi-radicals, that is, independent of the type of the radical species and the π-topology (see Fig S2–S6 and Table S4, ESI
†). Here,
JEx0 corresponds to
J0 values on the multi-spin photoexcited states. These
β values in the photoexcited states were close to the reported ones of exchange interactions between neutral bi-radicals (
β = 0.24 Å
−1) and the single molecular conductance (
β = 0.27 Å
−1) of the oligophenylethynyl linker. This result indicates that the
β value through the π-linker is independent of whether the excited state or ground state and their spin quantum numbers.
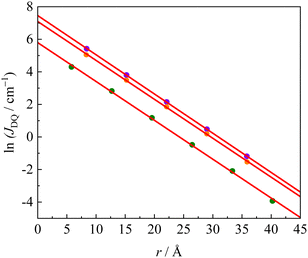 |
| | Fig. 6 Linker length dependence on JDQ; pINnb (orange), pOVna (green) and pOVnb (violet) series. The red lines are linear fittings using eqn (3). | |
On the other hand, JEx0 depended on the radical species, π-topology and linker structure. The dependence is already explained in the previous section. The JEx0 values of oxo-verdazyl radical series, pOVna, pOVnb, mOVna, and mOVnb were 329, 1737, −144 and −593 cm−1, respectively. Although pOV0b and mOV0b have a π-linker longer than those of pOV0a and mOV0a, they show larger JDQ values. In the case of the direct orbital-conjugation, the propagation of the exchange interaction via the π-linker is expected to be related to the overlap integral of the two molecular-atomic orbitals (probably natural orbitals) at the joint position between the linker and triplet chromophore. Therefore, intramolecular exchange interaction depends on the dihedral angle (ϕ), as follows:63–65
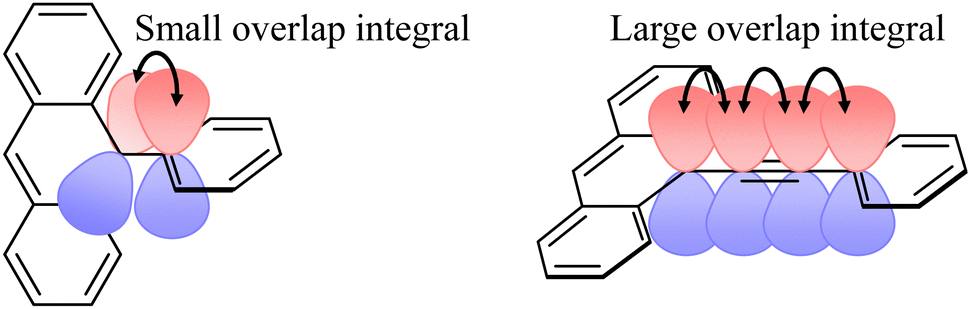
As shown in
Fig. 7, the overlap integral of the phenyl anthracene unit is expected to be smaller than that of the phenylethynyl anthracene unit because of the larger dihedral angle.
p(m)OVna shows cos
2ϕ = 0.28 because of the large dihedral angle (
ϕ = 57.9°) of the Q state. In contrast, in
p(m)OVnb, shows cos
2ϕ ∼ 1 because of the small dihedral angle (
ϕ = 0.1°). Matsuda
et al. reported almost the same β as ours, and the linker length dependence was also a similar exponential decay (
eqn (3)). When
J0 was calculated backward according to
eqn (3) using their data,
J0 ∼ −370 cm
−1.
52 This magnitude is of the same order as
JEX0 of
pOVna. Since the unpaired electron in the SOMO of the radical moiety interacts with both the unpaired electrons in the HOMO and the LUMO of the chromophore in the excited state,
JEX0 is expected to become larger than
J0 of the ground-state biradical systems, but the smaller value for
pOVna is probably due to the larger dihedral angle.
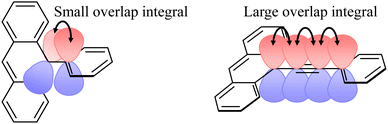 |
| | Fig. 7 Schematic view of π-orbital overlap in phenyl anthracene (left) and phenylethynyl anthracene (right). | |
ESR parameters of Q state on the anthracene π-conjugated stable radical systems
When the molecule has a spin quantum number of S ≥ 1, the ESR spectrum shows zero-field splitting due to the magnetic interactions between unpaired electrons. They are characterized by the effective spin Hamiltonian given bywhere μB, g, and D are Bohr magneton, the electron g tensor, and fine-structure tensor, respectively. Although the higher order term in S is a group theoretically allowed in the fine-structure term, they are negligible in the molecules constructed from only light atoms. The g and D are important factors that determine not only the shape of the ESR spectrum but also the behaviour of the polarization transfer and the magnetic field effects in the excited states. In the anthracene – radical π-conjugated spin systems, the anisotropy of the g tensor is sufficiently small compared to the zero-field splitting. Here, we treated the systems as an isotropic g value, which is the averaged value, (gxx + gyy + gzz)/3. The fine structure tensor can be characterized in terms of the zero-field splitting parameters (D and E). The spin sublevels of the spin systems with S ≥ 1 are, therefore, resolved by the fine-structure term even without the external magnetic field, known as zero-field splitting.
When the quartet state is constructed from the radical–triplet pair, the g tensor is given by the following equation:66
| | 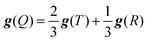 | (6) |
A comparison of the
g values obtained by the DFT calculation and by
eqn (6) is shown in
Fig. 8. The
g values for the triplet-state of anthracene and each radical are summarized in Table S7 (ESI
†). The results of the
g value calculations using DFT and
eqn (6) were in good agreement with the experimental results. However, the estimations using
eqn (6) were slightly better than the
g values calculated directly using the DFT calculation of the whole molecule. In this DFT calculation, the following multiple terms contribute to the
g tensor.
| | | g = ge1 + ΔgRMC + ΔgDSO + ΔgPSO | (7) |
where
ge, Δ
gRMC, Δ
gDSO and Δ
gPSO are the isolated electron's
g factor, relativistic mass correction, diamagnetic spin–orbit term, and paramagnetic spin–orbit term, respectively. In general, the largest contribution to the shift from
ge (free electron
g value; 2.0023) is the Δ
gPSO term. The large
g value shift of the
TZ radical can be explained by the sulphur atom, in which the spin–orbit interaction is larger than that of the other constructed elements, resulting in a larger Δ
gPSO in the
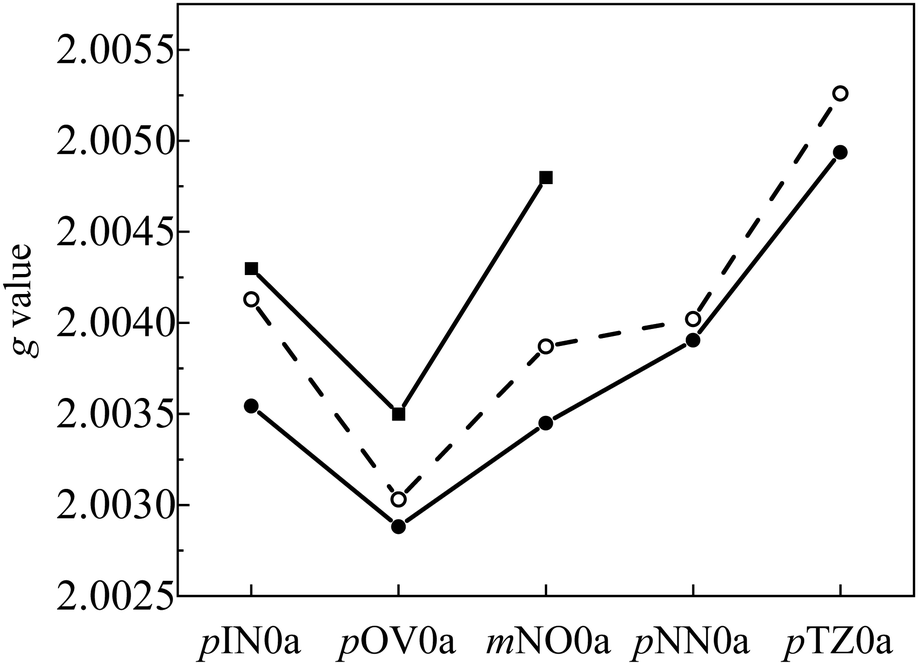
g value. The linker dependence of the calculated
g value of the Q state is shown in
Fig. 9. The
g values of the
p(m)OVna series are similar to those of the
p(m)OVnb series. The same finding holds between
p(m)INna and
p(m)INnb series. Thus, the
g values are independent of the linker structure (length and dihedral angle) and only dependent on the radical species. This means that the effect of the spin delocalization on the linker is negligible in the
g value, even in the type
b structures. In such cases, the
g value is explained by
eqn (6), in which
g(Q) depends only on
g(R) because
g(T) is dominantly determined by the triplet state of the anthracene chromophore.
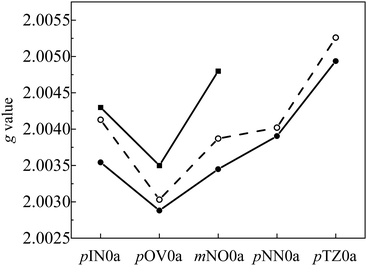 |
| | Fig. 8 Calculated g values of the Q states of the anthracene–radical π-conjugated spin systems; DFT method (closed circle), eqn (6) (open circle) and experimental (closed cube). | |
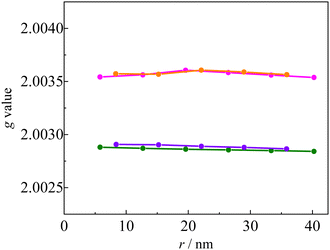 |
| | Fig. 9 Calculated g values of the Q states of pINna (pink), pOVna (green), pINnb (orange), and pOVnb (violet) | |
The fine-structure tensor consists of DSS and DSO terms that are due to magnetic dipole–dipole interactions (spin–spin interactions) and spin–orbit coupling, respectively. The contribution of the DSO term is small in organic materials composed only of light elements such as H, C, N and O because the spin–orbit coupling is proportional to the fourth power of the atomic number of the constituent elements. The pq matrix-element in the DSS tensor term is given by
| | 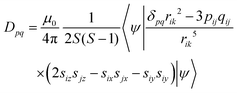 | (8) |
When the wavefunction is approximated by a single electron configuration, it can be obtained from the spin density matrix.67
| | 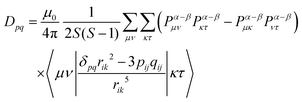 | (9) |
Here,
Pα–β is the spin density matrix obtained by the DFT calculation. It is known that, in the estimation of the
DSS term, the wavefunction obtained by the restricted open-shell method (RODFT) gives better results than the unrestricted method (UDFT).
68 The unrestricted natural local orbital (UNO) calculated by the diagonalization of the density matrix obtained from the wave function have been reported to give better agreement than the DIRECT method in UDFT.
68 It is reported that the calculated
DSS term by the UNO method were close to that of the RODFT.
69 The RODFT method has poor convergence in SCF calculations; in this work, we chose the UNO method. It is also known that the
DSS term obtained by the DFT calculation is underestimated when the unpaired electrons are distributed in the same π electron framework, such as the ππ* excited states of aromatic hydrocarbons.
69 When the quartet state is constructed from the radical–triplet pair, the
D tensor of the Q state is given by the following:
66| | 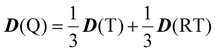 | (10) |
D(T) and
D(RT) are the fine-structure tensors for the triplet excited state and the magnetic dipolar–dipolar interaction between the radical and the triplet moieties. We denote that
D(T) and
D(RT) correspond to the contributions from
D(T) and
D(RT) in the zero-field splitting parameter (
D), respectively.
D(RT) is proportional to
r−3 when a point dipole approximation of the triplet anthracene and the radical is used:
| | 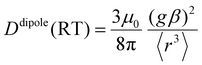 | (11) |
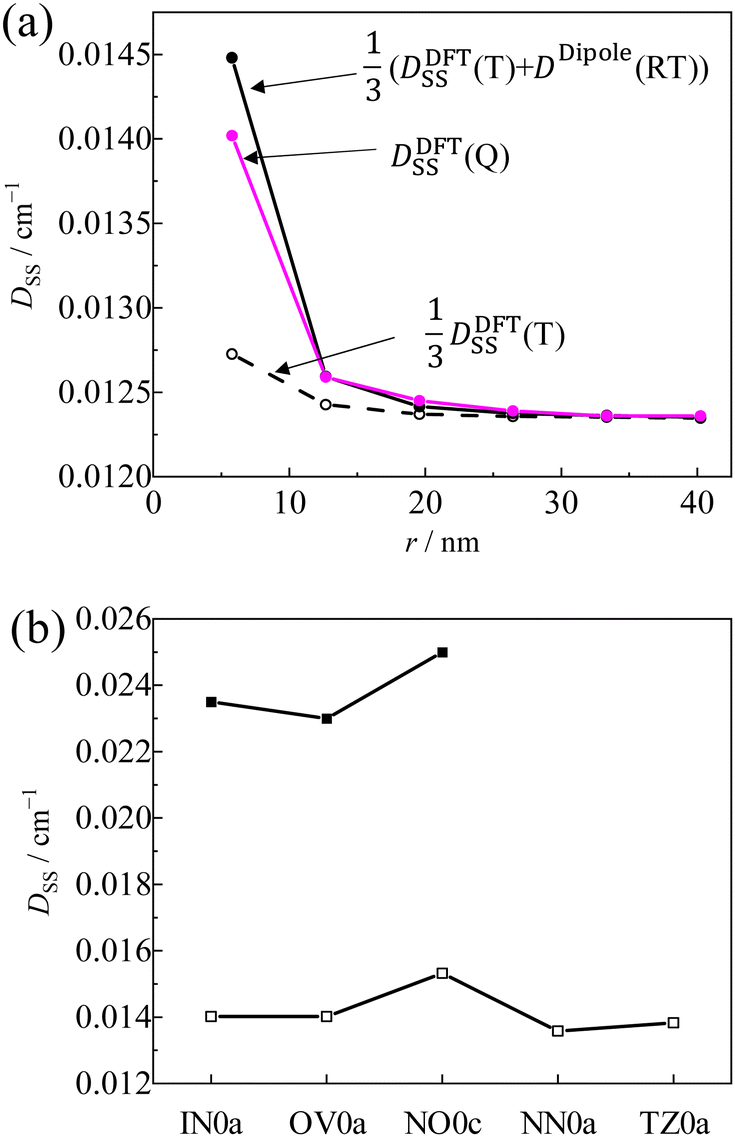
As shown in
Fig. 10a,
D(Q) obtained by the DFT calculation for the
pINna series agreed well with the estimated values using
eqn (10), in which
D(T) and
D(RT) were obtained by the DFT calculation for the molecules without radical substituents and by the point dipole approximation, respectively. However, the calculated
DSS(Q) value (the contribution from
DSS term in the
D value) of
pIN0a was only about 60% of the experimental one because the
D(T) term was under-estimated by half (
Fig. 10b). Although the CASSCF method using a large active space can estimate the
DSS tensor more accurately,
70 it is too computationally expensive for the large-size molecules as treated in this work. Therefore, although the magnitudes are underestimated, we discuss only a trend in magnetic dipole interactions of radicals
via the π-linker to the triplet-state of the anthracene chromophore by DFT calculations.
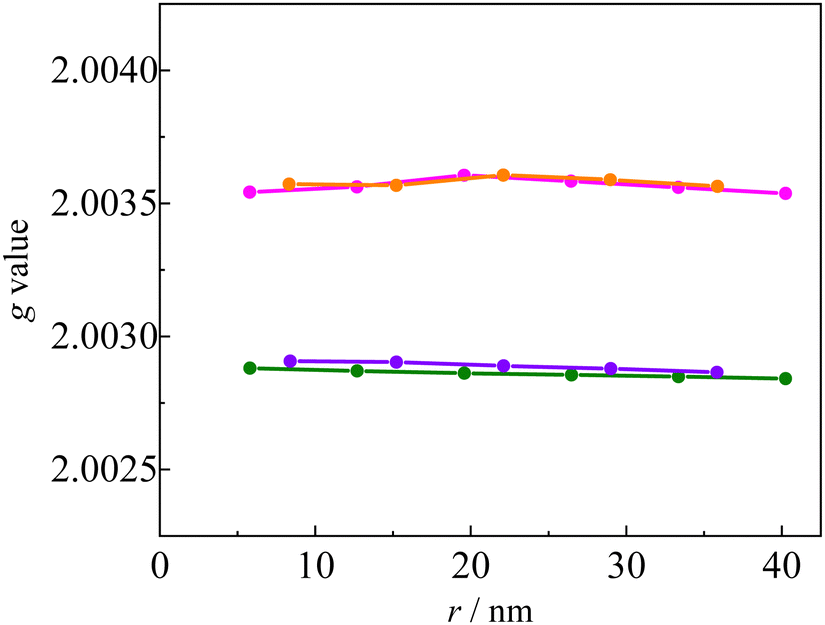
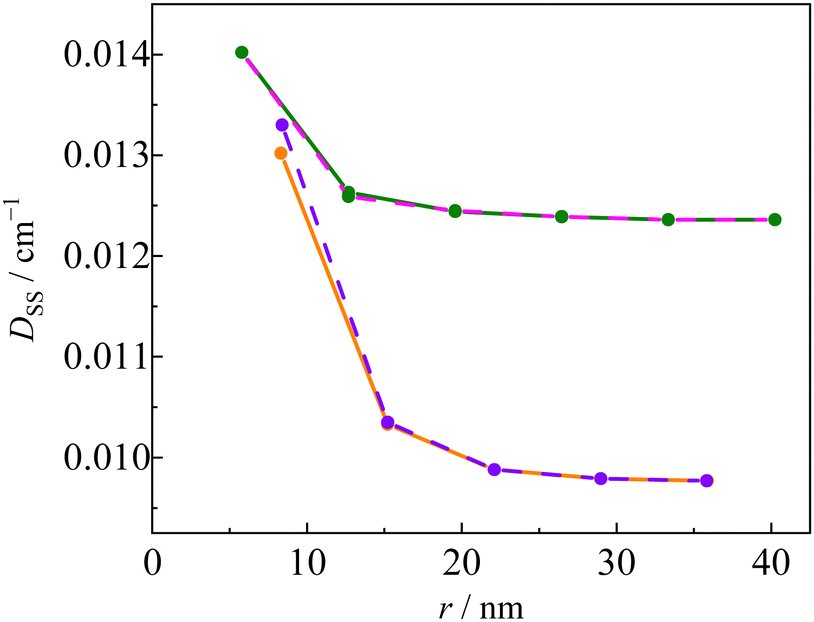
Fig. 11 shows the linker dependence of the
DSS value obtained by the restricted B3LYP method using 6-31G(d,p) basis sets. Similar length dependence was observed in
DSS calculations with other basis sets, such as restricted open BP/EPR-II and unrestricted CAM-B3LYP/def2-TZVP (Fig. S7, ESI
†). The
D values of the
pOVna(b) series were similar to those of
pINna(b). This behaviour is the opposite of the structure dependence of the
g value, that is, the
D values are independent of the radical species and only influenced by the linker structure (length and dihedral angle). For example, the
DSS value of
pOV5a was larger than that of
pOV4b despite the fact that it has a longer linker length. Thus,
DSS depends not only on the linker length
r, but also on the dihedral angle
ϕ. In the
pOVnb and
pINnb series, the unpaired electrons in the triplet-state of the anthracene chromophore are expected to delocalize to the linker due to the small dihedral angle
ϕ between the anthracene and phenyl groups. The
D(T) in
eqn (10) of the type
b linker series is smaller in magnitude than that of the type
a linker sers because the molecular orbital of the triplet chromophore is delocalized, and consequently, the
D(Q) of the type
b linker series becomes smaller.
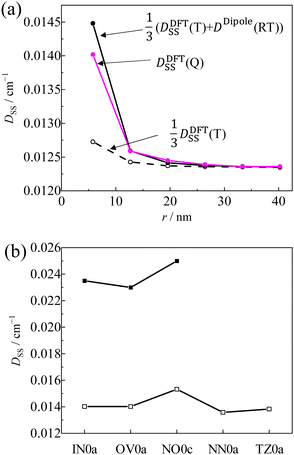 |
| | Fig. 10 (a) Linker dependence on the D value of pINna series by DFT (pink), eqn (10) (closed circle) and one third of the D value of T1 state of without radical moiety by the DFT (open circle), (b) calculated D value (open cube) and experimental D value (closed cube) of the Q states. | |
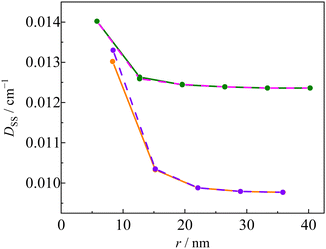 |
| | Fig. 11 Calculated D value of the Q states of INna (pink), OVna (green), INnb (orange), and OVnb (violet) series. | |
Conclusion
The exchange interactions, g values, and fine-structure interactions in the quartet (Q) excited states of the anthracene–radical π-conjugated systems were systematically investigated by DFT calculations. Based on the results of structure optimizations for the ground and excited states and JDQ calculations by the broken symmetry DFT for the excited states, the smaller Δϕ leads to the larger exchange interaction between the triplet anthracene and the radical. The steric hindrance was removed by inserting the ethynyl group between the anthracene and phenyl groups, and the ϕ of the ground and excited states were close to zero, resulting in a large JDQ. The decay constant β. of the exchange interaction via the π-linker between anthracene and the stable radical was almost identical to the ground state. The g values of the Q state depended only on the radical type, which was independent of the linker structure. Conversely, the fine-structure tensor of the Q state was independent of the radical type and depended on the linker length and dihedral angle.
We reported that, in the triplet chromophore–radical linked systems, the enhanced intersystem crossing led to significant photo-chemical stabilization for pure organic materials decomposed by visible light.18–20 Recently, it has been reported that the enhanced intersystem crossing rate and efficiency depend on their π-topology and linker length.20,71 The spin polarization generated by tuning the exchange interaction and zero-field splitting in the excited states of the triplet chromophore–radical linked system has been applied to quantum technology as optical qubit initialization.72,73 In addition, in the triplet-radical pair systems, a generation of the giant magnetization to nuclear spins in the solid matrix using the photo-induced polarization transfer has been proposed by tuning the exchange interaction and zero-field splitting of the triplet excited states of the chromophore.74 The present study will also give useful knowledge for the structural optimization of the chromophore–radical π-conjugated systems toward applications, such as photo-DNP solid-state magic angle spinning NMR, information science, and so forth.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Grant-in-Aid for Scientific Research (B) (No. 20H02715) from the Japan Society for the Promotion of Science (JSPS).
Notes and references
- Y. Teki, Chem. – Eur. J., 2020, 26, 980–996 CrossRef CAS PubMed.
- M. R. Wasielewski, M. D. E. Forbes, N. L. Frank, K. Kowalski, G. D. Scholes, J. Yuen-Zhou, M. A. Baldo, D. E. Freedman, R. H. Goldsmith, T. Goodson, 3rd, M. L. Kirk, J. K. McCusker, J. P. Ogilvie, D. A. Shultz, S. Stoll and K. B. Whaley, Nat. Rev. Chem., 2020, 4, 490–504 CrossRef CAS PubMed.
- T. Quintes, M. Mayländer and S. Richert, Nat. Rev. Chem., 2023, 7, 75–90 CrossRef PubMed.
-
H. Hayashi, Introduction To Dynamic Spin Chemistry, World Scientific Publishing Co Pte Ltd., Singapore, 2004 Search PubMed.
- M. L. Kirk, D. A. Shultz, J. Chen, P. Hewitt, D. Daley, S. Paudel and A. van der Est, J. Am. Chem. Soc., 2021, 143, 10519–10523 CrossRef CAS PubMed.
- M. L. Kirk, D. A. Shultz, P. Hewitt and A. van der Est, J. Phys. Chem. Lett., 2022, 13, 872–878 CrossRef CAS PubMed.
- M. L. Kirk, D. A. Shultz, P. Hewitt, J. Chen and A. van der Est, J. Am. Chem. Soc., 2022, 144, 12781–12788 CrossRef CAS PubMed.
- A. Kawai, T. Okutsu and K. Obi, J. Phys. Chem., 1991, 95, 9130–9134 CrossRef CAS.
- Y. M. Kobori, M. Masaaki, K. Akio and O. Kinichi, Chem. Phys. Lett., 1996, 252, 355–361 CrossRef.
- M. T. Colvin, R. Carmieli, T. Miura, S. Richert, D. M. Gardner, A. L. Smeigh, S. M. Dyar, S. M. Conron, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2013, 117, 5314–5325 CrossRef CAS PubMed.
- E. T. Chernick, R. Casillas, J. Zirzlmeier, D. M. Gardner, M. Gruber, H. Kropp, K. Meyer, M. R. Wasielewski, D. M. Guldi and R. R. Tykwinski, J. Am. Chem. Soc., 2015, 137, 857–863 CrossRef CAS PubMed.
- V. Rozenshtein, A. Berg, E. Stavitski, H. Levanon, L. Franco and C. Corvaja, J. Phys. Chem. A, 2005, 109, 11144–11154 CrossRef CAS PubMed.
- D. F. Evans, J. Chem. Soc., 1957, 3885–3888 RSC.
- R. L. Ake and M. Gouterman, Theor. Chim. Acta, 1969, 15, 20–42 CrossRef CAS.
- Y. Teki, S. Miyamoto, M. Nakatsuji and Y. Miura, J. Am. Chem. Soc., 2001, 123, 294–305 CrossRef CAS PubMed.
- Y. E. Kandrashkin and A. van der Est, J. Chem. Phys., 2019, 151, 184301 CrossRef PubMed.
- Y. E. Kandrashkin and A. van der Est, J. Phys. Chem. Lett., 2021, 12, 7312–7318 CrossRef CAS PubMed.
- Y. Kawanaka, A. Shimizu, T. Shinada, R. Tanaka and Y. Teki, Angew. Chem., Int. Ed., 2013, 52, 6643–6647 CrossRef CAS PubMed.
- A. Shimizu, A. Ito and Y. Teki, Chem. Commun., 2016, 52, 2889–2892 RSC.
- N. Minami, K. Yoshida, K. Maeguchi, K. Kato, A. Shimizu, G. Kashima, M. Fujiwara, C. Uragami, H. Hashimoto and Y. Teki, Phys. Chem. Chem. Phys., 2022, 24, 13514–13518 RSC.
- Z. Wang, J. Zhao, A. Barbon, A. Toffoletti, Y. Liu, Y. An, L. Xu, A. Karatay, H. G. Yaglioglu, E. A. Yildiz and M. Hayvali, J. Am. Chem. Soc., 2017, 139, 7831–7842 CrossRef CAS PubMed.
- B. K. Rugg, M. D. Krzyaniak, B. T. Phelan, M. A. Ratner, R. M. Young and M. R. Wasielewski, Nat. Chem., 2019, 11, 981–986 CrossRef CAS PubMed.
- M. Maylander, S. Chen, E. R. Lorenzo, M. R. Wasielewski and S. Richert, J. Am. Chem. Soc., 2021, 143, 7050–7058 CrossRef PubMed.
- Y. Teki, S. Miyamoto, K. Iimura, M. Nakatsuji and Y. Miura, J. Am. Chem. Soc., 2000, 122, 984–985 CrossRef CAS.
- Y. Teki, M. Nakatsuji and Y. Miura, Mol. Phys., 2002, 100, 1385–1394 CrossRef CAS.
- Y. Teki, T. Toichi and S. Nakajima, Chem. – Eur. J., 2006, 12, 2329–2336 CrossRef CAS PubMed.
- Y. Teki, S. Miyamoto and K. Koide, Phys. Chem. Chem. Phys., 2015, 17, 31646–31652 RSC.
- M. Asano, Y. Kaizu and H. Kobayashi, J. Chem. Phys., 1988, 89, 6567–6576 CrossRef CAS.
- A. Ito, M. Hinoshita, K. Kato and Y. Teki, Chem. Lett., 2016, 45, 1324–1326 CrossRef CAS.
- I. Ciofini, P. P. Laine, M. Zamboni, C. A. Daul, V. Marvaud and C. Adamo, Chem. – Eur. J., 2007, 13, 5360–5377 CrossRef CAS PubMed.
- I. Ciofini, C. Adamo, Y. Teki, F. Tuyeras and P. P. Laine, Chem. – Eur. J., 2008, 14, 11385–11405 CrossRef CAS PubMed.
- T. Steenbock, L. L. M. Rybakowski, D. Benner, C. Herrmann and G. Bester, J. Chem. Theory Comput., 2022, 18, 4708–4718 CrossRef CAS PubMed.
- M. Franz, F. Neese and S. Richert, Chem. Sci., 2022, 13, 12358–12366 RSC.
- F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12, e1606 CrossRef.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- T. Soda, Y. Kitagawa, T. Onishi, Y. Takano, Y. Shigeta, H. Nagao, Y. Yoshioka and K. Yamaguchi, Chem. Phys. Lett., 2000, 319, 223–230 CrossRef CAS.
- D. Cho, K. Chul Ko, Y. Ikabata, K. Wakayama, T. Yoshikawa, H. Nakai and J. Y. Lee, J. Chem. Phys., 2015, 142, 024318 CrossRef PubMed.
-
Y. Teki, et al., unpublished work.
-
Y. Teki and K. Itoh, in Magnetic Properties of Organic Materials, Marcel Dekker Inc., New York-Basel, 1999, pp. 237–265 Search PubMed.
- S. S. Kim and S. I. Weissman, J. Magn. Reson., Ser. A, 1976, 24, 167–169 Search PubMed.
- J. Fujisawa, K. Ishii, Y. Ohba, S. Yamauchi, M. Fuhs and K. Möbius, J. Phys. Chem. A, 1999, 103, 213–216 CrossRef CAS.
- J. Fujisawa, Y. Iwasaki, Y. Ohba, S. Yamauchi, N. Koga, S. Karasawa, M. Fuhs, K. Möbius and S. Weber, Appl. Magn. Reson., 2001, 21, 483–493 CrossRef CAS.
- H. Moons, E. Goovaerts, V. P. Gubskaya, I. A. Nuretdinov, C. Corvaja and L. Franco, Phys. Chem. Chem. Phys., 2011, 13, 3942–3951 RSC.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- R. Ahmed and A. K. Manna, J. Phys. Chem. A, 2022, 126, 6594–6603 CrossRef CAS PubMed.
- W. T. Borden and E. R. Davidson, J. Am. Chem. Soc., 1977, 99, 4587–4594 CrossRef CAS.
- K. Itoh, T. Takui, Y. Teki and T. Kinoshita, Mol. Cryst. Liq. Cryst., 1989, 176, 49–65 CrossRef CAS.
- Y. Teki and K. Itoh, ACS Symp. Ser., 1996, 644, 16–29 CrossRef CAS.
- J. J. De Kanter, R. Kaptein and R. A. Van Santen, Chem. Phys. Lett., 1977, 45, 575–579 CrossRef.
- K. Higashiguchi, K. Yumoto and K. Matsuda, Org. Lett., 2010, 12, 5284–5286 CrossRef CAS PubMed.
- M. Shinomiya, K. Higashiguchi and K. Matsuda, J. Org. Chem., 2013, 78, 9282–9290 CrossRef CAS PubMed.
- S. Nishizawa, J. Hasegawa and K. Matsuda, Chem. Phys. Lett., 2013, 555, 187–190 CrossRef CAS.
- S. Nishizawa, J. Hasegawa and K. Matsuda, J. Phys. Chem. C, 2013, 117, 26280–26286 CrossRef CAS.
- S. Hasegawa and K. Matsuda, J. Phys. Chem. C, 2015, 119, 5117–5121 Search PubMed.
- S. Hasegawa and K. Matsuda, J. Phys. Chem. C, 2015, 119, 20169–20178 CrossRef.
- K. Tomfohr and O. F. Sankey, Phys. Status Solidi B, 2002, 233, 59–69 CrossRef.
- Q. Lu, K. Liu, H. Zhang, Z. Du, X. Wang and F. Wang, ACS Nano, 2009, 3, 3861–3868 CrossRef CAS PubMed.
- V. Kaliginedi, P. Moreno-Garcia, H. Valkenier, W. Hong, V. M. Garcia-Suarez, P. Buiter, J. L. Otten, J. C. Hummelen, C. J. Lambert and T. Wandlowski, J. Am. Chem. Soc., 2012, 134, 5262–5275 CrossRef CAS PubMed.
- A. M. Scott, T. Miura, A. B. Ricks, Z. E. Dance, E. M. Giacobbe, M. T. Colvin and M. R. Wasielewski, J. Am. Chem. Soc., 2009, 131, 17655–17666 CrossRef CAS PubMed.
- T. Miura, A. M. Scott and M. R. Wasielewski, J. Phys. Chem. C, 2010, 114, 20370–20379 CrossRef CAS.
- A. B. Ricks, K. E. Brown, M. Wenninger, S. D. Karlen, Y. A. Berlin, D. T. Co and M. R. Wasielewski, J. Am. Chem. Soc., 2012, 134, 4581–4588 CrossRef CAS PubMed.
- T. Miura, M. Aikawa and Y. Kobori, J. Phys. Chem. Lett., 2014, 5, 30–35 CrossRef CAS PubMed.
- D. A. Shultz, R. M. Fico, H. Lee, J. W. Kampf, K. Kirschbaum, A. A. Pinkerton and P. D. Boyle, J. Am. Chem. Soc., 2003, 125, 15426–15432 CrossRef CAS PubMed.
- S. Nishizawa, J. Hasegawa and K. Matsuda, Chem. Lett., 2014, 43, 530–532 CrossRef CAS.
- C. Shu, Z. Yang and A. Rajca, Chem. Rev., 2023, 123, 11954–12003 CrossRef CAS PubMed.
-
A. Bencini and D. Gatteschi, EPR of Exchange Coupled Systems, Springer-Verlag Berlin, Heidelberg, 1990 Search PubMed.
- R. McWeeny and Y. Mizuno, Proc. R. Soc. London, Ser. A, 1961, 259, 554–577 CAS.
- F. Neese, J. Am. Chem. Soc., 2006, 128(31), 10213–10222 CrossRef CAS PubMed.
- S. Sinnecker and F. Neese, J. Phys. Chem. A, 2006, 110, 12267–12275 CrossRef CAS PubMed.
- K. Sugisaki, K. Toyota, K. Sato, D. Shiomi, M. Kitagawa and T. Takui, Phys. Chem. Chem. Phys., 2011, 13, 6970–6980 RSC.
- M. Maylander, T. Quintes, M. Franz, X. Allonas, A. Vargas Jentzsch and S. Richert, Chem. Sci., 2023, 14, 5361–5368 RSC.
- H. Mao, R. M. Young, M. D. Krzyaniak and M. R. Wasielewski, J. Phys. Chem. B, 2022, 126, 10519–10527 CrossRef CAS PubMed.
- Y. Qiu, A. Equbal, C. Lin, Y. Huang, P. J. Brown, R. M. Young, M. D. Krzyaniak and M. R. Wasielewski, Angew. Chem., Int. Ed., 2023, 62, e202214668 CrossRef CAS PubMed.
- K. Kundu, T. Dubroca, V. Rane and F. Mentink-Vigier, J. Phys. Chem. A, 2022, 126, 2600–2608 CrossRef CAS PubMed.
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Yoshio
Teki
*a and
Yoshio
Teki