
Progress on enhancing the charge separation efficiency of carbon nitride for robust photocatalytic H2 production
Mengmeng
Shao
 *a,
Yangfan
Shao
b and
Hui
Pan
*cd
*a,
Yangfan
Shao
b and
Hui
Pan
*cd
aSchool of Materials Science and Engineering, Dongguan University of Technology, Dongguan 523808, China. E-mail: shaomm@dgut.edu.cn
bInstitute of Materials Research, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen 518055, China
cInstitute of Applied Physics and Materials Engineering, University of Macau, Macao 999078, China. E-mail: huipan@um.edu.mo
dDepartment of Physics and Chemistry, Faculty of Science and Technology, University of Macau, Macao 999078, China
First published on 19th March 2024
Abstract
Solar-driven H2 production from water splitting with efficient photocatalysts is a sustainable strategy to meet the clean energy demand and alleviate the approaching environmental issues caused by fossil fuel consumption. Among various semiconductor-based photocatalysts, graphitic carbon nitride (g-C3N4) has attracted much attention due to its advantages of long term-stability, visible light response, low cost, and easy preparation. However, the intrinsic Coulombic attraction between charge carriers and the interlayer electrostatic barrier of bulk g-C3N4 result in severe charge recombination and low charge separation efficiency. This perspective summarizes the recent progress in the development of g-C3N4 photocatalytic systems, and focuses on three main modification strategies for promoting charge transfer and minimizing charge recombination, including structural modulation, heterojunction construction, and cocatalyst loading. Based on this progress, we provide conclusions regarding the current challenges of further improving photocatalytic efficiency to fulfill commercial requirements, and propose some recommendations for the design of novel and satisfactory g-C3N4 photocatalysts, which is expected to progress the solar-to-hydrogen conversion.
1. Introduction
Fuel is an indispensable driving force for the development of human society. In the ancient world, humans bade farewell to a life of devouring raw meat by drilling wood to make fire, and began the era of using firewood as the main energy source. Since the 18th century, with the outbreak of the industrial revolution, fossil fuels, such as coal, oil, and natural gas, have emerged as the main energy source in the world. Nowadays the increasing demand for fossil fuels and various environmental issues (e.g., global warming, acid rain, and smog) caused by their excessive consumption have posed great challenges to the current energy system. Among emerging energy sources, hydrogen has the highest energy density, is eco-friendly, abundant, and carbon-neutral, and is considered a promising alternative to fossil fuels.1,2In recent decades, solar-driven H2 production from water splitting has received great attention due to the green process only involving sustainable solar energy, water, and photocatalysts.3–5 Starting from the pioneering work of Fujishima and Honda about water-splitting on TiO2,6 numerous active semiconductor photocatalysts have been developed, such as CdS, g-C3N4, BiOBr, CdxZn1−xS, GaN–ZnO solid solution, and ZnIn2S4.7–17 The typical process of photocatalytic water-splitting mainly includes three steps (Fig. 1a):18,19 (1) the generation of photo-induced electron and hole pairs, where the electrons in the valence band (VB) are excited to the conduction band (CB) under light irradiation with higher energy than the band gap of photocatalysts; (2) the separation and transfer of photo-induced charge carriers, which are always accompanied by recombination; (3) the transferred charges participate in the surface catalytic reaction, producing H2 and O2 by H+ reduction and H2O oxidation, where the redox potentials of H+/H2 and O2/H2O should be located between the CB minimum and VB maximum. Based on the above steps, the development of robust photocatalysts is focused on increasing the light-harvesting range, improving charge separation efficiency, and accelerating surface redox reactions. Currently, to promote the practical application of photocatalytic H2 production, the photocatalysts with the visible light response and high apparent quantum efficiency (AQE) attain significant interest.
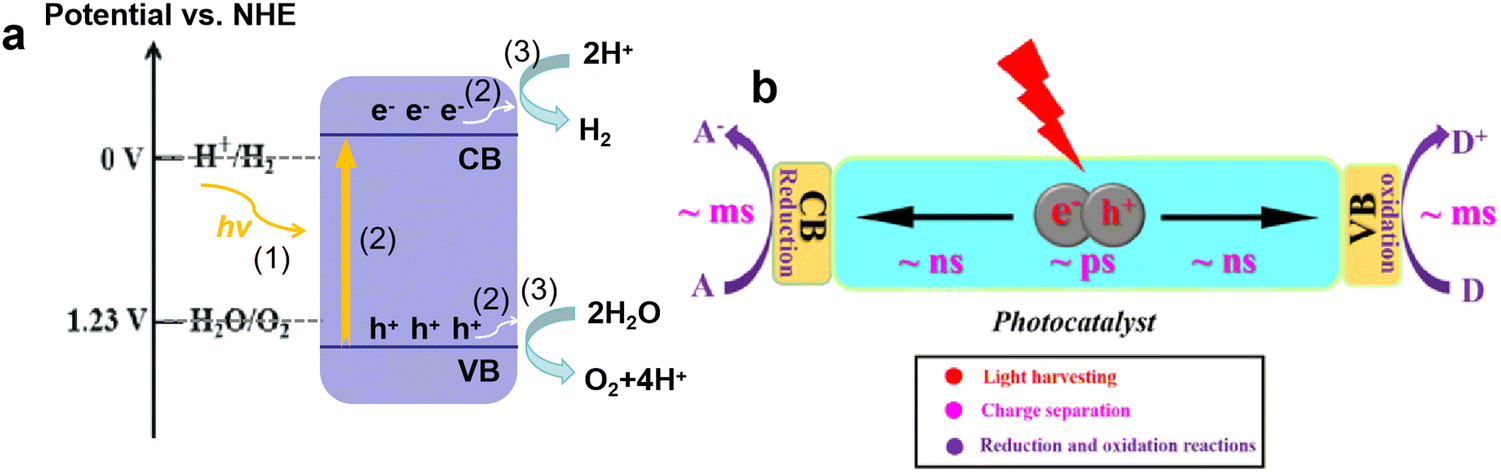 | ||
| Fig. 1 Photocatalytic H2 evolution process (a). The kinetic times for separation, transfer, and reaction of photogenerated charge carriers (b). Reproduced with permission from ref. 31. Copyright 2021, Wiley-VCH. | ||
As reported, the polymeric semiconductor, graphitic carbon nitride (g-C3N4), is well known as a photocatalyst for H2 production because of its advantageous layered structure, long-term stability, ease of synthesis, low cost, and eco-friendly nature. Especially, g-C3N4 with a narrow band gap (∼2.7 eV) is capable of harvesting visible light, and its band edge positions are suitable for water splitting.20–22 In 2009, Wang et al.23 first showed photocatalytic H2 production from water using g-C3N4 as the photocatalyst under visible light irradiation. The result of Wang's study motivated the researchers to further explore and improve the photocatalytic H2 production performance of g-C3N4. For instance, Zhu and Zhang et al.24 prepared 3D porous g-C3N4 through a bottom-up self-assembly method, achieving photocatalytic overall water splitting with ∼1.4% AQE at 420 nm. Liu and Lu et al.25 designed a ZnIn2S4/g-C3N4 heterojunction composite, which displayed excellent photocatalytic activity with a H2 production rate of about 87 µmol h−1 and AQE of 10.74% at 420 nm. Our group also explored the photocatalytic performance of different cocatalyst loaded g-C3N4,26–28 and found the H2 production rates of VS2/g-C3N4 and W2C/g-C3N4 both exceeded 80 µmol h−1, while the highest quantum efficiency achieved on VS2/g-C3N4 was just 5.5%.
However, although g-C3N4 is a very promising photocatalyst for H2 production from water splitting, and related studies have made some progress, the quantum efficiency is still too low to meet the solar-to-hydrogen conversion efficiency of 10% required for commercialization. The main reason lies in the high recombination rate of photo-induced charges, leading to less electron involvement in the H2 evolution reaction.29,30 For photocatalysis, “no charge, no reaction” is clear, the charge separation and transfer are crucial for photocatalytic performance. As shown in Fig. 1b, due to the strong Coulombic interaction between electron and hole pairs, the charge carriers undergo severe recombination within the bulk and surface phases (several ps < t < dozens ps), which is faster than the separation of these charges (∼ns) and the involved surface reactions (∼ms).31,32 Another challenge for charge separation and transfer is the existence of the interlayer electrostatic barrier in bulk g-C3N4, which hinders the transfer of electrons from the bulk to surface active sites, and results in severe bulk recombination and poor photocatalytic activity.33,34 Thus, the efficient charge separation and opposite transfer to surface redox sites are of the utmost importance in photocatalytic reactions.
During the last decade, a series of papers have been published revealing the advances in the field of photocatalytic H2 production over g-C3N4.35–37 As we know, the separation and transfer of charges is a crucial step for photocatalysis. Currently, various approaches have been developed to modify g-C3N4 and improve charge separation efficiency, such as morphological control, metal/non-metal doping, heterojunction, etc.22 Considering the unique characteristics of each strategy, it is necessary to compare different modification methods and summarize their advantages in a review, which is beneficial for further understanding the mechanism of charge separation and transfer, providing strategies for developing highly active g-C3N4-based photocatalysts for H2 production.
Therefore, in this perspective, following the introduction, our focus is to discuss the general strategies for strengthening charge separation and transfer in g-C3N4, and analyze their respective characteristics. We then extend our discussion to the synergistic effect of combining multiple methods on the improvement of photocatalytic efficiency. Finally, we conclude with some recommendations for the design of benchmark g-C3N4-based photocatalysts for practical applications.
2. Structural modulation of g-C3N4
As discussed in the introduction, the poor charge separation efficiency of photocatalysts essentially lies in the high Coulombic attraction of photo-induced charges, which makes the dissociation of electron and hole pairs into free charges difficult. For the g-C3N4 photocatalyst, another added problem is the presence of electrostatic barriers between layers, which results in the trapping of charge carriers in the bulk phase and few electrons being transferred to the surface. Thus, to facilitate charge separation and transfer in g-C3N4, the modulation of intrinsic structure is a commonly adopted strategy. In this section, we focus on discussing several important published studies about structural modulation of g-C3N4via crystal structure regulation, morphological tuning and doping engineering. A comparison of the photocatalytic H2 evolution activity of the recent g-C3N4 systems based on structural modulation is given in Table 1.| Photocatalyst | Light source | Reaction conditions | H2 evolution rate | AQE | Ref. |
|---|---|---|---|---|---|
| a Poly(triazine imide) (PTI) based carbon nitride. b The precursors of thiourea (T) and melamine (M) for g-C3N4 (CN). c Thiophene-2,5-dicarbaldehyde (TDA). d Aromatized terminal. e 5-aminotetrazole (ATZ) is used to prepare g-C3N4 in NaCl/KCl molten salt. f The precursor of 2-aminothiophene-3-carbonitrile (A) for poly-heptazine-imide (PHI) based carbon nitride. g Triethanolamine. | |||||
| Crystalline g-C3N4 nanosheets | 300 W Xe lamp (λ > 420 nm) | 25 mg catalyst, TEOAg solution, Pt cocatalyst | 20.9 mmol h−1 g−1 | 73.6% at 420 nm | 44 |
| Single-crystalline PTIa nanosheets | 300 W Xe lamp (λ > 300 nm) | 100 mg catalyst, pure water, CoOx and PtCrOx cocatalysts | 6100 µmol h−1 g−1 | 25% at 365 nm | 45 |
| CN-TMb | 300 W Xe lamp (λ > 420 nm) | 50 mg catalyst, TEOA solution, Pt cocatalyst | 3135 µmol h−1 g−1 | 21.03% at 420 nm | 47 |
| Prism-like PTI | 300 W Xe lamp (λ >300 nm) | 100 mg catalyst, pure water, Pt/Co cocatalysts | 1890 µmol h−1 g−1 | 8% at 365 nm | 48 |
| g-C3N4 nanosheets | 300 W Xe lamp (λ > 400 nm) | 50 mg catalyst, TEOA solution, Pt cocatalyst | 10.14 mmol h−1 g−1 | 7.34% at 400 nm | 49 |
| g-C3N4 nanosheets | 300 W Xe lamp (λ > 400 nm) | 20 mg catalyst, TEOA solution, Pt cocatalyst | 2590 µmol h−1 g−1 | 10.4% at 400 nm | 50 |
| g-C3N4 nanorods | 300 W Xe lamp (λ > 420 nm) | 10 mg catalyst, TEOA solution, Pt cocatalyst | 578.5 µmol h−1 g−1 | — | 54 |
| g-C3N4 nanorods | 300 W Xe lamp (λ > 420 nm) | 25 mg catalyst, TEOA solution, Pt cocatalyst | 732 µmol h−1 g−1 | 7.1% at 420 nm | 56 |
| g-C3N4 nanotubes | 300 W Xe lamp (λ > 420 nm) | 20 mg catalyst, TEOA solution, Pt cocatalyst | 4779.8 µmol h−1 g−1 | — | 60 |
| g-C3N4 tubes | 350 W Xe lamp | 30 mg catalyst, TEOA solution, Ni2P cocatalyst | 19.25 mmol h−1 g−1 | 5.8% at 400 nm | 62 |
| C3N4-TDAc | 300 W Xe lamp (λ > 420 nm) | 20 mg catalyst, TEOA solution, Pt cocatalyst | 12.35 mmol h−1 g−1 | 40.5% at 400 nm | 69 |
| g-C2N3 | 350 W Xe lamp | 50 mg catalyst, Na2S/Na2SO3 solution | 14.9 mmol h−1 g−1 | 19.9% at 420 nm | 70 |
| Ard-C3N4 | Xe lamp AM1.5 (λ > 420 nm) | 50 mg catalyst, TEOA solution, Pt cocatalyst | 10.8 mmol h−1 g−1 | 10.35% at 420 nm | 71 |
| CN-ATZ-NaKe | 50 W LED (λ > 420 nm) | 50 mg catalyst, TEOA and K2HPO4 solution, Pt cocatalyst | 13 mmol h−1 g−1 | 65% at 420 nm | 73 |
| PHI-Af | 300 W Xe lamp | 50 mg catalyst, TEOA solution, Pt cocatalyst | 4240 µmol h−1 g−1 | 12% at 420 nm | 74 |
| Li-g-C3N4 | 350 W Xe lamp | 30 mg catalyst, TEOA solution | 14 mmol h−1 g−1 | 22.9% at 420 nm | 78 |
| In-g-C3N4 | 300 W Xe lamp (λ > 420 nm) | 100 mg catalyst, TEOA solution, Pt cocatalyst | 1.35 mmol h−1 g−1 | — | 80 |
| Cu-g-C3N4 | 300 W Xe lamp (λ > 420 nm) | 20 mg catalyst, methanol solution, Pt cocatalyst | 10.6 mmol h−1 g−1 | 9.2% at 420 nm | 81 |
2.1. Crystal structure regulation
g-C3N4 is typically synthesized through the traditional thermal polymerization of organic compounds, such as melamine, dicyandiamide, urea, thiourea, etc.38 The simple one-step polymerization method often causes incomplete polycondensation, a low degree of polymerization, and poor crystallinity of g-C3N4, which bring about many internal and surface defects, thereby increasing the charge recombination centers and resulting in poor photocatalytic activity (Fig. 2).39,40 Therefore, it is necessary to prepare highly crystalline g-C3N4 with less defects for enhanced charge separation efficiency and efficient photocatalytic reaction.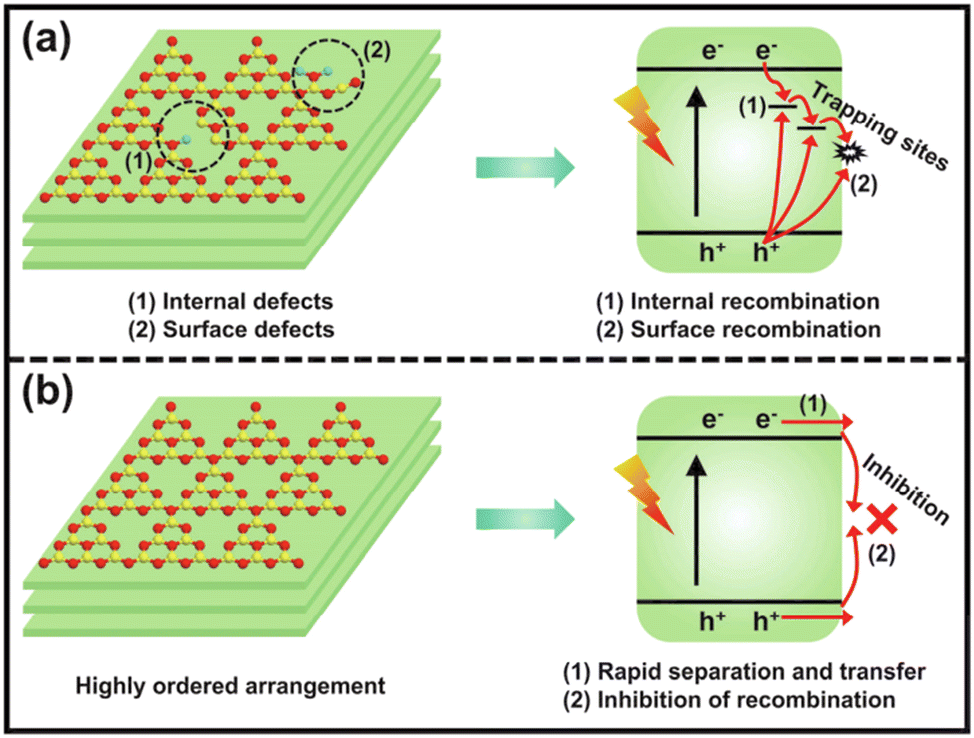 | ||
| Fig. 2 The charge separation and transfer of conventional poorly-crystalline g-C3N4 (a) and highly ordered g-C3N4 (b). Reproduced with permission from ref. 39. Copyright 2023, Elsevier. | ||
Many strategies have been developed to promote the polymerization of precursors and synthesize the highly crystalline g-C3N4, including salt-assisted methods, microwave synthesis, two-step calcination, etc.41–43 For example, Song et al.44 first prepared ultrathin g-C3N4 nanosheets by air-assisted thermal exfoliation, then the resulting nanosheets were treated in the NaCl/KCl molten salts to obtain the highly crystalline g-C3N4 nanosheets based on the heptazine–triazine units (donor–acceptor motifs, D–A). The high crystallinity and formed D–A structure promoted charge separation and transfer, resulting in a much higher H2 evolution activity of 20.9 mmol h−1 g−1 with an AQE of 73.6% at 420 nm compared with the original g-C3N4 nanosheets (1.0 mmol h−1 g−1). Wang and co-workers synthesized a single-crystalline carbon nitride, poly(triazine imide) nanosheets (PTI), through ternary molten salt treatment (LiCl/KCl/NaCl).45 Compared to binary molten salt systems (LiCl/KCl or NaCl/LiCl), the ternary eutectic salts could provide a more mild polymerization environment with higher melting points, which was beneficial to the in-plane polymerization and extension of the conjugated structure, leading to the formation of single-crystalline PTI nanosheets with less structure defects and a shorter charge migration distance. Thus, the obtained PTI nanosheets exhibited a high photocatalytic activity with an AQE of 25% at 365 nm for overall water splitting. In addition, compared with the traditional calcination process, the microwave method can quickly increase the temperature and trigger strong collisions between the reactants, thereby promoting the polymerization reaction and resulting in highly crystalline and active g-C3N4.46 Chen and Yang et al.47 combined the molten-salt strategy and microwave method to prepare highly active triazine-heptazine-based g-C3N4 with a H2 evolution rate of 3135 µmol h−1 g−1 (AQE of 21.03% at 420 nm), which was obviously higher than that of g-C3N4 treated with traditional high-temperature calcination (2591 µmol h−1 g−1).
Apart from crystallinity regulation, recently, Wang and co-workers investigated the influence of crystal planes on photocatalytic water splitting.48 They prepared the highly crystalline polytriazine imides-based carbon nitride (PTI) with a prism-like shape and different facets via the KCl/LiCl molten salt-assisted method. The results of in-situ photodeposition of the Pt cocatalyst demonstrated that the prismatic planes (1010) were the main reactive facets for catalytic reaction, as the Pt cocatalyst tended to accumulate on the (1010) planes. Furthermore, the calculation results also indicated that the electrons could easily transfer toward the prismatic (1010) planes. Based on this discovery, PTI catalysts with different surface area ratios of the (1010) planes were designed, and it was found that the photocatalytic activity increased with the increasing surface area ratio of the (1010) planes, and the optimal AQE of overall water splitting reached 8% at 365 nm with a H2 evolution rate of 1890 µmol h−1 g−1. Thus, the development of the crystalline g-C3N4-based system with less internal defects and more reactive facets exposed can improve charge separation efficiency and achieve the desired photocatalytic activity.
2.2. Morphological tuning
For the severe charge recombination in bulk g-C3N4, the design of ultrathin g-C3N4 nanosheets with expanded layer spacing is an effective method to weaken or break the shackle of interlayer electrostatic barriers and promote electron transfer from the bulk to the surface. For example, Yang and Kong et al.49 prepared a series of porous g-C3N4 nanosheets with different thicknesses (2–20 nm) through a thermal-triggering in-situ gas-shocking method. The most exfoliated g-C3N4 nanosheets presented an ultrathin structure with only 2 nm thickness, resulting in a larger interlayer spacing. The results showed that the ultrathin g-C3N4 nanosheets (∼2 nm) exhibited the highest photocatalytic activity with a H2 production rate up to 10.14 mmol h−1 g−1 (AQE of 7.34% at 400 nm), which was about 57 times that of bulk g-C3N4. The obviously enhanced photocatalytic activity was attributed to the fact that the ultrathin structure not only weakens the interlayer electrostatic barrier and promotes charge transfer to the surface, but also shortens the charge migration distance, thereby achieving a high charge separation efficiency. Shi et al.50 also reported the significantly improved photocatalytic H2 production activity over g-C3N4 nanosheets. They developed an available bottom-up acidification method to prepare the ultrathin g-C3N4 nanosheets with an average thickness of ∼3 nm (Fig. 3a–d). As a result, the as-prepared g-C3N4 nanosheets not only showed a high AQE at 400 nm (10.4%) and 420 nm (8.4%), but also exhibited an enhanced photocatalytic H2 evolution rate of 2590 µmol h−1 g−1, which was nearly 10-fold higher than that of bulk g-C3N4. The combined characterizations of electrochemical impedance spectra (EIS), photocurrent responses, and time-resolved PL spectra clearly demonstrated that the excellent photocatalytic activity of g-C3N4 nanosheets was due to the improved charge transfer and separation as well as the prolonged charge lifetime.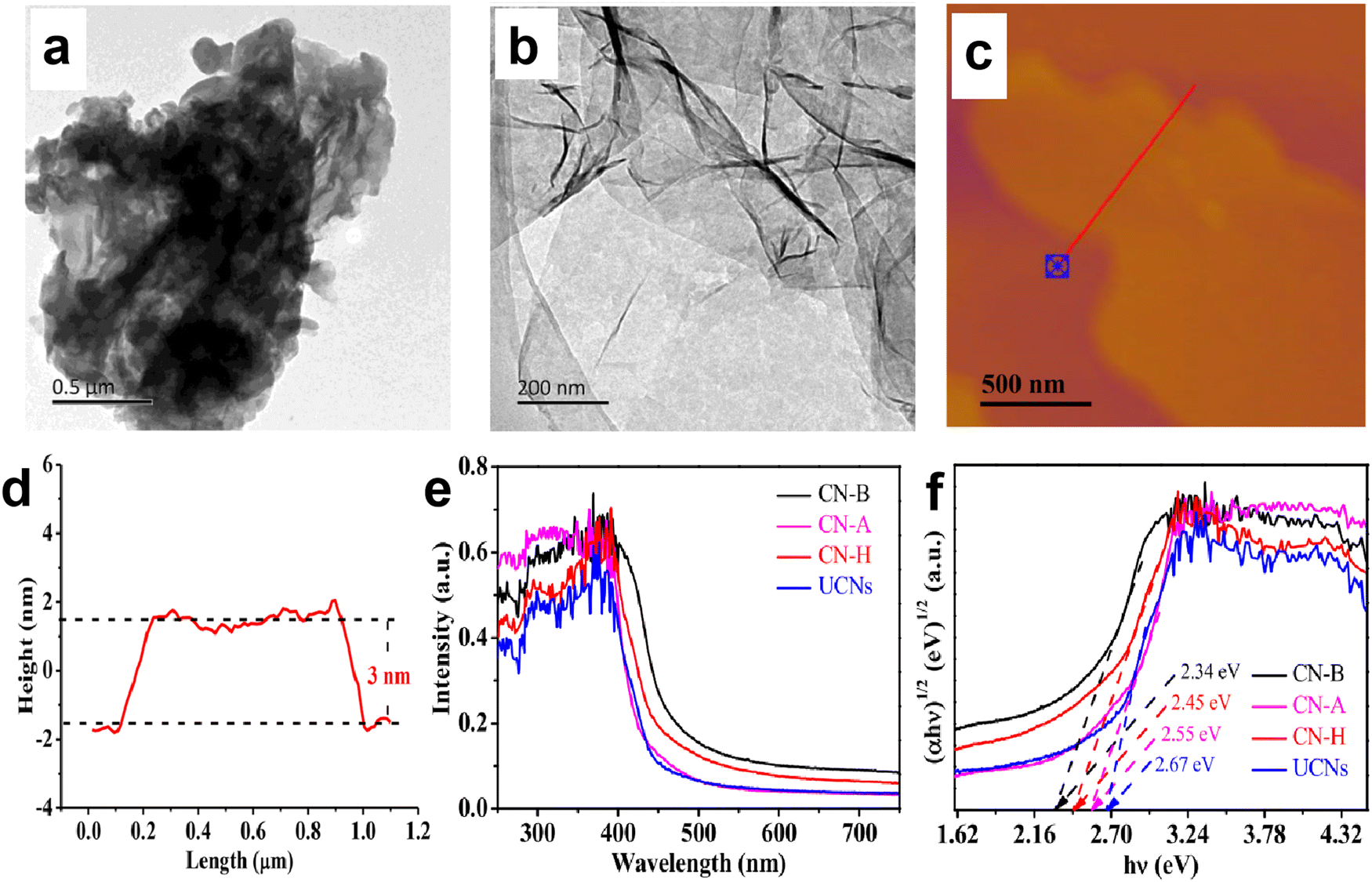 | ||
| Fig. 3 TEM images of bulk g-C3N4 (a) and ultrathin g-C3N4 nanosheets (b). AFM image of g-C3N4 nanosheets (c) and the corresponding height curves determined along the red line (d). UV-vis absorption spectra (e) and Tauc plots of the as-prepared g-C3N4 materials (f), where CN-B and UCNs correspond to bulk g-C3N4 and ultrathin g-C3N4 nanosheets, respectively. Reproduced with permission from ref. 50. Copyright 2020, Elsevier. | ||
Although the nanosheet structure can effectively reduce the charge recombination in the bulk phase of g-C3N4, and shorten the charge migration path to facilitate the subsequent catalytic reaction, the quantum confinement effect of nanoscale g-C3N4 can lead to an increase in its bandgap, which weakens the visible light response ability of g-C3N4 and limits the improvement of photocatalytic performance.50–52 For instance, the light absorption edges of the g-C3N4 nanosheets prepared by Shi et al.50 displayed a blue shift (Fig. 3e and f), implying the decreased light absorption range and enlarged bandgap of nanoscale g-C3N4. The corresponding Tauc plots clearly showed that the bandgap of the nanosheet structure (2.67 eV) was larger than that of the bulk structure (2.34 eV).
In addition to 2D nanosheet structures, 1D g-C3N4 nanorods or nanotubes have also received extensive attention due to their structural advantages in facilitating charge transfer along the axis.53–55 Zhu's group developed a network structure assembled by g-C3N4 nanorods for H2 evolution and phenol degradation.54 The higher intensity of photocurrent response and smaller arc radius in the Nyquist plots of the g-C3N4 nanorod network determined the obviously enhanced charge separation and transfer efficiency. Thus, compared to bulk g-C3N4, the g-C3N4 nanorod system displayed higher photocatalytic activity with a H2 evolution rate of 578.5 µmol h−1 g−1, a nearly six-fold increase. Liu et al.56 prepared the oxygen doped porous g-C3N4 nanorods, and found that the obtained g-C3N4 nanorods with Pt loading displayed an excellent photocatalytic H2 evolution rate of 732 µmol h−1 g−1, nearly 6 times higher than the bulk g-C3N4 system. Furthermore, an AQE of 7.1% was achieved at 420 nm in TEOA solution, 0.29% in pure water. Compared with g-C3N4 nanorods, the nanotubes not only have the advantages of a 1D structure, but also promote the directional migration of photo-induced electrons from concave to convex due to the formation of apparent potential difference between the inner and outer surface of the tubular structure.57–59 Meanwhile, the hollow tubular structure can enhance light scattering and improve the light response of g-C3N4 nanotubes.60,61 For example, Song et al.62 prepared g-C3N4 square tubes and loaded Ni2P as a cocatalyst on the surface of the g-C3N4 tubes (Fig. 4). The calculation and characterization results confirmed the directional electron transfer from the inner to the outer surface of the g-C3N4 tubes, and the increased light response range. Thus, the photocatalytic H2 evolution activity of the Ni2P loaded g-C3N4 tube was much higher than that of pristine g-C3N4 supported with noble metals.
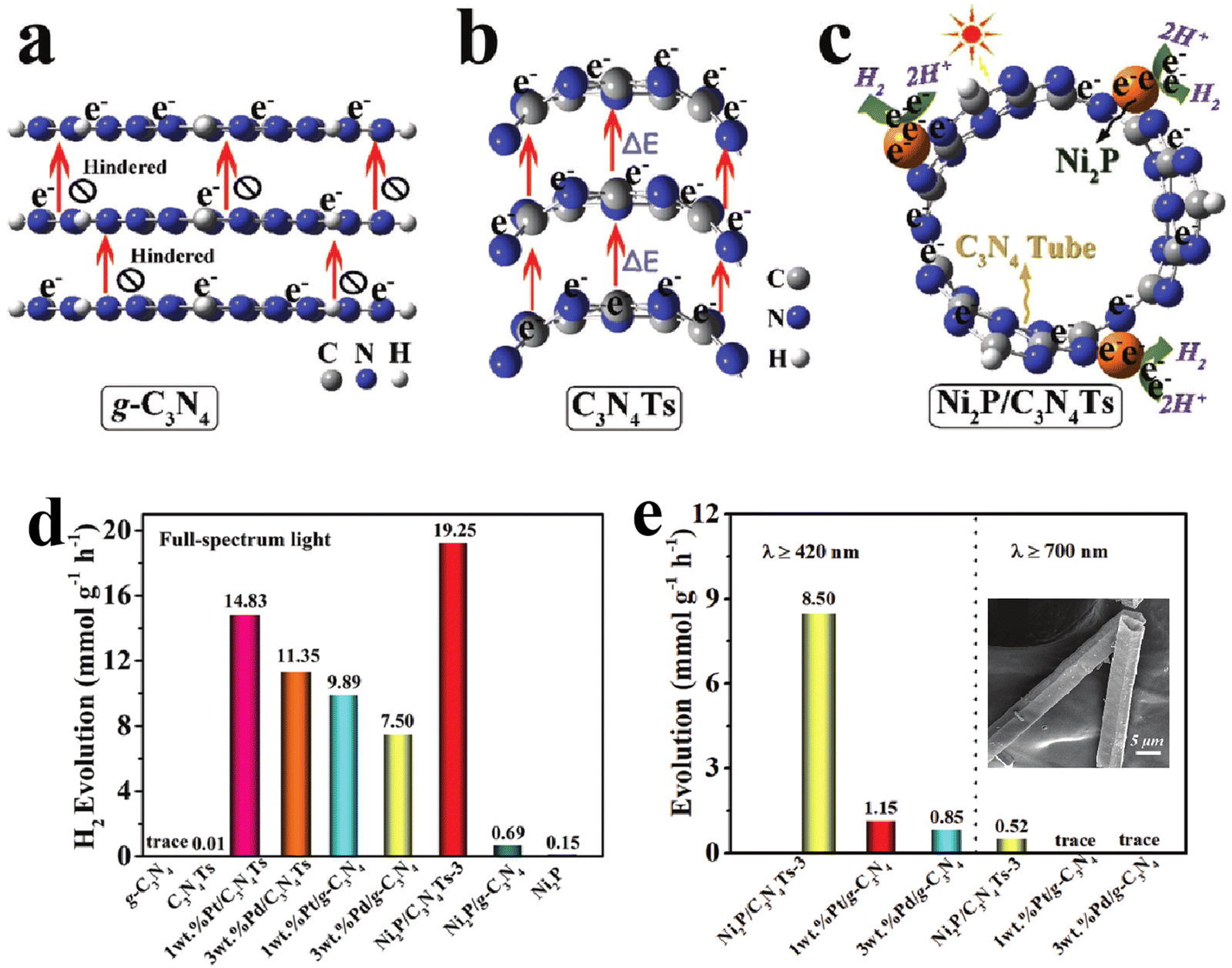 | ||
| Fig. 4 Schematic illustration of spatial separation and utilization of photo-induced electrons in bulk g-C3N4 (a), g-C3N4 tube (b), and Ni2P/g-C3N4 tube (c); photocatalytic H2 evolution activities at different wavelengths for bulk g-C3N4 and g-C3N4 tube systems (d) and (e); the inset of (e) is a FESEM image of the g-C3N4 tube. Reproduced with permission from ref. 42. Copyright 2020, Wiley-VCH. | ||
2.3. Doping engineering
Doping engineering is another general strategy for regulating the intrinsic structure of g-C3N4. The incorporation of heteroatoms in g-C3N4 or grafting other delocalization groups would damage the planar π-conjugated structure and lead to differentiation of delocalized π-electron distribution, thereby promoting the directional transfer of photogenerated charges along the potential difference.63,64 Based on the typical 2D stacking structure of g-C3N4, there are two main doping configurations, in-plane doping and interlayer doping.It is known that the high geometrical symmetry of the planar tri-s-triazine structure of g-C3N4 brings about the uniform π-electron distribution,65 which results in random charge transfer within the plane. Thus, grafting g-C3N4 with diverse delocalization groups by replacing the tri-s-triazine structures or triazine hexagons can induce the in-plane π-electron redistribution and the formation of the intrinsic polarization electric field to favor directional transfer of photo-induced charge carriers. The widely investigated grafting groups include aromatic motifs, carbon nitride allotropes, and C-chains.66–68 Xu et al.69 constructed a D-A system by incorporating thiophene-2,5-dicarbaldehyde (TDA) into the g-C3N4 network, and found that the modified g-C3N4 displayed an obviously improved photocatalytic performance for bisphenol A degradation and H2 evolution from water-splitting. The experimental and calculation results (Fig. 5) showed that the grafted TDA molecule could act as an electron acceptor to promote intermolecular charge transfer, and the incorporated aromatic ring from TDA enhanced the π-delocalization as well as the light response, which were responsible for the remarkable improvement of photocatalytic activity. Song et al.70 designed an allotrope of g-C3N4, g-C2N3, where the aromatic azide pentagons were incorporated into the triazine hexagons. The theoretical simulations indicated that the positive and negative charge centers demonstrated significant dislocation, which resulted in the formation of the intrinsic polarization electric field to break the Coulomb interaction of electron–hole pairs. Consequently, the designed g-C2N3 exhibited a very high photocatalytic H2 evolution rate of 14.9 mmol h−1 g−1 without any cocatalysts (AQE = 19.9% at 420 nm), which was even 2.6 times that of the g-C3N4 loaded with Pt. Moreover, Li and Wang et al.71 reported a gas–solid grafting method to prepare an asymmetric structure of g-C3N4 with aromatized terminals. The research demonstrated that the aromatization of the terminals enhanced the asymmetry of π-electron distribution, thereby providing a driving force to achieve the efficient charge transport and separation and improving the photocatalytic activity by 15.4 fold. Recently, Zhang and Wang et al.72 developed the ionothermal method via a molten salt mixture to provide a mild liquid environment for polymerization, then obtained the well-organized and highly active carbon nitride. Based on this, they reported the copolymerization of 5-amino-tetrazole with nucleobases in salt melts, and synthesized the allotropes of g-C3N4, poly-heptazine-imide (PHI) based carbon nitrides.73 The incorporation of nucleobases induced the formation of the D–A structure, which greatly promoted charge transfer and separation, thus, the PHI-based carbon nitrides showed higher H2 evolution activity than the melon-based counterparts. Then they further combined the copolymerization and ionothermal method to polymerize urea with a thiophene derivative (2-aminothiophene-3-carbonitrile) and obtained the condensed allotrope of g-C3N4 with separated positive and negative charge centers, which displayed enhanced light absorption and charge separation efficiency. The optimal photocatalytic H2 evolution activity was about 212.3 µmol h−1 with an AQE of 12% at 420 nm, much higher than the pure urea-derived counterpart (81.8 µmol h−1).74
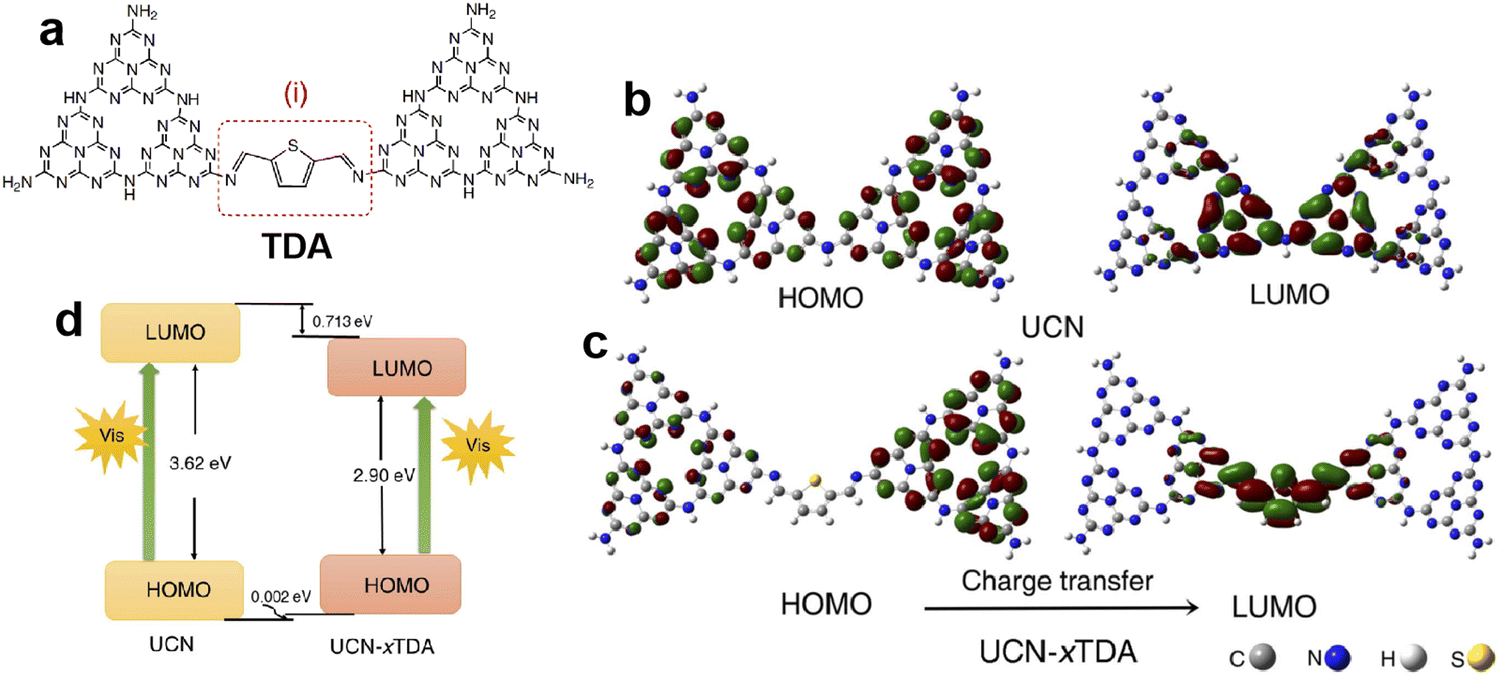 | ||
| Fig. 5 The possible structure of TDA grafted g-C3N4 (a); the electronic structures of the HOMO and LUMO for (b) g-C3N4 derived from urea (UCN) and (c) TDA grafted g-C3N4 (UCN-xTDA); (d) the HOMO and LUMO levels for UCN and UCN-xTDA. Reproduced with permission from ref. 69. Copyright 2022, Royal Society of Chemistry. | ||
In addition to the modification of molecules or functional groups, the incorporation of metals into the cave structure among heptazine rings is also frequently applied to regulate the electronic structure of g-C3N4. Currently, the reported doping metals mainly include transition metals, alkali metals, alkaline earth metals, and rare earth metals.75–77 According to coordination and orbital theory, the lone electron pairs of sp2-hybridized N atoms at the cave site can couple with the unoccupied orbital of metals, which can induce the in-plane π-electron redistribution and promote charge directional transfer. For instance, Song et al.78 incorporated Li ions into the cave sites of g-C3N4 to change the uniform π-electron distribution (Fig. 6a and b). The mülliken charge results showed that the electrons preferentially concentrated around Li and C atoms at the triangular cave, implying the formation of an in-plane coupling electric field, which favored charge directional transfer. As expected, the Li doped g-C3N4 exhibited a much lower charge recombination rate and higher H2 evolution activity of 0.418 mmol h−1 (∼14000 µmol h−1 g−1, AQE = 22.9% at 420 nm) compared with pristine g-C3N4 (0.009 mmol h−1).
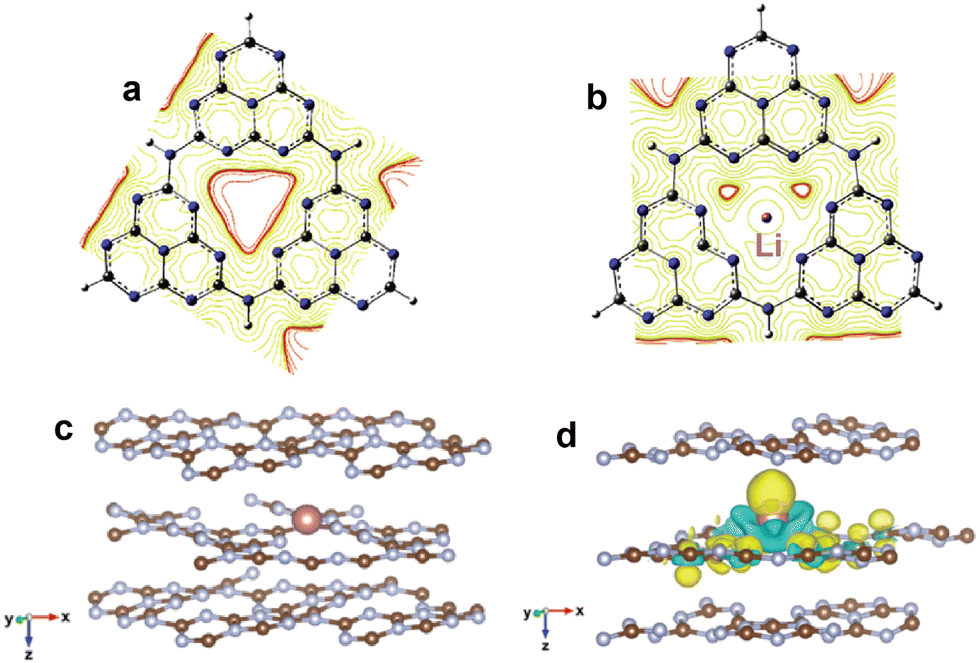 | ||
| Fig. 6 Equipotential curve distribution on g-C3N4 (a) and Li doped g-C3N4 (b). Reproduced with permission from ref. 78. Copyright 2020, Elsevier. The optimized structure of g-C3N4 with interlayer In doping (c); differential charge density of In doped g-C3N4 (d), where the yellow and green isosurfaces represent the gain and loss of electrons, respectively. Reproduced with permission from ref. 80. Copyright 2021, Royal Society of Chemistry. | ||
It is noted that the size of the triangular cave among the tri-s-triazine units of g-C3N4 is about 0.47 nm,79 which is suitable for accommodating metals with smaller atomic diameters, such as Li and Na, etc. The large-sized alkali/alkaline earth metals, such as K, Rb, and Cs, would be incorporated into the interlayers of g-C3N4. Meanwhile, the transition metals with different shapes and directions of d orbitals can also be doped between layers instead of the triangular cave, due to the exclusion between the complex d orbitals and N 2pz orbital at the cave site. Thus, the electronic interaction between the interlayer metals and the layers would induce the formation of an interlayer charge transport channel, which favors charge transfer among the layers of g-C3N4. Yang et al.80 reported a series of In3+ doped g-C3N4via a facile in-situ thermal copolymerization approach, and found that the In3+ ions were more easily doped into the interlayer of g-C3N4 due to the lower calculated energy of the out-of-plane configuration (Fig. 6c and d). The differential charge density result showed an obvious difference in electron density between doped In and g-C3N4, which facilitated the charge migration in g-C3N4. Therefore, the optimal In3+ doped g-C3N4 exhibited an excellent photocatalytic H2 evolution activity of 1.35 mmol h−1 g−1, 17 fold that of pristine g-C3N4. Fu et al.81 incorporated g-C3N4 with single-atom Cu, which was bonded with compositional N in two ways, in-plane bonding and interlayer bonding. The characterization results demonstrated that two groups of Cu-Nx as different charge transfer channels could promote the in-plane and interlayer charge transport, respectively, thereby greatly improving photocatalytic performance. Consequently, the Cu decorated g-C3N4 displayed a robust H2 production rate of 10.6 mmol h−1 g−1 (AQE = 9.2% at 420 nm), which was about 30 times higher than that of bulk g-C3N4.
3. Heterojunction construction
With regard to structural modulation of g-C3N4, e.g. the morphology and doping engineering discussed above, a driving force based on the structural potential difference is introduced to fight against the Coulomb interaction rather than directly weakening the Coulomb field strength. In fact, it is also considered as an efficient strategy to promote the dissociation of excitons (electron–hole pairs) into free charge carriers through weakening the Coulomb effect. According to the Coulomb field equation, Fc = kqeqh/r2, the Coulombic attraction of electron–hole pairs decreases with increasing the bandgap of semiconductor photocatalysts (i.e., increase in r value).82 Meanwhile, the semiconductors with large bandgap usually possess higher CB and lower VB positions, which facilitate the redox reactions. Thus, the wide bandgap property of photocatalysts is desired for weakening the Coulomb field strength and enhancing the redox ability, thereby improving the photocatalytic efficiency. However, a wide bandgap also leads to a weakened light response, and reduces the generation of photo-induced charges. Therefore, it is incompatible for single-component photocatalysts to possess a weakened Coulomb effect, sufficient redox capability, and large light-harvesting range, noticeably which are expected to be achieved by designing heterojunction photocatalysts.For heterojunction systems, they are typically composed of two semiconductor materials with different band structures, which can be categorized as a straddling gap heterojunction (type I), broken gap heterojunction (type III), and staggered gap heterojunction (type II and Z-scheme).83,84 In type I heterojunctions (Fig. 7a), the two semiconductors exhibit nested band alignment. The photo-induced electrons and holes on semiconductor A migrate to semiconductor B, resulting in charge accumulation on semiconductor B, which in turn triggers the high recombination of electron–hole pairs. For type III heterojunctions (Fig. 7b), a broken gap alignment is observed, and the photogenerated electrons and holes cannot migrate or transfer to any semiconductor, which also leads to a high charge recombination rate. As for type II heterojunctions (Fig. 7c), the staggered gap between two semiconductors facilitates the spatial separation of electron–hole pairs. As we see, semiconductor A possesses higher CB and VB levels, its photo-induced electrons can transfer to the CB of semiconductor B, while the holes on the VB transfer in the opposite direction. Thus, the transferred electrons and holes accumulate on semiconductor B and semiconductor A, respectively, which realizes the spatial separation of charges. However, in type II heterojunctions, the original electrons and holes would repel the transferred charge carriers, and the work function difference of semiconductors also influences charge transfer, thus, a Z-scheme heterojunction with a band structure similar to type II heterojunctions is proposed as well (Fig. 7d). Typically, a built-in electric field (BIEF) is formed at the interface of semiconductors due to the work function difference, then the useless photogenerated electrons and holes recombine driven by the BIEF.85 In this way, the remaining electrons and holes are left on semiconductor A and semiconductor B, respectively, achieving the effective separation of charge carriers and favorable redox capability for reaction. In this section, we focus on reviewing the charge separation and transfer processes in two heterojunction systems, Type II heterojunction and Z-scheme heterojunction. A comparison of the photocatalytic H2 evolution activity of recent g-C3N4-based heterojunctions is given in Table 2.
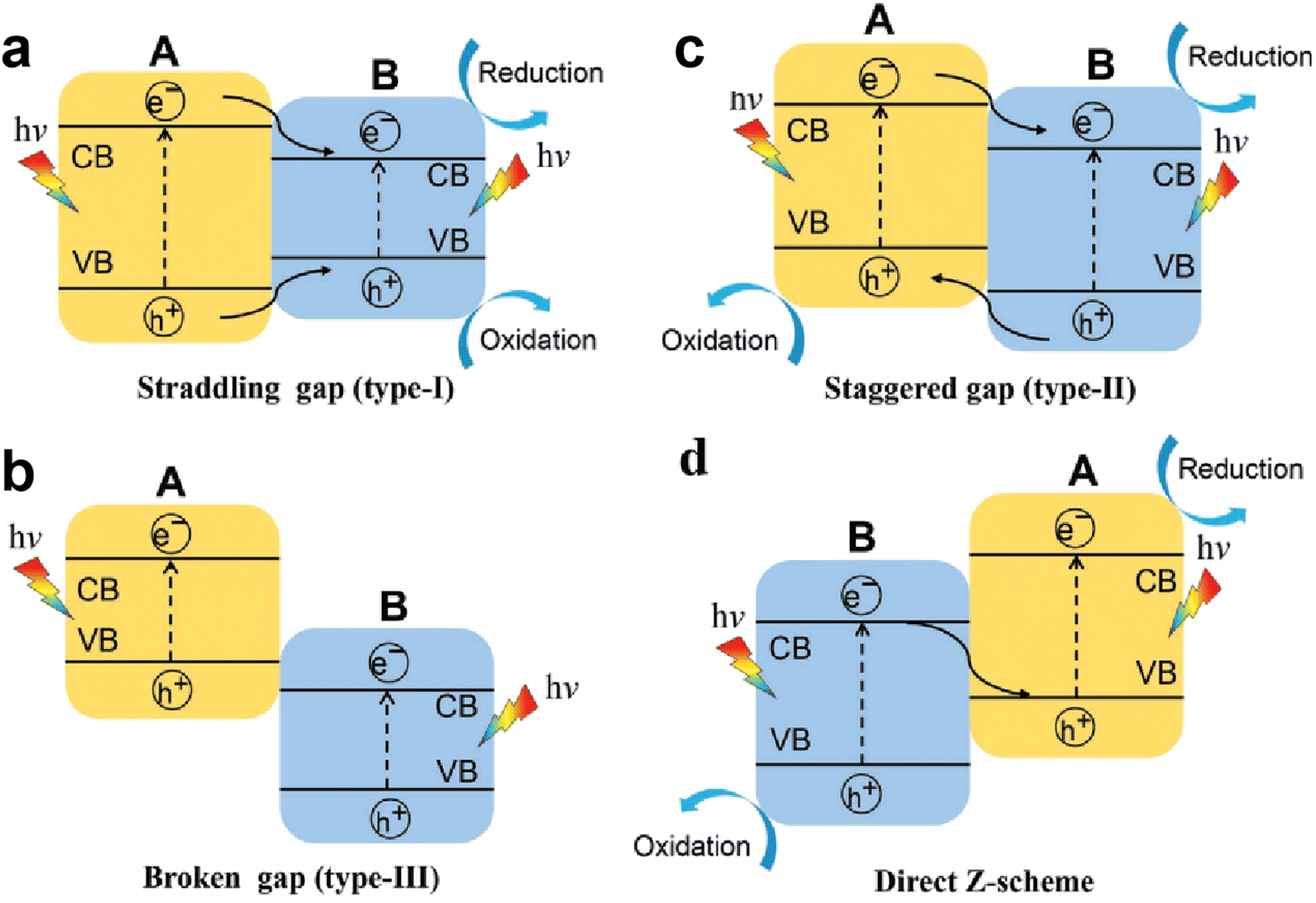 | ||
| Fig. 7 Schematic illustration of the transfer paths of photo-induced charge carriers in different heterojunction systems: (a) straddling gap heterojunction (type I), (b) broken gap heterojunction (type III), and (c) and (d) staggered gap heterojunction (type II and Z-scheme). Reproduced with permission from ref. 83. Copyright 2020, Wiley-VCH. | ||
| Photocatalyst | Type | Light source | Reaction conditions | H2 evolution rate | AQE | Ref. |
|---|---|---|---|---|---|---|
| a Triethylamine. b Boron-doped and nitrogen-deficient g-C3N4 nanosheets. c Triazine-heptazine-based carbon nitride. d Poly(heptazine–triazine) imides. e BST: C–N compound broken by s-triazine units. | ||||||
| CuFe2O4/g-C3N4 | Type II | 200 W Xe lamp | 20 mg catalyst, TEOA solution | 700.3 µmol h−1 g−1 | 25% at 450 nm | 88 |
| ZnIn2S4/g-C3N4 | Type II | 300 W Xe lamp (λ > 420 nm) | 50 mg catalyst, TEOA solution, Pt cocatalyst | 1.63 mmol h−1 g−1 | 0.90% at 420 nm | 89 |
| CoO/g-C3N4 | Type II | 300 W Xe lamp (λ > 420 nm) | 40 mg catalyst, TEAa solution | 263 µmol h−1 g−1 | 1.9% at 420 nm | 95 |
| CdSe/WS2/g-C3N4 | Type II | White LED | 5 mg catalyst, TEOA solution | 1.29 mmol h−1 g−1 | — | 101 |
| BaTiO3/Au/g-C3N4 | Z-scheme | 300 W Xe lamp (AM 1.5G) | 20 mg catalyst, TEOA solution | 1769.3 µmol h−1 g−1 | — | 107 |
| g-C3N4/W18O49 | Z-scheme | 300 W Xe lamp (λ > 400 nm) | 50 mg catalyst, TEOA solution, Pt cocatalyst | 5231 µmol h−1 g−1 | 23.1% at 420 nm | 116 |
| CoSeO3/g-C3N4 | Z-scheme | 300 W Xe lamp | 20 mg catalyst, TEOA solution | 1459.2 µmol h−1 g−1 | 39.9% at 584 nm | 117 |
| ZnIn2S4/g-C3N4 | Z-scheme | 300 W Xe lamp (λ > 400 nm) | 2 mg catalyst, TEOA solution | 14.8 mmol g−1 h−1 | 0.516% at 400 nm | 119 |
| g-C3N4/Co-MOF | Z-scheme | 225 W Xe lamp | 5 mg catalyst, ascorbic acid solution, Pt cocatalyst | 33.17 mmol h−1 g−1 | — | 120 |
| Ti3C2/TiO2/g-C3N4 | S-scheme | 300 W Xe lamp (λ > 420 nm) | 50 mg catalyst, TEOA solution Ti3C2 cocatalyst | 5540 µmol h−1 g−1 | 5.81% at 420 nm | 125 |
| CuInS2/g-C3N4 | S-scheme | 350 W Xe lamp (λ > 365 nm) | 20 mg catalyst, TEOA solution | 102.4 µmol h−1 g−1 | — | 126 |
| Mn0.2Cd0.8S/g-C3N4 | S-scheme | 300 W Xe lamp (λ > 420 nm) | TEOA solution, Pt cocatalyst | 11.42 mmol h−1 g−1 | — | 127 |
| CNN/BDCNNb | Homojunction | 300 W Xe lamp (λ > 300 nm) | 40 mg catalyst, pure water, Pt and Co(OH)2 cocatalyst | 823.5 µmol h−1 g−1 | 5.95% at 400 nm | 130 |
| S-C3N4/C3N4 | Homojunction | 300 W Xe lamp (λ > 420 nm) | 20 mg catalyst, TEOA solution, Pt cocatalyst | 5548.1 µmol h−1 g−1 | 0.43% at 420 nm | 131 |
| CN-NaKc (triazine-heptazine) | Homojunction | 50 W LED (λ > 420 nm) | 50 mg catalyst, TEOA solution, Pt cocatalyst | 11.7 mmol h−1 g−1 | 60% at 420 nm | 132 |
| PHI/PTId | Homojunction | 300 W Xe lamp (λ > 420 nm) | 50 mg catalyst, TEOA solution, Pt cocatalyst | 6970 µmol h−1 g−1 | 42% at 420 nm | 133 |
| g-C3N4/BSTe | Homojunction | 225 W Xe lamp | 5 mg catalyst, TEOA solution, Pt cocatalyst | 12.47 mmol h−1 g−1 | — | 134 |
3.1. Type II heterojunction
Among various g-C3N4 heterojunctions, type II g-C3N4 heterojunctions are the most common system. According to the charge transfer mechanism of the type II system,86,87 the CB and VB positions of g-C3N4 should be both higher or lower than those of another semiconductor in heterojunctions, thereby forming a staggered band structure that thermodynamically favors the subsequent charge transfer and realizes the spatial separation of electron–hole pairs. As a result, there are numerous studies about developing g-C3N4 heterojunctions with type II alignment to promote charge transfer during photocatalytic reaction, e.g., CuFe2O4/g-C3N4,88 ZnIn2S4/S-doped g-C3N4,89 g-C3N4/r-TiO2,90 CsCu2I3/g-C3N4,91 β-AsP/g-C3N4,92 Cu2O/g-C3N4,93 B-doped C3N4/ZnO,94 CoOx/g-C3N4,95 CeO2/g-C3N4,96 CdS/g-C3N4,97 ZnxCd1−xIn2S4/g-C3N4,98 g-C3N4/SnS2,99 g-C3N4/Zn-MOF,100 and CdSe/WS2/g-C3N4.101 Ahmad et al.88 developed a type II heterostructure of g-C3N4 combined with CuFe2O4, and found the CuFe2O4/g-C3N4 showed a 2.5-fold enhancement in photocatalytic activity and high AQE for H2 evolution (25% at 450 nm). The obviously improved photocatalytic activity was mainly attributed to the thermodynamically optimal band alignment that facilitated directional charge transfer and obtained spatially separated electrons (on g-C3N4) and holes (on CuFe2O4) for efficient catalytic reaction. Xie and Luo et al.89 utilized a simple in-situ hydrothermal method to construct S-doped g-C3N4 with ZnIn2S4 as a type II photocatalyst for splitting H2O into H2. Compared to single S-doped g-C3N4 and ZnIn2S4, the constructed heterojunction displayed a much higher photocatalytic H2 production performance of 1.63 mmol h−1 g−1 (AQE = 0.90% at 420 nm), which was respectively an 8-fold and 3-fold enhancement in photocatalytic activity for S-doped g-C3N4 and ZnIn2S4. Additionally, Jiang et al.95 reported a tunable heterojunction photocatalyst of cobalt oxide (CoOx) confined in g-C3N4 nanotubes, where the type II heterojunction of CoO/g-C3N4 and type I heterojunction of Co3O4/g-C3N4 were obtained. The results showed that the prepared two heterojunctions of CoOx/g-C3N4 both exhibited better photocatalytic performance than the CoOx and g-C3N4. And the CoO/g-C3N4 with type II band alignment exhibited higher photocatalytic H2 evolution activity than the Co3O4/g-C3N4 counterpart, which was mainly due to the longer lifetime of charge carriers, faster charge transfer, and higher charge separation efficiency in the type II heterojunction of CoO/g-C3N4. Recently, ternary heterojunctions with band coupling structures of type II/type II were designed to further accelerate the charge separation and transfer, and thus enhance the photocatalytic efficiency. For instance, a ternary hybrid photocatalyst of CdSe/WS2/g-C3N4 was prepared by Liang and Chen et al.101 According to the band structures of pristine CdSe, WS2, and g-C3N4, the type II/type II heterostructure was concluded to form at the interfaces of CdSe/WS2/g-C3N4, which could not only expand the light absorption range but also greatly promote charge separation and restrain recombination. As a result, the designed ternary CdSe/WS2/g-C3N4 photocatalyst displayed significantly enhanced photocatalytic performance, achieving the maximum H2 evolution rate of 1.29 mmol h−1 g−1, which was 1.7 and 1.3 times greater than binary WS2/g-C3N4 and CdSe/g-C3N4, respectively.As discussed above, the type II band alignment can indeed accelerate the migration and transfer of photogenerated charge carriers, and thus enhance the photocatalytic efficiency. However, as indicated in Fig. 7c, the spatial separation of charge carriers in type II heterojunctions is at the expense of their redox abilities when the electrons and holes are accumulated in more positive and negative energy levels, respectively, resulting in a weakened driving force for the following redox reaction.
3.2. Z-scheme heterojunction
Compared to type II heterojunctions, the Z-scheme heterojunction not only favors the spatial separation of charge carriers and inhibits charge recombination, but also retains a high catalytic activity with the photogenerated electrons and holes at higher chemical potentials of CB and VB, respectively.102 Typically, in a Z-scheme photocatalyst (Fig. 7d), the close contact of two semiconductors induces the recombination of ineffective charges at the interface, resulting in an enlarged distance for the active charge carriers, which effectively weakens the Coulombic attraction of electron–hole pairs and in turns achieves a high charge separation efficiency. Therefore, among different heterojunction photocatalysts, Z-scheme heterojunctions have received more attention and research due to their combined advantages of enhanced charge separation efficiency, weakened Coulomb field strength, and favorable redox ability.The first generation of Z-scheme heterojunctions based on mimicking the natural photosynthesis process was proposed by Bard in 1979,103 where the electron acceptor/donor ion pairs (A/D) were used as the redox mediators (Fig. 8a), e.g., I−/IO3−, Fe2+/Fe3+, and Co(bpy)32+/Co(bpy)33+.104,105 Although the introduction of the redox couples is beneficial for charge separation, it also brings several issues that the reversible redox couples would compete with the reactants for reaction and their application is limited to the liquid phase systems. In order to address the drawbacks of the conventional Z-scheme heterojunctions, an all-solid-state Z-scheme heterojunction was developed by Tada et al.,106 where the redox ion pairs were substituted by solid electron mediators (Fig. 8b), such as noble metals, carbon nanotubes/nanofibers, graphene, and other conductive materials. Li et al.107 constructed an all-solid-state Z-scheme photocatalyst by combining BaTiO3 and g-C3N4 as the main catalytic components, using Au nanoparticles as the electron mediator. The prepared Z-scheme BaTiO3/Au/g-C3N4 was able to show enhanced charge separation efficiency while retaining the sufficient redox capability for reaction. Compared to g-C3N4, the BaTiO3/Au/g-C3N4 exhibited higher photocatalytic activity toward H2 production and RhB degradation, particularly, an 18-fold enhancement in H2 evolution rate was achieved. Wang and Li et al.108 also designed an all-solid-state ternary Z-scheme photocatalyst by coupling g-C3N4, carbon dots (CDs) and TiO2, where the CDs were adopted as an electron mediator. They found that the g-C3N4/CDs/TiO2 exhibited a high H2 evolution activity of 580 µmol h−1 g−1, and surpassed the binary g-C3N4/TiO2 by 1.6 times, which was attributed to the effective spatial separation of charge carriers and their fast transfer assisted by the CDs mediator.
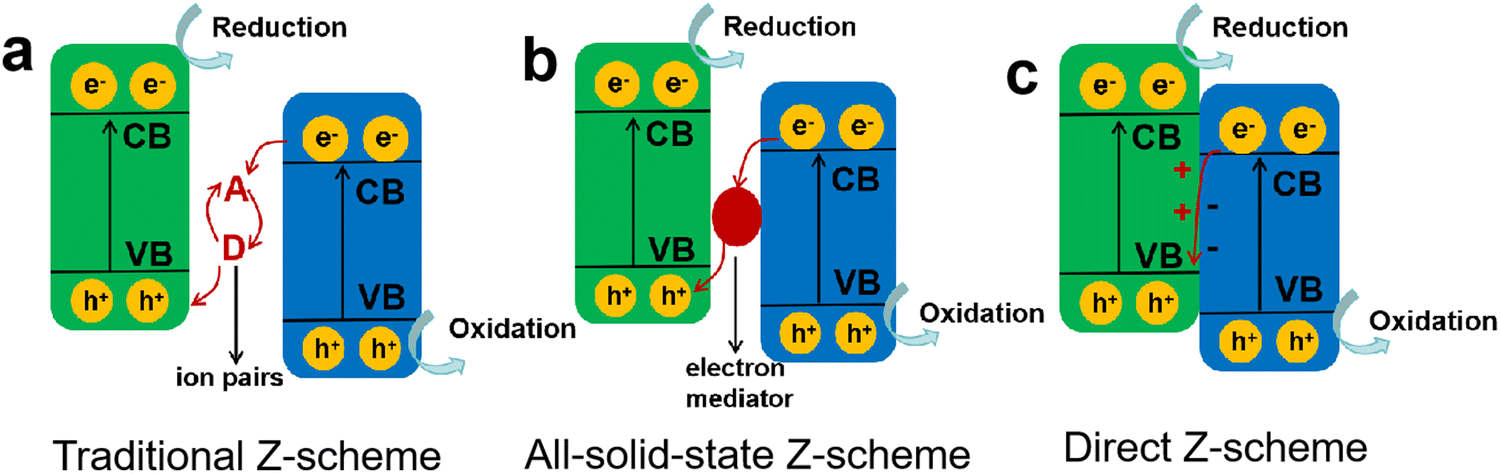 | ||
| Fig. 8 Different types of Z-scheme heterojunctions: (a) traditional Z-scheme, (b) all-solid-state Z-scheme, (c) direct Z-scheme. | ||
Obviously, as the second-generation Z-scheme heterojunctions, all-solid-state photocatalysts can effectively solve the problems of the ion pair-mediated Z-scheme photocatalytic system. However, the loading of solid electron mediators on the surface of semiconductors can trigger a shielding effect, reducing the light absorption and utilization, and the widely used noble metal mediators would also increase the costs and limit the wide application. Subsequently, a mediator-free Z-scheme heterojunction of ZnO/CdS was designed to further optimize the charge transfer process in photocatalytic reaction,109 and then Yu's group proposed the third-generation Z-scheme heterojunction without any mediators (Fig. 8c), namely the direct Z-scheme heterojunction or S-scheme heterojunction.110,111 In a direct Z-scheme photocatalytic system, the shortcomings of the above two Z-scheme heterojunctions can be overcome, such as the light-shielding effect induced by solid mediators, high cost caused by the loading of mediators, and the backward reactions due to the presence of redox ion pairs.
We can see that the band structure of the direct Z-scheme heterojunction is similar to that of type II heterojunctions, whereas the migration and transfer paths of charge carriers are different (Fig. 7c and 8c). In direct Z-scheme heterojunction photocatalysts, the charge reorganization would occur at the interface of two closely contacted semiconductors due to their differences in work function or Fermi level, that is, electrons can autonomously move from one semiconductor with a lower work function to another with a higher work function, forming a built-in electric field (BIEF) at the interface.112 Subsequently, under light irradiation, the BIEF drives the separation and transfer of photogenerated electrons and holes, resulting in the recombination of useless charge carriers and leaving the active charge carriers at more favorable redox potentials.113,114 Therefore, the direct Z-scheme heterojunction with a spontaneously formed BIEF at the interface exhibits a high charge separation efficiency and redox capability, while avoiding the defects of the first and second generation Z-scheme heterojunction photocatalysts (i.e., electron mediator-coupled systems).
Due to these advantages, the development of g-C3N4-based direct Z-scheme heterojunctions has received more attention than those traditional Z-scheme systems. Lately, numerous direct Z-scheme heterojunctions of g-C3N4-coupled composites with robust photocatalytic activities have been reported, such as g-C3N4/Bi4NbO8Cl,115 g-C3N4/W18O49,116 CoSeO3/g-C3N4,117 Bi4O5Br2/g-C3N4,118 ZnIn2S4/g-C3N4,119 g-C3N4/Co-MOF,120 CoS/g-C3N4,121 LaCoO3/g-C3N4,122 hBN/g-C3N4,123 g-C3N4/Bi2MoO6,124 Ti3C2/TiO2/g-C3N4,125 CuInS2/g-C3N4,126 and Mn0.2Cd0.8S/g-C3N4.127 Typically, Zou et al.116 constructed a C3N4/W18O49 composite and found that the Fermi level of g-C3N4 could be uplifted through the adsorption of triethanolamine (TEOA), thereby transforming the composite from type II to direct Z-scheme (Fig. 9a and b). The C3N4/W18O49 composite with efficient Z-scheme charge transfer exhibited much higher photocatalytic activity compared with pure g-C3N4 and type-II C3N4/W18O49, resulting in a H2 evolution rate of 5231 µmol h−1 g−1 and AQE of 23.1% at 420 nm (Fig. 9c). Dong and Cao et al.117 reported a novel CoSeO3 assisted g-C3N4 composite with a Z-scheme structure. After combining with CoSeO3, the CoSeO3/g-C3N4 displayed an expanded light absorption range, reduced charge transfer resistance and accelerated charge separation, and the optimal photocatalytic H2 evolution activity of CoSeO3/g-C3N4 was about 65 times that of pristine g-C3N4. Similarly, Dai and Zhang et al.127 designed an S-scheme heterojunction photocatalyst of Mn0.2Cd0.8S/g-C3N4 through an in-situ hydrothermal growth process. The characterization results showed that the Mn0.2Cd0.8S/g-C3N4 hybrid possessed an intimate interface and favorable S-scheme band alignment, which could promote the charge transfer at the interface and increase the lifetime of reactive charge carriers, as well as retain the high redox capacity. The photocatalytic H2 production activity of Mn0.2Cd0.8S/g-C3N4 reached 11.42 mmol h−1 g−1, which was much higher than that of the original g-C3N4 and Mn0.2Cd0.8S.
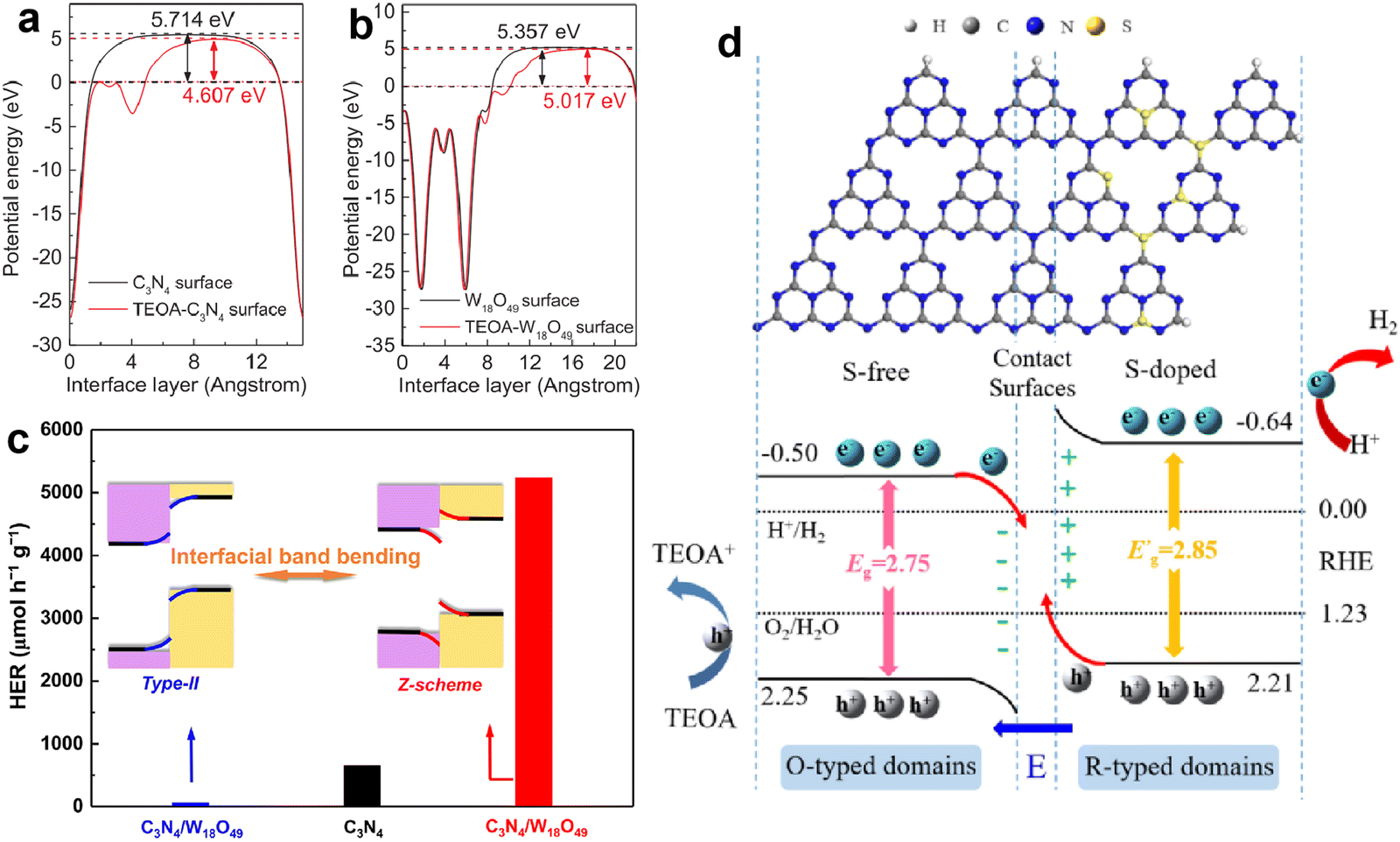 | ||
| Fig. 9 The calculated work functions of g-C3N4 (a) and W18O49 (b) surfaces with and without TEOA; photocatalytic H2 evolution activities of pure g-C3N4, type II g-C3N4/W18O49, and Z-scheme g-C3N4/W18O49 (c). Reproduced with permission from ref. 116. Copyright 2017, Elsevier. The structural configuration and charge transfer mechanism of S-doped g-C3N4/g-C3N4 isotype heterojunction (d). Reproduced with permission from ref. 131. Copyright 2022, Elsevier. | ||
Besides these, recently, the isotype heterojunctions of g-C3N4-based composites have also been discussed and investigated due to their better lattice matching and compatibility, which are beneficial to the separation and transfer of photo-induced charge carriers at the interface.128–131 Shen et al.130 prepared a series of g-C3N4 nanosheets with different contents of B dopants and N defects, resulting in the formation of g-C3N4 with controllable band structures, which could be applied as an O2-evolving component and combined with the original g-C3N4 as a H2-evolving component to construct an isotype Z-scheme heterojunction for overall water splitting. The obtained g-C3N4 self-based heterojunction displayed excellent photocatalytic activity, and the optimal H2 and O2 production rates achieved stoichiometric values of 32.94 and 16.42 µmol h−1 (AQE of 5.95% at 400 nm). The superior photocatalytic performance was attributed to the strong interfacial interaction, favorable redox ability, and efficient charge transfer through the interfacial Z-scheme route. Jiang et al.131 also reported a novel isotype heterostructure of S-doped g-C3N4/g-C3N4 with S-scheme band alignment (Fig. 9d), which exhibited high photocatalytic H2 evolution activity of 5548.1 µmol h−1 g−1 (AQE of 0.43% at 420 nm) and remarkable durability for 24 h, nearly 49 times that of pure g-C3N4. Zhang and Wang et al.132 prepared a triazine-heptazine based copolymer with the internal donor–acceptor heterostructure, which greatly accelerated the interface charge transfer and achieved high photocatalytic H2 evolution activity with an AQE of 60% at 420 nm. Afterwards, they also fabricated binary poly(heptazine–triazine) imides with semi-coherent interfaces. The fine lattice compatibility and intimate contact interface induced the formation of the built-in electric field, which facilitated rapid charge separation and transfer, resulting in a high AQE of 42% at 420 nm, even 64% by using K2HPO4 as the charge mediator.133 Additionally, Shi et al.134 also developed a g-C3N4 homojunction by decorating the broken s-triazine unit C–N compound, which was tightly anchored on the g-C3N4 and could extract photogenerated electrons from g-C3N4. Furthermore, the broken s-triazine units provided active sites to grab H+, thereby promoting the H+ reduction and achieving a fourfold improvement in H2 evolution activity.
4. Cocatalyst loading
In addition to the heterojunction of coupling two semiconductors, loading metal-based cocatalysts on the surface of photocatalysts can also greatly promote the separation and transfer of photo-induced charge carriers, and then improve photocatalytic H2 production activity, such as the reported Pt loaded g-C3N4, CdS and TiO2, and they all exhibited better photocatalytic performance than their original counterparts.135–137 The role of cocatalyst in improving photocatalytic activity is as follows:138 on the one hand, the cocatalyst can serve as an electron trap to capture photogenerated electrons from the CB level of the semiconductor photocatalyst and retain the holes on the semiconductor, which achieve spatial separation of charge carriers and effectively suppress the recombination; on the other, the cocatalyst can provide abundant catalytic sites and reduce the activation energy of the H2 evolution reaction. Fig. 10a shows a schematic diagram of the role of cocatalysts in photocatalytic water splitting. In order to achieve state-of-the-art photocatalytic activity, the cocatalyts should display suitable band alignment with the semiconductor, strong conductivity, and high catalytic activity toward H2 evolution.139,140 According to the requirements of cocatalysts, H2-evolution electrocatalysts with excellent catalytic activity are commonly considered as promising cocatalysts for photocatalytic H2 production.141–143 Most electrocatalysts have lower Fermi levels (larger work functions), which is conducive to capturing photogenerated electrons, and their high catalytic activity can also boost the kinetics of surface H2 evolution reaction. We previously prepared the VS2 electrocatalyst with excellent catalytic activity toward H2 evolution, and loaded it on g-C3N4 as a cocatalyst.27,144 Compared to pure g-C3N4, the VS2 supported g-C3N4 displayed much higher photocatalytic H2 evolution activity, a nearly 30 times increase. Our characterization and calculation results clearly revealed that the loading of VS2 obviously improved the charge separation efficiency and reduced the energy barrier for H2 evolution.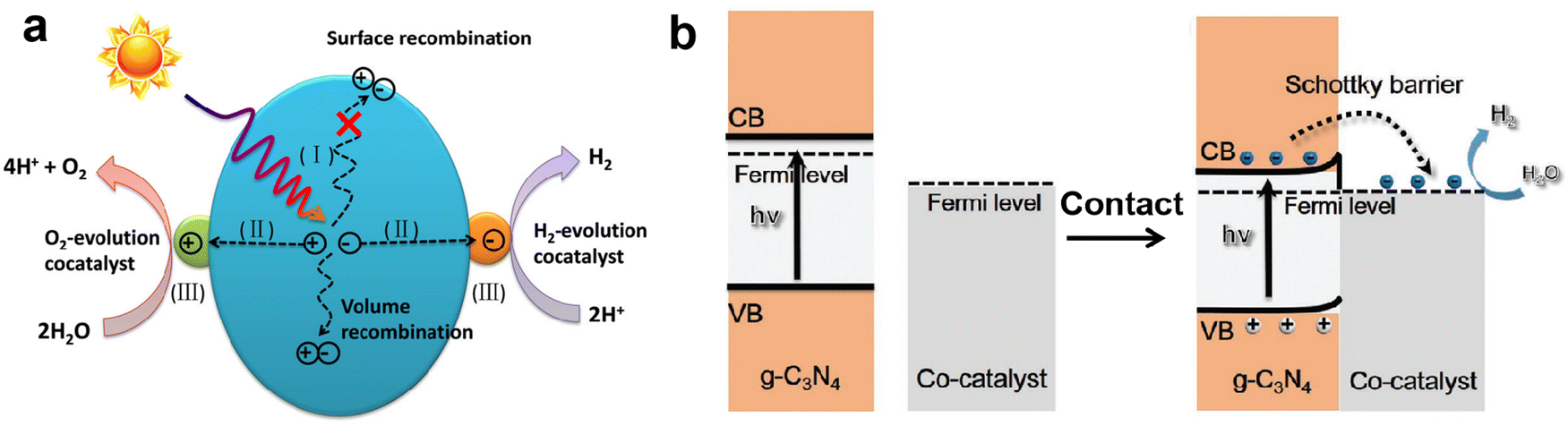 | ||
| Fig. 10 The role of cocatalyst in photocatalytic water splitting (a). Reproduced with permission from ref. 139. Copyright 2020, Wiley-VCH. The band alignment between metallic cocatalysts and n-type g-C3N4 after intimate contact (b). Reproduced with permission from ref. 140. Copyright 2021, Wiley-VCH. | ||
For the n-type g-C3N4 semiconductor, its Fermi level is close to the CB and generally higher than that of the metal-based cocatalysts, that is, the smaller work function of g-C3N4 (ϕg-C3N4 < ϕcocatalyst). When the g-C3N4 surface is in intimate contact with the cocatalyst, the interfacial electrons transfer and lead to the formation of a Helmholtz double layer at the cocatalyst-g-C3N4 junction, where the g-C3N4 is positively charged and the cocatalyst is negatively charged. The established electric field between g-C3N4 and cocatalyst causes the band edges of g-C3N4 to bend upwards, forming an energy barrier called the Schottky barrier.145,146 Although the presence of a Schottky barrier at the interface would affect the charge transfer, the photogenerated electrons with sufficient energy can still flow from g-C3N4 to the cocatalyst through the Schottky barrier. Meanwhile, the rectification characteristic of the Schottky barrier suppresses the back recombination of charge carriers, ensuring the directional charge transfer and improving charge separation efficiency.147,148 Therefore, a perfect match of g-C3N4 and cocatalyst and the appropriate Schottky barrier are necessary to facilitate the interfacial charge transfer, prolong the lifetime of charge carriers, and improve photocatalytic H2 production activity.
In this section, typical noble metal-based cocatalysts and transition metal-derived cocatalysts are discussed, with a focus on the role of cocatalysts in charge separation and transfer. A comparison of the photocatalytic H2 evolution activity of recent g-C3N4 loaded with different cocatalyst is given in Table 3.
| Photocatalyst | Cocatalyst | Light source | Reaction conditions | H2 evolution rate | AQE | Ref. |
|---|---|---|---|---|---|---|
| a PTI: poly(triazine imide) based carbon nitride. | ||||||
| Au/g-C3N4 | Au | 300 W Xe lamp (λ > 420 nm) | 20 mg catalyst, TEOA solution | 159.9 µmol h−1 g−1 | — | 154 |
| Au/g-C3N4 | Au nanospheres | 300 W Xe lamp (λ > 420 nm) | 20 mg catalyst, TEOA solution | 540 µmol h−1 g−1 | 1.2% at 380 nm | 156 |
| Rh/Cr2O3-CoOx/PTIa | Rh/Cr2O3-CoOx | 300 W Xe lamp | 100 mg catalyst, pure water | 4500 µmol h−1 g−1 | 20.2% at 365 nm | 159 |
| Pt-CoOx/PTI | Pt-CoOx | 300 W Xe lamp | 100 mg catalyst, pure water | 2730 µmol h−1 g−1 | 12% at 365 nm | 160 |
| TiO2@Pt/C3N4 | TiO2@Pt | 300 W Xe lamp | 100 mg catalyst, pure water | 7.63 µmol h−1 g−1 | 0.11% at 365 nm | 161 |
| PtAu/g-C3N4, | PtAu | 300 W Xe lamp | 50 mg catalyst, Na2S/Na2SO3 solution | 1009 µmol h−1 g−1 | 0.45% at 420 nm | 164 |
| PtNi/g-C3N4 | PtNi | 300 W Xe lamp (λ > 420 nm) | 25 mg catalyst, TEOA solution | 9528 µmol h−1 g−1 | 10.6% at 370 nm | 165 |
| Ni/TiO2/g-C3N4 | Ni | 300 W Xe lamp | 50 mg catalyst, TEOA solution | 134 µmol h−1 g−1 | 15% at 420 nm | 170 |
| NiMo/g-C3N4 | NiMo | 300 W Xe lamp (λ > 420 nm) | 20 mg catalyst, TEOA solution | 1785 µmol h−1 g−1 | ∼0.25% at 365 nm | 172 |
| NiCu/g-C3N4 | NiCu | 5 W LED | 10 mg catalyst, TEOA solution | 2088 µmol h−1 g−1 | 6.83% at 475 nm | 173 |
| Mo2C@C/g-C3N4 | Mo2C | 300 W Xe lamp (λ > 420 nm) | 80 mg catalyst, TEOA solution | 651 µmol h−1 g−1 | 1.44% at 400 nm | 178 |
| WC/g-C3N4 | WC | 300 W Xe lamp | 50 mg catalyst, TEOA solution | 146.1 µmol h−1 g−1 | — | 180 |
| Ni2P/rGO/g-C3N4 | Ni2P | 300 W Xe lamp (λ > 420 nm) | 10 mg catalyst, TEOA solution | 2922 µmol h−1 g−1 | 5.6% at 420 nm | 187 |
| Cu3P/g-C3N4 | Cu3P | 300 W Xe lamp (λ > 420 nm) | 20 mg catalyst, TEOA solution | 343 µmol h−1 g−1 | — | 188 |
| MoP/g-C3N4 | MoP | 300 W Xe lamp | 10 mg catalyst, TEOA solution | 3868 µmol h−1 g−1 | 21.6% at 405 nm | 189 |
| NiCoP/g-C3N4 | NiCoP | 300 W Xe lamp | 20 mg catalyst, TEOA solution | 5162 µmol h−1 g−1 | 18.5% at 400 nm | 190 |
| Cu-Ni(OH)2/g-C3N4 | Cu-Ni(OH)2 | 300 W Xe lamp (AM 1.5 G) | 20 mg catalyst, TEOA solution | 2037 µmol h−1 g−1 | — | 194 |
| Pt/Ni(OH)2/g-C3N4 | Pt/Ni(OH)2 | 300 W Xe lamp | 10 mg catalyst, TEOA solution | 3005 µmol h−1 g−1 | 11.2% at 420 nm | 196 |
| Pt/NVC/g-C3N4 | Pt/NVC | 300 W Xe lamp (λ > 400 nm) | 50 mg catalyst, TEOA solution | 800 µmol h−1 g−1 | 0.20% at 420 nm | 197 |
4.1. Noble metal-based cocatalysts
Noble metals such as Pt, Pd, Ru, Ir, Rh, and Au have often been utilized as the cocatalysts for photocatalytic H2 production, due to their strong metallicity, high work function for electron transfer, and robust catalytic activity for H2 evolution.149–151 Among them, Pt can form an ideal Pt-H bond with appropriate strength and then significantly reduce the activation energy of H2 evolution, so that Pt shows the highest H2 evolution activity.152 Consequently, Pt is the most commonly used cocatalyst for improving photocatalytic H2 production performance. Wang and Domen et al.153 compared the photocatalytic H2 production activities of g-C3N4 loaded with various noble metal cocatalysts, including Ru, Rh, Pd, Ir, Pt, and Au, and found the Pt loaded g-C3N4 exhibited the highest photocatalytic activity toward H2 evolution. Moreover, nanoscale noble metals with a strong plasmonic effect, such as Au, Ag, and Pt, can improve the absorption of visible light and provide many high-energy hot electrons for reaction, thus further enhancing the photocatalytic performance.154,155 Wang et al.154 deposited Au nanoparticles on the surface of g-C3N4 and obtained the obviously enhanced photocatalytic H2 evolution activity. The characterization revealed that the loaded Au nanoparticles played a dual role in the photocatalytic reaction. On one hand, the Au nanoparticles could improve the charge separation efficiency through capturing electrons from g-C3N4; on the other hand, the Au plasma could provide additional hot electrons for H+ reduction. Hence, the accelerated charge transfer and increased number of electrons contributed to the enhanced photocatalytic activity of Au/g-C3N4. Yang and Wang et al.156 also investigated the effect of different sizes and shapes of Au nanocrystals on the photocatalytic performance of g-C3N4. Among the prepared Au/g-C3N4, the 18-nm-sized Au nanosphere loaded g-C3N4 showed an excellent photocatalytic H2 production activity of 540 µmol h−1 g−1 (AQE = 1.2% at 380 nm), far exceeding the larger-sized Au nanosphere and Au nanorod loaded g-C3N4 system. The superior H2 evolution activity was attributed to the optimized Au nanocrystals in promoting charge transfer and extending the light-harvesting range. Although noble metals can effectively promote proton reduction, they are also active sites for the backward reaction of H2 and O2. Thus, a noble metal/metal oxide cocatalyst with a core–shell structure was proposed to suppress backward reaction.157,158 For example, Zhang and Wang et al.159 decorated poly(triazine imide)-based carbon nitride with a Rh/Cr2O3 core–shell cocatalyst for H2 evolution, and CoOx as the O2 evolution cocatalyst. The presence of a Cr2O3 layer prevented the O2 transfer but facilitated the proton penetration, thereby hindering the backward reaction over Rh and achieving an AQE of 20.2% at 365 nm for overall water splitting, which was much higher than the Pt and CoOx modified one (AQE = 12%).160 Su et al.161 modified Pt/C3N4 with a TiO2 thin layer, which effectively prevented the reverse reaction and photocorrosion. Compared with naked Pt/C3N4, TiO2@Pt/C3N4 exhibited 130% enhanced overall efficiency of water splitting (AQE = 0.11% at 365 nm). In addition to pure noble metals, noble metal-based bimetallic alloys have also been extensively studied as cocatalysts to improve photocatalytic performance.162,163 The advantages of bimetallic alloys are related to the synergistic effect of two metals in tuning the electronic structure, work function and catalytic activity, which lead to efficient charge transfer and photoreduction reaction. For example, Pradhan et al.164 synthesized pure metals (Pt, Au) and PtAu alloy loaded g-C3N4, respectively, and investigated their photocatalytic H2 production activities. The PtAu alloy modified g-C3N4 exhibited better photocatalytic activity than the monometallic counterparts. A similar phenomenon of the improved photocatalytic performance was also observed in PtNi alloy loaded g-C3N4.165Although the loading of noble metal-based cocatalysts can significantly boost the photocatalytic activity of H2 production, the high cost and scarcity of noble metal materials limit their large-scale applications. Therefore, the development of low-cost, abundant, and highly active non-noble metal-based cocatalysts is highly desired for photocatalytic H2 production. During the next part, various transition metal-based cocatalysts are discussed.
4.2. Transition metal-based cocatalysts
Currently, there are many studies on transition metals as cocatalysts for photocatalytic H2 production, such as Ni, Cu, and Co, etc.166–168 Among various transition metals, Ni has been widely studied as a cocatalyst for g-C3N4 because its work function (ϕ = 5.4 eV) is close to that of Pt (ϕ = 5.7 eV) and its Fermi level is more positive than the CB of g-C3N4, which facilitate the capture of photogenerated electrons from g-C3N4.169 In the meantime, the Fermi level of Ni is more negative than the reduction potential of H+/H2, which promotes the transferred electrons to participate in the reduction reaction. Wu et al.170 deposited the single-atom Ni on TiO2/g-C3N4 as a cocatalyst, which could capture photogenerated electrons from g-C3N4 and accelerate the H+ reduction, resulting in higher H2 production activity (AQE of 15% at 420 nm) than the TiO2/g-C3N4 without Ni cocatalyst. Do et al.171 also reported the obviously improved photocatalytic H2 evolution activity of g-C3N4 and S-doped g-C3N4 when Ni was applied as the cocatalyst. Besides individual transition metals, transition metal-based alloys have also received widespread attention as cocatalysts for photocatalytic reaction. For example, Zhang and Wang et al.172 showed the drastically enhanced photocatalytic performance of g-C3N4 with NiMo alloy as the cocatalyst. The optimal H2 production rate of NiMo alloy modified g-C3N4 was found to be 1785 µmol h−1 g−1 (AQE of ∼0.25% at 365 nm), which was even comparable to the Pt/g-C3N4 (2250 µmol h−1 g−1). A similar enhancement in photocatalytic H2 production performance was also observed in Ni–Cu bimetallic cocatalyst loaded g-C3N4, which exhibited excellent photocatalytic activity, about 24 and 2 times that of g-C3N4 and Ni/g-C3N4, respectively.173 As mentioned above, the photocatalytic activity of g-C3N4 can be obviously improved by loading transition metals and their alloys. However, compared with the benchmark cocatalyst Pt, although most transition metals can effectively capture photogenerated electrons and improve charge separation efficiency, their catalytic activities toward H2 evolution are lower than Pt, resulting in the lower photocatalytic performance of transition metal cocatalyst modified g-C3N4 than that of Pt loaded g-C3N4.As we know, the intrinsic activity of catalysts is closely related to their electronic structures; exploring transition metal-derived cocatalysts with Pt-like electronic properties is necessary for further boosting photocatalytic H2 evolution. Levy et al.174 reported that carbonized tungsten (WC) exhibited Pt-like catalytic activity in hydrogenolysis and isomerization reactions. Subsequently, Bennett and coworkers carried out theoretical calculations to investigate the WC and found that the introduction of carbon into the tungsten changed its electronic distribution, resulting in the density of electronic states near the Fermi level close to Pt, which contributed to the Pt-like catalytic activity of WC.175 Therefore, the transition metal-derived compounds as the substitute for Pt cocatalyst have received increasing attention in the field of photocatalytic H2 production, including transition metal carbides, nitrides, phosphides, sulfides, etc.176–179 Li et al.180 loaded the WC onto g-C3N4 and studied the effect of WC on photocatalytic H2 production activity. It was demonstrated that the loaded WC as an efficient cocatalyst not only enhanced the surface H2-evolution kinetics, but also served as an electron transfer channel to promote interfacial charge transfer. We previously studied the role of tungsten carbide (W2C), tungsten sulfide (WS2), and tungsten nitride (W2N) in photocatalytic H2 production and compared their catalytic activities.28 We found that the W2C loaded g-C3N4 exhibited the highest H2 evolution activity. Based on the above study, we also designed a carbonized MoS2 (MoS2/Mo2C) as the cocatalyst for g-C3N4, and demonstrated its superior photocatalytic activity to that of the pure MoS2 loaded one, nearly 6 times higher in the rate of H2 evolution.181 Thus, transition metal carbides as the alternative for noble metal Pt have prominent advantages and potential application value.
Similar to metal carbides, many metal phosphides display high electrocatalytic activity toward H2 evolution, such as Ni2P, CoP, MoP, Cu3P, etc.182 And the use of metal phosphides as cocatalysts can effectively improve the surface H2 evolution kinetics and charge separation efficiency, thus achieving efficient photocatalytic H2 production activity.183–186 For example, Huang and coworkers compared the effect of different Ni-based compounds as cocatalysts on rGO/g-C3N4, and revealed that the Ni2P loaded rGO/g-C3N4 displayed better photocatalytic H2 evolution activity than the NiS, Ni3C, and Ni3N loaded system.187 Among these photocatalysts, the suitable band alignment between the Ni2P and rGO/g-C3N4 obviously promoted electron transfer from g-C3N4 to Ni2P via the rGO medium, then the H2 was produced on the Ni2P reaction site. This work clearly demonstrated the advantages of metal phosphide cocatalysts in suppressing charge recombination and accelerating H2 evolution. Furthermore, Liu et al.188 synthesized two types of copper phosphide loaded g-C3N4, Cu3P/g-C3N4 and Cu97P3/g-C3N4. The loading of copper phosphide significantly boosted the separation and transfer of photogenerated charge carriers, leading to an improved photocatalytic activity of Cu3P and Cu97P3 loaded g-C3N4. Shi et al.189 deposited the MoP as a cocatalyst on g-C3N4, the close interface induced the bonding state of Mo-N, which promoted photogenerated electron transfer to MoP and greatly improved the charge separation efficiency. Meanwhile, the loading of MoP also reduced the energy barrier for H2 production. The designed MoP/g-C3N4 displayed a H2 evolution activity of 3868 µmol h−1 g−1 with an AQE of 21.6% at 405 nm. Moreover, Liu and Fan et al.190 also designed a bimetallic phosphide NiCoP as the cocatalyst for g-C3N4 to further improve the photocatalytic performance. It was demonstrated that the rate of photocatalytic H2 evolution over NiCoP/g-C3N4 was obviously higher than that over Ni2P/g-C3N4 and Co2P/g-C3N4, even better than that over Pt anchored g-C3N4. The improved activity of g-C3N4 was attributed to the formation of an appropriate Schottky barrier at the interface of NiCoP/g-C3N4, which could promote the directional migration of photoexcited electrons to the NiCoP cocatalyst and inhibit the back recombination. Meanwhile, the synergistic effect of bimetals in NiCoP could further reduce the activation energy of H2 evolution, thus accelerating the H+ reduction.
Recently, on the basis of single-component cocatalysts, dual cocatalysts and composite cocatalysts have gradually been developed.191–193 In dual cocatalyst decorated g-C3N4, the spatially separated dual cocatalysts serve as the reduction site and oxidation site to trap the electrons and holes from g-C3N4, respectively, thus greatly minimizing charge recombination. For example, the use of Cu and Ni(OH)2 as dual cocatalysts for g-C3N4 showed better photocatalytic activity with the H2 evolution amount exceeding the sum of single Cu and Ni(OH)2 counterparts.194 The improvement in photocatalytic activity was mainly attributed to the coupling of Cu and Ni(OH)2 cocatalysts, which could drive the transfer of photogenerated electrons and holes to Cu and Ni(OH)2 respectively, achieving the spatial separation of charge carriers. A combination of Pt and MnOx was also reported as the dual cocatalysts to promote the separation, transfer, and utilization of photo-induced electron–hole pairs in CdS/g-C3N4.195 It is known that loading cocatalysts with good metallicity and matched Fermi level can indeed favor the directional transfer of charge carriers from g-C3N4 to cocatalysts. In the subsequent catalytic reaction, most cocatalysts can display high catalytic activity toward the reduction of H+ to H2, but confront an obstacle regarding the large energy barrier for water dissociation, which is the first step in H2 evolution reaction under neutral or alkaline conditions. Thus, the composite cocatalysts capable of promoting charge separation and improving reaction kinetics of proton reduction and water dissociation are developed. Zou and coworkers designed Pt cluster decorated Ni(OH)2 as a composite cocatalyst for g-C3N4, where the electron sink (Pt) and hole sink (Ni(OH)2) were included in one cocatalyst.196 It was found that the g-C3N4 with spatially separated Pt–Ni(OH)2 cocatalyst exhibited a lower charge recombination rate than the composite counterpart, which was attributed to the effective spatial separation of electron–hole pairs. While in terms of the redox reaction, the close contact of Pt and Ni(OH)2 in the composite cocatalyst could synergistically promote H2O dissociation and H+ reduction, which could not be achieved in the spatially separated Pt–Ni(OH)2 system. As a result, the g-C3N4 with Pt/Ni(OH)2 composite cocatalyst displayed higher photocatalytic activity than the Pt, Ni(OH)2, and spatially separated Pt-Ni(OH)2 loaded g-C3N4. We also investigated a composite cocatalyst of Pt/N doped VC (Pt/NVC) and found an improvement in the photocatalytic H2 production activity of Pt/NVC/g-C3N4 compared to NVC/g-C3N4 and Pt/g-C3N4.197 Therefore, the highly efficient cocatalysts are expected to achieve a high charge separation efficiency and robust catalytic activity simultaneously, and the multifunctional composite cocatalyst with strong synergistic effect is a promising cocatalyst for photocatalytic reactions.
5. Summary and outlook
5.1. Summary
The easy preparation, low cost, visible-light response, suitable band structure, and high stability of metal-free g-C3N4 make it one of the most promising photocatalysts for H2 production from water reduction reactions. However, the inherent Coulomb interaction of photogenerated electron–hole pairs and the existence of an interlayer potential barrier in g-C3N4 induce the severe bulk recombination of charge carriers, thus greatly reducing the number of active electrons involved in the surface reduction reaction. To oppose the charge recombination and improve the charge separation efficiency, various approaches have been adopted to promote the separation and transfer of photoexcited charge carriers through weakening their Coulomb attraction.In this perspective, we summarized several common and effective strategies for assisting charge separation in the bulk and surface of g-C3N4, including structural modulation, heterojunction construction, and cocatalyst loading. In terms of structural modulation, we discussed the effects of crystal structure, morphology and doping on charge separation and migration. The g-C3N4 prepared by traditional high-temperature calcination shows poor crystallinity and many defects, resulting in a high charge recombination rate. Hence, many strategies have been developed to promote the polymerization of precursors and synthesize the highly crystalline g-C3N4, such as salt-assisted methods, microwave synthesis, and two-step calcination. Meanwhile, for bulk g-C3N4, the electrostatic barrier between layers can hinder charge transfer to the surface active sites and induce severe bulk recombination. Thus, many works about morphological tuning of g-C3N4 have been carried out to change the original structure of layer-by-layer stacking and facilitate the charge transfer across the interlayers. Designing ultrathin g-C3N4 nanosheets effectively enlarges the distance between layers, thereby breaking the constraint of interlayer electrostatic barrier and reducing the bulk recombination of photogenerated charge carriers. Otherwise, the tubular morphology of g-C3N4 also favors charge transfer and separation within the bulk phase. When the 2D g-C3N4 bends to form the hollow tubular structure, an apparent potential difference is formed between the inner and outer surfaces of tubular g-C3N4, which could drive the electron transfer from the inner to the outer surface and greatly improve the bulk charge separation efficiency. Besides the morphological tuning, the doping strategy has also been widely used to regulate the structure of g-C3N4, thus facilitating the charge migration and achieving a high charge separation efficiency. In particular, the incorporation of metals and delocalization groups would break the planar π-conjugated structure of g-C3N4 and induce the redistribution of in-plane delocalized charges, which result in the formation of an intrinsic polarization electric field to promote the in-plane charge transfer along the potential difference. Therefore, the appropriate structural modulation of bulk g-C3N4via crystallinity regulation, morphology and doping engineering can obviously suppress the charge recombination within the bulk and surface and then improve the charge separation efficiency.
Additionally, the design of g-C3N4-based composites, including semiconductor combination and cocatalyst loading, is another effective approach to improve surface charge separation efficiency by promoting charge migration and transfer at the interface of composites. Among various g-C3N4 composites, the Z-scheme heterojunction has attracted much attention due to its advantage of the directional charge transfer driven by a built-in electric field. In a Z-scheme g-C3N4 heterojunction, the intimate contact of two semiconductors with different work functions induces interfacial charge reorganization and the formation of a built-in electric field at the interface, which can drive the photogenerated charges across the interface and consume ineffective charges, thus achieving the spatial separation of charge carriers and significantly minimizing the surface recombination. Similarly, for the cocatalyst loaded g-C3N4 system, the difference in work function also causes the charge rearrangement at the interface and results in the formation of a Schottky barrier, which controls the flow of the photogenerated electrons to the cocatalyst and suppresses the back recombination of charge carriers.
5.2. Outlook
This progress can indeed improve the separation efficiency of photogenerated electron–hole pairs and achieve robust photocatalytic H2 evolution activity. However, the solar-to-hydrogen conversion efficiency of the g-C3N4 system still struggles to meet the commercial level, due to the difficulty in synergistically regulating the separation and transfer of whole charge carriers, including bulk phase separation and surface separation. For the above discussed studies, the crystallinity regulation, doping engineering, heterojunction design, and cocatalyst loading can greatly promote the charge transfer within the plane or at the interface, which effectively suppress the surface recombination of charge carriers. While the morphological tuning mainly favors the charges to transfer from the bulk to surface, its role in promoting the surface charge separation is not so prominent. Therefore, although the above modification approaches have achieved many positive results, there are still certain limitations in improving the photocatalytic performance of g-C3N4 based on a single strategy since each strategy has its own advantages and weaknesses. To facilitate the charge transfer and separation both in the bulk phase and surface and achieve an excellent whole charge separation efficiency, it is essential to combine multiple methods to synergistically modify the pristine g-C3N4.Furthermore, there is another issue that is often overlooked in the charge transfer process, the randomness of charge transfer paths. Although the introduction of a potential difference or built-in electric field in g-C3N4 can induce directional transfer of charge carriers from one component to another, the charge movement and transfer paths are random, which can easily trigger charge recombination during the charge migration. Thus, constructing charge transport channels to achieve ordered charge migration is another key to further improving charge separation efficiency and photocatalytic activity.
In the meantime, the transfer mechanism of photogenerated charge carriers and photocatalytic mechanism still need detailed and in-depth understanding and research, which directly affect the structural design of the g-C3N4 system. For instance, the type II and direct Z-scheme heterojunction systems possess similar band structures; precisely probing interfacial charge transfer is crucial to distinguishing the heterojunction types. Hence, it is necessary to combine various characterization techniques to investigate the behavior of photogenerated charge carriers in photocatalytic reactions, including the surface photovoltage technique, transient photocurrent response, electrochemical impedance spectroscopy (EIS), photoluminescence (PL), X-ray photoelectron spectroscopy (XPS), etc. In particular, the advanced characterizations of time-resolved technology and in-operando technology need to be developed to explore the dynamics and specific migration pathways of photogenerated charge carriers.
In summary, continuous efforts are required to develop novel and robust g-C3N4 photocatalytic systems for practical application. There are some recommendations for the design of g-C3N4 photocatalysts: (1) combining the advantages of multiple methods to modify g-C3N4 and improve the overall charge separation efficiency, including bulk phase and surface separation; (2) constructing efficient in-plane and interlayer joint charge transport channels to achieve ordered migration of overall photogenerated charge carriers; (3) developing various and advanced characterizations to clearly reveal the charge transfer and separation mechanism, thereby constructing the desired g-C3N4 system.
Author contributions
Mengmeng Shao: investigation, writing – original draft. Yangfan Shao: data curation, writing – review & editing. Hui Pan: conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (12204263) and the Guangdong Basic and Applied Basic Research Foundation (2022A1515010628).References
- Q. Hassan, A. Z. Sameen, H. M. Salman, M. Jaszczur and A. K. Al-Jiboory, J. Energy Storage, 2023, 72, 108404 CrossRef.
- H. Ishaq, I. Dincer and C. Crawford, Int. J. Hydrogen Energy, 2022, 47, 26238–26264 CrossRef CAS.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- Y. Zhao, C. Ding, J. Zhu, W. Qin, X. Tao, F. Fan, R. Li and C. Li, Angew. Chem., Int. Ed., 2020, 59, 9653–9658 CrossRef CAS PubMed.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- R. Shen, D. Ren, Y. Ding, Y. Guan, Y. H. Ng, P. Zhang and X. Li, Sci. China Mater., 2020, 63, 2153–2188 CrossRef CAS.
- C. Xu, D. Li, X. Liu, R. Ma, N. Sakai, Y. Yang, S. Lin, J. Yang, H. Pan, J. Huang and T. Sasaki, Chem. Eng. J., 2022, 430, 132861 CrossRef CAS.
- Z. Luo, X. Ye, S. Zhang, S. Xue, C. Yang, Y. Hou, W. Xing, R. Yu, J. Sun, Z. Yu and X. Wang, Nat. Commun., 2022, 13, 2230 CrossRef CAS PubMed.
- B. Debnath, S. Dhingra and C. M. Nagaraja, Sol. RRL, 2021, 5, 2100226 CrossRef CAS.
- L. Chen, X. Yu, Z. Hua, Q. Liu, V. An, L. Feng, J. Guo, X. Zhang, J. Li and B. Liu, ACS Appl. Energy Mater., 2023, 6, 3769–3777 CrossRef CAS.
- D. Wei, Y. Ding and Z. Li, Int. J. Hydrogen Energy, 2020, 45, 17320–17328 CrossRef CAS.
- B. Li, W. Wang, J. Zhao, Z. Wang, B. Su, Y. Hou, Z. Ding, W.-J. Ong and S. Wang, J. Mater. Chem. A, 2021, 9, 10270–10276 RSC.
- J. Xu, C. Sun, Z. Wang, Y. Hou, Z. Ding and S. Wang, Chem. – Eur. J., 2018, 24, 18512–18517 CrossRef CAS PubMed.
- S. Wang, Y. Wang, S. L. Zhang, S.-Q. Zang and X. W. Lou, Adv. Mater., 2019, 31, 1903404 CrossRef CAS PubMed.
- Z. Xiong, Y. Hou, R. Yuan, Z. Ding, W.-J. Ong and S. Wang, Acta Phys.-Chim. Sin., 2022, 38, 2111021 Search PubMed.
- Z. Wang, B. Su, J. Xu, Y. Hou and Z. Ding, Int. J. Hydrogen Energy, 2020, 45, 4113–4121 CrossRef CAS.
- S. San Martín, M. J. Rivero and I. Ortiz, Catalysts, 2020, 10, 901 CrossRef.
- H. Pan, Renewable Sustainable Energy Rev., 2016, 57, 584–601 CrossRef CAS.
- Y. Yang, W. Niu, L. Dang, Y. Mao, J. Wu and K. Xu, Front. Chem., 2022, 10, 955065 CrossRef CAS PubMed.
- G. Z. S. Ling, V. B.-Y. Oh, C. Y. Haw, L.-L. Tan and W.-J. Ong, Energy Mater. Adv., 2023, 4, 0038 CrossRef CAS.
- D. Ma, Z. Zhang, Y. Zou, J. Chen and J.-W. Shi, Coord. Chem. Rev., 2024, 500, 215489 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- X. Chen, R. Shi, Q. Chen, Z. Zhang, W. Jiang, Y. Zhu and T. Zhang, Nano Energy, 2019, 59, 644–650 CrossRef CAS.
- X. Dang, M. Xie, F. Dai, J. Guo, J. Liu and X. Lu, Adv. Mater. Interfaces, 2021, 8, 2100151 CrossRef CAS.
- M. Shao, Y. Shao, J. Chai, Y. Qu, M. Yang, Z. Wang, M. Yang, W. F. Ip, C. T. Kwok, X. Shi, Z. Lu, S. Wang, X. Wang and H. Pan, J. Mater. Chem. A, 2017, 5, 16748–16756 RSC.
- M. Shao, Y. Shao, S. Ding, J. Wang, J. Xu, Y. Qu, X. Zhong, X. Chen, W. F. Ip, N. Wang, B. Xu, X. Shi, X. Wang and H. Pan, Appl. Catal., B, 2018, 237, 295–301 CrossRef CAS.
- M. Shao, W. Chen, S. Ding, K. H. Lo, X. Zhong, L. Yao, W. F. Ip, B. Xu, X. Wang and H. Pan, ChemSusChem, 2019, 12, 3355–3362 CrossRef CAS PubMed.
- M. Li, Y. Gong, Y. Wang and T. He, Phys. Chem. Chem. Phys., 2022, 24, 19659–19672 RSC.
- Q. Yao, H. Li, J. Xue, S. Jiang, Q. Zhang and J. Bao, Angew. Chem., Int. Ed., 2023, 62, e202308140 CrossRef CAS PubMed.
- Y. Bao, S. Song, G. Yao and S. Jiang, Sol. RRL, 2021, 5, 2100118 CrossRef CAS.
- M. Z. Rahman and C. B. Mullins, Acc. Chem. Res., 2019, 52, 248–257 CrossRef CAS PubMed.
- Y. Kang, Y. Yang, L.-C. Yin, X. Kang, L. Wang, G. Liu and H.-M. Cheng, Adv. Mater., 2016, 28, 6471–6477 CrossRef CAS PubMed.
- W. Cui, P. Chen, L. Chen, J. Li, Y. Zhou and F. Dong, J. Phys.: Energy, 2021, 3, 032008 CAS.
- X. Chu, C. I. Sathish, J.-H. Yang, X. Guan, X. Zhang, L. Qiao, K. Domen, S. Wang, A. Vinu and J. Yi, Small, 2023, 19, 2302875 CrossRef CAS PubMed.
- G. Zhang, Y. Xu, J. Zhu, Y. Li, C. He, X. Ren, P. Zhang and H. Mi, Appl. Catal., B, 2023, 338, 123049 CrossRef CAS.
- N. P. Dharmarajan, D. Vidyasagar, J.-H. Yang, S. N. Talapaneni, J. Lee, K. Ramadass, G. Singh, M. Fawaz, P. Kumar and A. Vinu, Adv. Mater., 2024, 36, 2306895 CrossRef CAS PubMed.
- J. Wang and S. Wang, Coord. Chem. Rev., 2022, 453, 214338 CrossRef CAS.
- B. Zhao, W. Zhong, F. Chen, P. Wang, C. Bie and H. Yu, Chin. J. Catal., 2023, 52, 127–143 CrossRef CAS.
- Y. Li, D. Zhang, J. Fan and Q. Xiang, Chin. J. Catal., 2021, 42, 627–636 CrossRef CAS.
- F. He, Y. Hu, H. Zhong, Z. Wang, S. Peng and Y. Li, Chem. Commun., 2023, 59, 10476–10487 RSC.
- F. Lin, S. Zhou, G. Wang, J. Wang, T. Gao, Y. Su and C.-P. Wong, Nano Energy, 2022, 99, 107432 CrossRef CAS.
- S. An, Y. Guo, X. He, P. Gao, G. Hou, J. Hou, C. Song and X. Guo, Appl. Catal., B, 2022, 310, 121323 CrossRef CAS.
- H. Song, X. Liu, Y. Wang, L. Chen, J. Zhang, C. Zhao, F. He, P. Dong, B. Li, S. Wang, S. Wang and H. Sun, J. Colloid Interface Sci., 2022, 607, 1603–1612 CrossRef CAS PubMed.
- Q. Wang, G. Zhang, W. Xing, Z. Pan, D. Zheng, S. Wang, Y. Hou and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202307930 CrossRef CAS PubMed.
- A. Torres-Pinto, C. G. Silva, J. L. Faria and A. M. T. Silva, Catal. Today, 2023, 424, 113868 CrossRef CAS.
- H. Jing, M. You, S. Yi, T. Li, H. Ji, Y. Wang, Z. Zhang, R. Zhang, D. Chen and H. Yang, ChemSusChem, 2020, 13, 827–837 CrossRef CAS PubMed.
- L. Lin, Z. Lin, J. Zhang, X. Cai, W. Lin, Z. Yu and X. Wang, Nat. Catal., 2020, 3, 649–655 CrossRef CAS.
- S. Gao, X. Wang, C. Song, S. Zhou, F. Yang and Y. Kong, Appl. Catal., B, 2021, 295, 120272 CrossRef CAS.
- L. Wang, Y. Hong, E. Liu, X. Duan, X. Lin and J. Shi, Carbon, 2020, 163, 234–243 CrossRef CAS.
- Y.-J. Yuan, Z. Shen, S. Wu, Y. Su, L. Pei, Z. Ji, M. Ding, W. Bai, Y. Chen, Z.-T. Yu and Z. Zou, Appl. Catal., B, 2019, 246, 120–128 CrossRef CAS.
- C. Wu, S. Xue, Z. Qin, M. Nazari, G. Yang, S. Yue, T. Tong, H. Ghasemi, F. C. R. Hernandez, S. Xue, D. Zhang, H. Wang, Z. M. Wang, S. Pu and J. Bao, Appl. Catal., B, 2021, 282, 119557 CrossRef CAS.
- Z. Jiang, X. Zhang, H.-S. Chen, X. Hu and P. Yang, ChemCatChem, 2019, 11, 4558–4567 CrossRef CAS.
- W. Luo, X. Chen, Z. Wei, D. Liu, W. Yao and Y. Zhu, Appl. Catal., B, 2019, 255, 117761 CrossRef CAS.
- H. Pan, Y.-W. Zhang, V. B. Shenoy and H. Gao, ACS Catal., 2011, 1, 99–104 CrossRef CAS.
- Y. Zeng, X. Liu, C. Liu, L. Wang, Y. Xia, S. Zhang, S. Luo and Y. Pei, Appl. Catal., B, 2018, 224, 1–9 CrossRef CAS.
- X. Pan and X. Bao, Acc. Chem. Res., 2011, 44, 553–562 CrossRef CAS PubMed.
- X. Pan and X. Bao, Chem. Commun., 2008, 6271–6281 RSC.
- X. Blase, L. X. Benedict, E. L. Shirley and S. G. Louie, Phys. Rev. Lett., 1994, 72, 1878–1881 CrossRef CAS PubMed.
- Z. Jiang, C. Jia, B. Wang, P. Yang and G. Gao, J. Alloys Compd., 2020, 826, 154145 CrossRef CAS.
- G. Jiang, X. You, B. An, F. Liu, X. Duan, Y. Wang, C. Liu and C. Zhao, Appl. Catal., B, 2022, 305, 121018 CrossRef CAS.
- Z. Lin, Y. Zhao, J. Luo, S. Jiang, C. Sun and S. Song, Adv. Funct. Mater., 2020, 30, 1908797 CrossRef CAS.
- C. Xu, X. Liu, D. Li, Z. Chen, J. Yang, J. Huang and H. Pan, ACS Appl. Mater. Interfaces, 2021, 13, 20114–20124 CrossRef CAS PubMed.
- J. Xie, C. Wang, N. Chen, W. Chen, J. Xu, P. Bai, B. Liu, L. Zhang and H. Wang, J. Mater. Chem. C, 2021, 9, 4378–4384 RSC.
- H. Che, C. Li, P. Zhou, C. Liu, H. Dong and C. Li, Appl. Surf. Sci., 2020, 505, 144564 CrossRef CAS.
- C. Xu, H. Liu, D. Wang, D. Li, Y. Zhang, X. Liu, J. Huang, S. Wu, D. Fan, H. Liu and H. Pan, Appl. Catal., B, 2023, 334, 122835 CrossRef CAS.
- H. Fang, J. Gao, J. Wang, J. Xu and L. Wang, Sep. Purif. Technol., 2023, 314, 123565 CrossRef CAS.
- N. Wang, L. Cheng, Y. Liao and Q. Xiang, Small, 2023, 19, 2300109 CrossRef CAS PubMed.
- C. Xu, X. Liu, H. Liu, D. Li, Y. Yang, S. Lin, D. Fan and H. Pan, J. Mater. Chem. A, 2022, 10, 21031–21043 RSC.
- D. Tang, C. Shao, S. Jiang, C. Sun and S. Song, ACS Nano, 2021, 15, 7208–7215 CrossRef CAS PubMed.
- W. Luo, Y. Li, J. Wang, J. Liu, N. Zhang, M. Zhao, J. Wu, W. Zhou and L. Wang, Nano Energy, 2021, 87, 106168 CrossRef CAS.
- G. Zhang, G. Li, T. Heil, S. Zafeiratos, F. Lai, A. Savateev, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 3433–3437 CrossRef CAS PubMed.
- G. Zhang, M. Liu, T. Heil, S. Zafeiratos, A. Savateev, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 14950–14954 CrossRef CAS PubMed.
- M. Chang, Z. Pan, D. Zheng, S. Wang, G. Zhang, M. Anpo and X. Wang, ChemSusChem, 2023, 16, e202202255 CrossRef CAS PubMed.
- S. Wu, Y. Yu, K. Qiao, J. Meng, N. Jiang and J. Wang, J. Photochem. Photobiol., A, 2021, 406, 112999 CrossRef CAS.
- J. Shen, C. Luo, S. Qiao, Y. Chen, Y. Tang, J. Xu, K. Fu, D. Yuan, H. Tang, H. Zhang and C. Liu, ACS Catal., 2023, 13, 6280–6288 CrossRef CAS.
- S. Tasleem and M. Tahir, Int. J. Hydrogen Energy, 2021, 46, 20995–21012 CrossRef CAS.
- J. Luo, J. He, S. Jiang, C. Sun and S. Song, Chem. Eng. J., 2020, 396, 125365 CrossRef CAS.
- M. Zhou, G. Dong, F. Yu and Y. Huang, Appl. Catal., B, 2019, 256, 117825 CrossRef CAS.
- X. Yang, Z. Guo, X. Zhang, Y. Han, Z. Xue, T. Xie and W. Yang, New J. Chem., 2021, 45, 544–550 RSC.
- X. Xiao, Y. Gao, L. Zhang, J. Zhang, Q. Zhang, Q. Li, H. Bao, J. Zhou, S. Miao, N. Chen, J. Wang, B. Jiang, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2003082 CrossRef CAS PubMed.
- J. He, Y. Zhao, S. Jiang and S. Song, Sol. RRL, 2021, 5, 2000446 CrossRef CAS.
- Q. Su, Y. Li, R. Hu, F. Song, S. Liu, C. Guo, S. Zhu, W. Liu and J. Pan, Adv. Sustainable Syst., 2020, 4, 2000130 CrossRef CAS.
- X. Xu, X. Feng, W. Wang, K. Song, D. Ma, Y. Zhou and J.-W. Shi, J. Colloid Interface Sci., 2023, 651, 669–677 CrossRef CAS PubMed.
- H. Dong, L. Tong, P. Zhang, D. Zhu, J. Jiang and C. Li, J. Mater. Sci. Technol., 2024, 179, 251–261 CrossRef.
- X. Kuang, X. Deng, Y. Ma, J. Zeng, B. Zi, Y. Zhang, J. Zhang, B. Xiao and Q. Liu, J. Mater. Chem. C, 2022, 10, 6341–6347 RSC.
- S. Yu, C. Li, Y. Lin, J. Zhang, Y. Liu and F. Yu, Sep. Purif. Technol., 2024, 341, 126888 CrossRef CAS.
- A. Mehtab, S. Banerjee, Y. Mao and T. Ahmad, ACS Appl. Mater. Interfaces, 2022, 14, 44317–44329 CrossRef CAS PubMed.
- Y. Wang, J. Li, S. Chen, Y. Xie, Y. Ma, Y. Luo, J. Huang, Y. Ling, J. Ye, Y. Liang and J. Du, J. Alloys Compd., 2022, 924, 166569 CrossRef CAS.
- S. Wei, F. Wang, P. Yan, M. Dan, W. Cen, S. Yu and Y. Zhou, J. Catal., 2019, 377, 122–132 CrossRef CAS.
- Y. Lv, D. Ma, C. Yang, K. Song, L. Shi, Y. Cheng, C. Niu and J.-W. Shi, Sep. Purif. Technol., 2023, 316, 123813 CrossRef CAS.
- H. Zhao, B. Jia, Z. Wang, L. Han, H. Song and P. Lu, Int. J. Hydrogen Energy, 2023, 48, 10051–10061 CrossRef CAS.
- A. Mehtab, Y. Mao, S. M. Alshehri and T. Ahmad, J. Colloid Interface Sci., 2023, 652, 1467–1480 CrossRef CAS PubMed.
- D. Kim and K. Yong, Appl. Catal., B, 2021, 282, 119538 CrossRef CAS.
- Y. Zhu, T. Wan, X. Wen, D. Chu and Y. Jiang, Appl. Catal., B, 2019, 244, 814–822 CrossRef CAS.
- X. Zhang, H. Liang, C. Li and J. Bai, Inorg. Chem. Commun., 2022, 144, 109838 CrossRef CAS.
- N. Güy, Appl. Surf. Sci., 2020, 522, 146442 CrossRef.
- Y. Zou, J.-W. Shi, L. Sun, D. Ma, S. Mao, Y. Lv and Y. Cheng, Chem. Eng. J., 2019, 378, 122192 CrossRef CAS.
- Y. Liu, C. Lv, J. Sun, X. Zhou, Y. Zhou and G. Chen, Adv. Mater. Interfaces, 2022, 9, 2200153 CrossRef CAS.
- D. Ma, J.-W. Shi, Z. Pu, S. Mao, X. Xu, D. He, R. Guo and F. Chen, Sol. RRL, 2022, 6, 2200714 CrossRef CAS.
- J. Pan, J. Liang, Z. Xu, X. Yao, J. Qiu, H. Chen, L. Qin, D. Chen and Y. Huang, Int. J. Hydrogen Energy, 2021, 46, 30344–30354 CrossRef CAS.
- J. Jia, Q. Zhang, K. Li, Y. Zhang, E. Liu and X. Li, Int. J. Hydrogen Energy, 2023, 48, 196–231 CrossRef CAS.
- A. J. Bard, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- B.-J. Ng, L. K. Putri, X. Y. Kong, Y. W. Teh, P. Pasbakhsh and S.-P. Chai, Adv. Sci., 2020, 7, 1903171 CrossRef CAS PubMed.
- Y. Kang, H. Qi, G. Wan, C. Zhen, X. Xu, L.-C. Yin, L. Wang, G. Liu and H.-M. Cheng, Joule, 2022, 6, 1876–1886 CrossRef CAS.
- H. Tada, T. Mitsui, T. Kiyonaga, T. Akita and K. Tanaka, Nat. Mater., 2006, 5, 782–786 CrossRef CAS PubMed.
- M. Wu, T. Ding, Y. Wang, W. Zhao, H. Xian, Y. Tian, T. Zhang and X. Li, Catal. Today, 2020, 355, 311–318 CrossRef CAS.
- Z. Hu, D. Shi, G. Wang, T. Gao, J. Wang, L. Lu and J. Li, Appl. Surf. Sci., 2022, 601, 154167 CrossRef CAS.
- X. Wang, G. Liu, Z.-G. Chen, F. Li, L. Wang, G. Q. Lu and H.-M. Cheng, Chem. Commun., 2009, 3452–3454 RSC.
- J. Yu, S. Wang, J. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS.
- Y. Lei, J. Ye, J. García-Antón and H. Liu, Chin. J. Catal., 2023, 53, 72–101 CrossRef CAS.
- H. Chen, S. Gao, G. Huang, Q. Chen, Y. Gao and J. Bi, Appl. Catal., B, 2024, 343, 123545 CrossRef CAS.
- Q. Tang, W. Tao, J. Hu, T. Gui, Z. Wang, Y. Xiao, R. Song, Y. Jiang and S. Guo, ACS Appl. Nano Mater., 2023, 6, 17130–17139 CrossRef CAS.
- Y. You, S. Wang, K. Xiao, T. Ma, Y. Zhang and H. Huang, ACS Sustainable Chem. Eng., 2018, 6, 16219–16227 CrossRef CAS.
- Z.-F. Huang, J. Song, X. Wang, L. Pan, K. Li, X. Zhang, L. Wang and J.-J. Zou, Nano Energy, 2017, 40, 308–316 CrossRef CAS.
- T. Feng, J. Jin, Y. Cao, H. Li, B. Dong and L. Cao, Int. J. Hydrogen Energy, 2022, 47, 5999–6010 CrossRef CAS.
- X. Zhao, Y. You, S. Huang, Y. Wu, Y. Ma, G. Zhang and Z. Zhang, Appl. Catal., B, 2020, 278, 119251 CrossRef CAS.
- M. Tan, Y. Ma, C. Yu, Q. Luan, J. Li, C. Liu, W. Dong, Y. Su, L. Qiao, L. Gao, Q. Lu and Y. Bai, Adv. Funct. Mater., 2022, 32, 2111740 CrossRef CAS.
- Z. Pu, B. Xiao, S. Mao, Y. Sun, D. Ma, H. Wang, J. Zhou, Y. Cheng and J.-W. Shi, J. Colloid Interface Sci., 2022, 628, 477–487 CrossRef CAS PubMed.
- Z.-x Bi, R.-t Guo, X.-y Ji, X. Hu, J. Wang, X. Chen and W.-g Pan, Int. J. Hydrogen Energy, 2022, 47, 34430–34443 CrossRef CAS.
- R. Wang, C. Ye, H. Wang and F. Jiang, ACS Omega, 2020, 5, 30373–30382 CrossRef CAS PubMed.
- L. Xu, J. Zeng, Q. Li, L. Xia, X. Luo, Z. Ma, B. Peng, S. X. Xiong, Z. Li, L.-L. Wang and Y. Lei, Appl. Surf. Sci., 2021, 547, 149207 CrossRef CAS.
- Y. Zhen, C. Yang, H. Shen, W. Xue, C. Gu, J. Feng, Y. Zhang, F. Fu and Y. Liang, Phys. Chem. Chem. Phys., 2020, 22, 26278–26288 RSC.
- G. Dong, Y. Zhang, Y. Wang, Q. Deng, C. Qin, Y. Hu, Y. Zhou and G. Tian, ACS Appl. Energy Mater., 2021, 4, 14342–14351 CrossRef CAS.
- J. Zhang, Y. Zhao, K. Qi and S.-Y. Liu, J. Mater. Sci. Technol., 2024, 172, 145–155 CrossRef.
- Z. Zhao, K. Dai, J. Zhang and G. Dawson, Adv. Sustainable Syst., 2023, 7, 2100498 CrossRef CAS.
- H. Sun, Y. Shi, W. Shi and F. Guo, Appl. Surf. Sci., 2022, 593, 153281 CrossRef CAS.
- Q. Xu, D. Ma, S. Yang, Z. Tian, B. Cheng and J. Fan, Appl. Surf. Sci., 2019, 495, 143555 CrossRef CAS.
- D. Zhao, Y. Wang, C.-L. Dong, Y.-C. Huang, J. Chen, F. Xue, S. Shen and L. Guo, Nat. Energy, 2021, 6, 388–397 CrossRef CAS.
- J. Jiang, Z. Xiong, H. Wang, G. Liao, S. Bai, J. Zou, P. Wu, P. Zhang and X. Li, J. Mater. Sci. Technol., 2022, 118, 15–24 CrossRef CAS.
- G. Zhang, L. Lin, G. Li, Y. Zhang, A. Savateev, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 9372–9376 CrossRef CAS PubMed.
- J. Zhang, X. Liang, C. Zhang, L. Lin, W. Xing, Z. Yu, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2022, 61, e202210849 CrossRef CAS PubMed.
- Y. Lv, D. Ma, K. Song, S. Mao, Z. Liu, D. He, X. Zhao, T. Yao and J.-W. Shi, J. Mater. Chem. A, 2023, 11, 800–808 RSC.
- Y. Hu, Y. Qu, Y. Zhou, Z. Wang, H. Wang, B. Yang, Z. Yu and Y. Wu, Chem. Eng. J., 2021, 412, 128749 CrossRef CAS.
- R. Feng, K. Wan, X. Sui, N. Zhao, H. Li, W. Lei, J. Yu, X. Liu, X. Shi, M. Zhai, G. Liu, H. Wang, L. Zheng and M. Liu, Nano Today, 2021, 37, 101080 CrossRef CAS.
- S. Qin, N. Denisov, J. Will, J. Kolařík, E. Spiecker and P. Schmuki, Sol. RRL, 2022, 6, 2101026 CrossRef CAS.
- L. Tian, X. Guan, S. Zong, A. Dai and J. Qu, Catalysts, 2023, 13, 355 CrossRef CAS.
- X. Liu and H. Zhuang, Int. J. Energy Res., 2021, 45, 1480–1495 CrossRef CAS.
- Q. Zhu, Z. Xu, B. Qiu, M. Xing and J. Zhang, Small, 2021, 17, 2101070 CrossRef CAS PubMed.
- R. Tong, Z. Sun, X. Wang, L. Yang, J. Zhai, S. Wang and H. Pan, Int. J. Hydrogen Energy, 2020, 45, 18912–18921 CrossRef CAS.
- M. Saruyama, C. M. Pelicano and T. Teranishi, Chem. Sci., 2022, 13, 2824–2840 RSC.
- R. Tong, K. W. Ng, X. Wang, S. Wang, X. Wang and H. Pan, J. Mater. Chem. A, 2020, 8, 23202–23230 RSC.
- Y. Qu, M. Shao, Y. Shao, M. Yang, J. Xu, C. T. Kwok, X. Shi, Z. Lu and H. Pan, J. Mater. Chem. A, 2017, 5, 15080–15086 RSC.
- S. A. Rawool, M. R. Pai, A. M. Banerjee, S. Nath, R. D. Bapat, R. K. Sharma, Jagannath, B. Dutta, P. A. Hassan and A. K. Tripathi, ACS Appl. Mater. Interfaces, 2023, 15, 39926–39945 CrossRef CAS PubMed.
- Z. Zhuang, Y. Li, Z. Li, F. Lv, Z. Lang, K. Zhao, L. Zhou, L. Moskaleva, S. Guo and L. Mai, Angew. Chem., Int. Ed., 2018, 57, 496–500 CrossRef CAS PubMed.
- G. Z. S. Ling, S.-F. Ng and W.-J. Ong, Adv. Funct. Mater., 2022, 32, 2111875 CrossRef CAS.
- Q. Wang, Q. Liu, Y.-Y. Ma, H.-X. Bi, J. Du and Z.-G. Han, Inorg. Chem. Front., 2024, 11, 1238–1251 RSC.
- T. Tong, B. Zhu, C. Jiang, B. Cheng and J. Yu, Appl. Surf. Sci., 2018, 433, 1175–1183 CrossRef CAS.
- Q. Yang, T. Wang, Z. Zheng, B. Xing, C. Li and B. Li, Appl. Catal., B, 2022, 315, 121575 CrossRef CAS.
- Z. Yu, Y. Li, A. Torres-Pinto, A. P. LaGrow, V. M. Diaconescu, L. Simonelli, M. J. Sampaio, O. Bondarchuk, I. Amorim, A. Araujo, A. M. T. Silva, C. G. Silva, J. L. Faria and L. Liu, Appl. Catal., B, 2022, 310, 121318 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 163–184 CrossRef CAS.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940–4947 CrossRef CAS.
- Z. Guo, F. Dai, H. Yin, M. Zhang, J. Xing and L. Wang, Colloid Interface Sci. Commun., 2022, 48, 100615 CrossRef CAS.
- X. Li, H. Jiang, C. Ma, Z. Zhu, X. Song, H. Wang, P. Huo and X. Li, Appl. Catal., B, 2021, 283, 119638 CrossRef CAS.
- Y. Guo, H. Jia, J. Yang, H. Yin, Z. Yang, J. Wang and B. Yang, Phys. Chem. Chem. Phys., 2018, 20, 22296–22307 RSC.
- Z. Li, R. Li, H. Jing, J. Xiao, H. Xie, F. Hong, N. Ta, X. Zhang, J. Zhu and C. Li, Nat. Catal., 2023, 6, 80–88 CrossRef CAS.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- M. Liu, G. Zhang, X. Liang, Z. Pan, D. Zheng, S. Wang, Z. Yu, Y. Hou and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202304694 CrossRef CAS PubMed.
- M. Liu, C. Wei, H. Zhuzhang, J. Zhou, Z. Pan, W. Lin, Z. Yu, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2022, 61, e202113389 CrossRef CAS PubMed.
- Z. Han, X. Ning, Z. Yin, W. Zhen, G. Lu and B. Su, Int. J. Hydrogen Energy, 2024, 59, 856–865 CrossRef CAS.
- Y. Liu, Z. Sun and Y. H. Hu, Chem. Eng. J., 2021, 409, 128250 CrossRef CAS.
- C. M. Pelicano, M. Saruyama, R. Takahata, R. Sato, Y. Kitahama, H. Matsuzaki, T. Yamada, T. Hisatomi, K. Domen and T. Teranishi, Adv. Funct. Mater., 2022, 32, 2202987 CrossRef CAS.
- K. Bhunia, M. Chandra, S. Khilari and D. Pradhan, ACS Appl. Mater. Interfaces, 2019, 11, 478–488 CrossRef CAS PubMed.
- R. Li, Y. Wang, C. Zuo, J. Wang, X. Sheng, Y. Huang, Y. Zhang and Y. Zhou, Int. J. Hydrogen Energy, 2023, 48, 28277–28288 CrossRef CAS.
- D. Ma, X. Zhang, C. Yang, X. Feng, Z.-F. Zhang, K. Song, S. Wu, L. Li, T. Jiang and J.-W. Shi, Sep. Purif. Technol., 2023, 327, 124996 CrossRef CAS.
- C. Wang, J. Xie, N. Chen, W. Chen, P. Bai and H. Wang, ACS Appl. Energy Mater., 2021, 4, 13796–13802 CrossRef CAS.
- T. Song, X. Zhang, K. Matras-Postolek and P. Yang, J. Environ. Chem. Eng., 2022, 10, 108747 CrossRef CAS.
- L. Ji, C. Lv, Z. Chen, Z. Huang and C. Zhang, Adv. Mater., 2018, 30, 1705653 CrossRef PubMed.
- S. Yang, K. Wang, Q. Chen and Y. Wu, J. Mater. Sci. Technol., 2024, 175, 104–114 CrossRef.
- M.-H. Vu, M. Sakar, C.-C. Nguyen and T.-O. Do, ACS Sustainable Chem. Eng., 2018, 6, 4194–4203 CrossRef CAS.
- X. Han, D. Xu, L. An, C. Hou, Y. Li, Q. Zhang and H. Wang, Appl. Catal., B, 2019, 243, 136–144 CrossRef CAS.
- Z. Jin and L. Zhang, J. Mater. Sci. Technol., 2020, 49, 144–156 CrossRef CAS.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CrossRef CAS PubMed.
- L. H. Bennett, J. R. Cuthill, A. J. McAlister, N. E. Erickson and R. E. Watson, Science, 1974, 184, 563–565 CrossRef CAS PubMed.
- A. Kumar Singh, C. Das and A. Indra, Coord. Chem. Rev., 2022, 465, 214516 CrossRef CAS.
- R. Tong, Z. Sun, X. Wang, S. Wang and H. Pan, J. Phys. Chem. C, 2019, 123, 26136–26144 CrossRef CAS.
- Y. Zou, D. Ma, D. Sun, S. Mao, C. He, Z. Wang, X. Ji and J.-W. Shi, Appl. Surf. Sci., 2019, 473, 91–101 CrossRef CAS.
- L.-f Hong, R.-t Guo, Y. Yuan, X.-y Ji, Z.-d Lin, Z.-s Li and W.-g Pan, ChemSusChem, 2021, 14, 539–557 CrossRef CAS PubMed.
- K. He, J. Xie, Z. Yang, R. Shen, Y. Fang, S. Ma, X. Chen and X. Li, Catal. Sci. Technol., 2017, 7, 1193–1202 RSC.
- M. Shao, Y. Shao, S. Ding, R. Tong, X. Zhong, L. Yao, W. F. Ip, B. Xu, X.-Q. Shi, Y.-Y. Sun, X. Wang and H. Pan, ACS Sustainable Chem. Eng., 2019, 7, 4220–4229 CrossRef CAS.
- Y. Li, Z. Dong and L. Jiao, Adv. Energy Mater., 2020, 10, 1902104 CrossRef CAS.
- J.-W. Shi, Y. Zou, L. Cheng, D. Ma, D. Sun, S. Mao, L. Sun, C. He and Z. Wang, Chem. Eng. J., 2019, 378, 122161 CrossRef CAS.
- L. Cheng, S. Xie, Y. Zou, D. Ma, D. Sun, Z. Li, Z. Wang and J.-W. Shi, Int. J. Hydrogen Energy, 2019, 44, 4133–4142 CrossRef CAS.
- B. Li, W. Guo, X. F. Lu, Y. Hou, Z. Ding and S. Wang, Mater. Rep.: Energy, 2023, 3, 100230 CAS.
- X. Jiang, Q. Liu, C. Cheng, F. Xing, C. Chen and C. Huang, Int. J. Hydrogen Energy, 2021, 46, 5197–5206 CrossRef CAS.
- J.-Q. Yan, W. Peng, S.-S. Zhang, D.-P. Lei and J.-H. Huang, Int. J. Hydrogen Energy, 2020, 45, 16094–16104 CrossRef CAS.
- H. Zhou, R. Chen, C. Han, P. Wang, Z. Tong, B. Tan, Y. Huang and Z. Liu, J. Colloid Interface Sci., 2022, 610, 126–135 CrossRef CAS PubMed.
- C. Cheng, S. Zong, J. Shi, F. Xue, Y. Zhang, X. Guan, B. Zheng, J. Deng and L. Guo, Appl. Catal., B, 2020, 265, 118620 CrossRef CAS.
- C. Jin, C. Xu, W. Chang, X. Ma, X. Hu, E. Liu and J. Fan, J. Alloys Compd., 2019, 803, 205–215 CrossRef CAS.
- Z. Li, Y. Ma, X. Hu, E. Liu and J. Fan, Chin. J. Catal., 2019, 40, 434–445 CrossRef CAS.
- B. Ma, X. Li, D. Li and K. Lin, Appl. Catal., B, 2019, 256, 117865 CrossRef CAS.
- B. Ma, X. Wang, K. Lin, J. Li, Y. Liu, H. Zhan and W. Liu, Int. J. Hydrogen Energy, 2017, 42, 18977–18984 CrossRef CAS.
- D. Zhan, J. Tian, Q. Fu, P. Liu, Y. Zhao, W. Liu, D. Li, Y. Huang and C. Han, Appl. Surf. Sci., 2023, 641, 158463 CrossRef CAS.
- X. Zhou, Y. Fang, X. Cai, S. Zhang, S. Yang, H. Wang, X. Zhong and Y. Fang, ACS Appl. Mater. Interfaces, 2020, 12, 20579–20588 CrossRef CAS PubMed.
- S. Sun, Y.-C. Zhang, G. Shen, Y. Wang, X. Liu, Z. Duan, L. Pan, X. Zhang and J.-J. Zou, Appl. Catal., B, 2019, 243, 253–261 CrossRef CAS.
- M. Shao, H. Chen, S. Hao, H. Liu, Y. Cao, Y. Zhao, J. Jin, H. Dang, Y. Meng, Y. Huo and L. Cui, Appl. Surf. Sci., 2022, 577, 151857 CrossRef CAS.
| This journal is © the Owner Societies 2024 |