
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTime-resolved EPR observation of blue-light-induced radical ion pairs in a flavin–Trp dyad†
Yoshimi
Oka
 *abc and
Katsuya
Inoue
c
*abc and
Katsuya
Inoue
c
aFrontier Research Core for Life Sciences, University of Toyama, 2630 Sugitani, Toyama 930-0194, Japan
bResearch Promotion Institute, Oita University, 700 Dannoharu, Oita 870-1192, Japan
cGraduate School of Advanced Science and Engineering, Chirality Research Center (CResCent), and International Institute for Sustainability with Knotted Chiral Meta Matter (WPI–SKCM2), Hiroshima University, 1-3-1 Kagamiyama, Higashihiroshima 739-8526, Japan. E-mail: yoshimo@hiroshima-u.ac.jp
First published on 29th May 2024
Abstract
A dyad of flavin and Trp bridged by a p-phenylamide linker was synthesized as an artificial model system to investigate molecular-based magnetic-field sensors relevant to blue-light photoreceptor proteins. The results demonstrated that intramolecular electron transfer generates a radical pair, only the triplet-born one of which has a microsecond lifetime at room temperature.
Many biological systems, such as photosynthetic systems, use photoinduced electron-transfer (ET) reactions and the resultant long-lived charge-separated states to take advantage of natural light.1 Inspired by these systems, extensive efforts have so far been devoted to developing electron donor (D)–acceptor (A) linked molecules.2 Recently, it has been suggested that blue-light photoreceptor proteins in the photolyase/cryptochrome family including a flavin cofactor have a strong possibility of acting as highly sensitive magnetic compasses in nature.3–5 The mechanism for sensing weak magnetic fields (i.e., a geomagnetic field of ∼50 μT) is presumed to be that when flavin adenine dinucleotide (FAD) in photolyase/cryptochrome is irradiated with blue light, ET occurs from tryptophan (Trp) with charge separation (CS) to generate a radical pair (RP) between flavin and Trp (Scheme 1a, intermolecular ET).5–7 Spin-state mixing of the RP, i.e., interconversion between singlet (S) and triplet (T0 and T±1) states, is induced by an internal magnetic field attributed to (electron–nuclear) hyperfine interactions, and is additionally influenced by a weak external magnetic field comparable in strength to the internal field that produces the hyperfine interactions (Scheme 1c, intermolecular ET).4–6 This process is thus regarded as key for highly sensitive magnetic-field sensors, and is allowed by the precise D–A arrangement in the protein.5–8 Lifetimes on the order of microseconds to milliseconds have been observed for the final RP states (including subsequent ET between Trp triads) at distances of 10–20 Å (Scheme 1d and e, intermolecular ET).9 The radical type and reaction mechanism can be directly detected by electron paramagnetic resonance (EPR) spectroscopy, in particular by time-resolved EPR (TREPR).6,10 This method is useful for studying short-lived paramagnetic intermediates, such as triplet states and singlet/triplet-born RPs, by direct measurement of the electron-spin polarization as a function of time under a fixed external magnetic field.
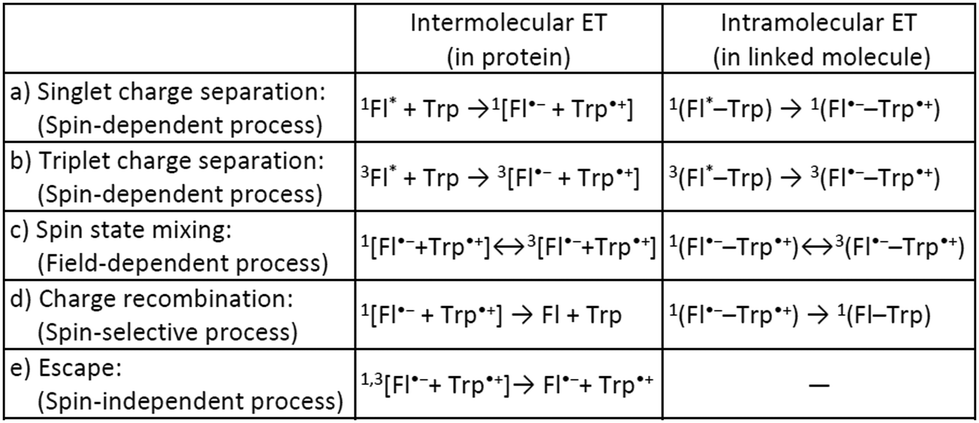 | ||
| Scheme 1 Photoinduced ET processes of Trp and flavin (Fl). | ||
As a model for magnetic field sensing proteins, the carotenoid–porphyrin–fullerene triad molecule, which is a well-known photosynthetic model, has been reported to produce RPs whose stability can be enhanced by a field of 50 μT.4 However, only a few attempts have been made to use flavin–Trp-linked molecules as magnetic-field sensors that can be controlled by spatial and orientational arrangements.11 On the other hand, there are examples of proteins, such as cryptochromes in migratory birds, for which it is difficult to express their functions even if reconstructed. Therefore, the correlation between structure and magnetic-field sensing function remains to be fully clarified. The present study focused on a dyad of flavin and Trp bridged by a linker, because such an approach was expected to allow the precise placement of D (i.e., Trp) and A (i.e., flavin). Herein, the synthesis, crystal structure, absorption and fluorescence properties and TREPR characterization of a flavin–Trp dyad bridged by a p-phenylamide linker (as illustrated in Scheme 2) are presented. We demonstrate the dynamics of a blue-light-induced CS reaction between flavin and Trp in the dyad by fluorescence quenching and TREPR measurements. The results obtained in this study are expected to shed light on the vague correlation between the structure and magnetosensing function in natural systems, and to contribute to the development of artificial D–A model systems that involve intramolecular vs. intermolecular interactions and singlet vs. triplet precursors.
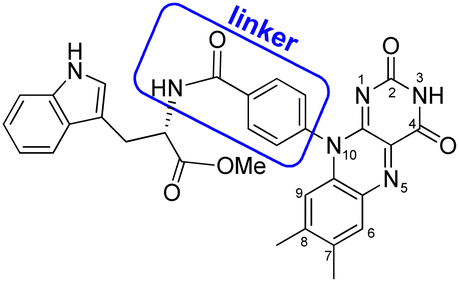 | ||
| Scheme 2 Molecular structure of flavin–Trp(OMe) dyad, 1. | ||
The flavin–Trp(OMe) dyad (1) bridged by a p-phenylamide linker, as shown in Scheme 2, was synthesized by a five-step reaction (Scheme S1 in ESI†). After the final dehydration step by the reaction between 4-(7′,8′-dimethylisoalloxazin-10′-yl)benzoic acid and L-tryptophan methyl ester hydrochloride, 1 was isolated by precipitation from a chloroform solution. 1 was then crystallized from a dimethyl sulfoxide (DMSO) solution at room temperature, and orange crystals suitable for X-ray structural analysis were obtained (see ESI†).
Compound 1 crystallizes in the space group P1, in which the S chirality of the tryptophan methyl ester is maintained. The crystal consists of two dyadic molecules of 1 that differ slightly in conformation, one water and six DMSO molecules per unit cell (Fig. 1 and Fig. S5–S7 in ESI†). The phenylamide linkers connect the flavin and Trp(OMe) units at the para positions, and the phenyl rings lie almost perpendicular to the isoalloxazine rings (dihedral angles: 73.8–108.7°). The resulting intramolecular distances between the centers of the isoalloxazine and indole rings and the nearest atoms to these rings are 11.4 Å and 7.6 Å, respectively. From the point of view of intermolecular interactions, π–π stacking and hydrogen bonding were found. A hydrogen bonding network is formed by the isoalloxazine C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at the 2-position and N–H at the 3-position to their inverted N–H and CO of the adjacent isoalloxazine, respectively, to produce an almost planar sheet (Fig. S8, ESI†). The indole ring in the dyad then interacts with the neighboring isoalloxazine, leading to head-to-tail stacking (Fig. 1), which results in a 3D network (Fig. S9, ESI†). The intermolecular separations of the centers of the isoalloxazine and indole rings are 4.3 and 4.4 Å, respectively, which are significantly shorter than the value of 8.5 Å reported for the cryptochrome of Arabidopsis thaliana.8 Furthermore, such intermolecular separations between the ring centers have been reported to be 3.3 and 3.4 Å in a 7,8-dimethylisoalloxazine-10-acetic acid
O at the 2-position and N–H at the 3-position to their inverted N–H and CO of the adjacent isoalloxazine, respectively, to produce an almost planar sheet (Fig. S8, ESI†). The indole ring in the dyad then interacts with the neighboring isoalloxazine, leading to head-to-tail stacking (Fig. 1), which results in a 3D network (Fig. S9, ESI†). The intermolecular separations of the centers of the isoalloxazine and indole rings are 4.3 and 4.4 Å, respectively, which are significantly shorter than the value of 8.5 Å reported for the cryptochrome of Arabidopsis thaliana.8 Furthermore, such intermolecular separations between the ring centers have been reported to be 3.3 and 3.4 Å in a 7,8-dimethylisoalloxazine-10-acetic acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L-tryptophan methylester(1:1)heptahydrate complex,12 whereas those in a synthetic flavin–Trp dyad linked by an alkyl linker were not observed at least crystallographically.13 Accordingly, the crystal structure of dyad 1 indicates that the intermolecular interaction of isoalloxazine–indole may be stronger due to the rigidity of the linker structure.
:L-tryptophan methylester(1:1)heptahydrate complex,12 whereas those in a synthetic flavin–Trp dyad linked by an alkyl linker were not observed at least crystallographically.13 Accordingly, the crystal structure of dyad 1 indicates that the intermolecular interaction of isoalloxazine–indole may be stronger due to the rigidity of the linker structure.
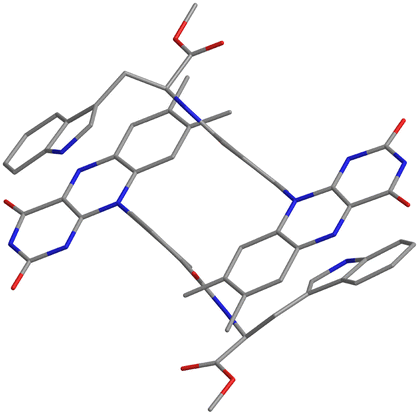 | ||
| Fig. 1 Crystal structure of 1 focusing on isoalloxazine and indole rings. Blue, red and gray represent N, O and C atoms, respectively. Solvent molecules and hydrogen atoms are omitted for simplicity. | ||
A spectroscopic study of 1 was conducted to obtain structural information in solution. The absorption maxima for 1 in DMSO solution at 298 K were located at 447 nm (S0–S1 of flavin, ε = 11200 M−1 cm−1) and 345 nm (S0–S2 of flavin, ε = 7300 M−1 cm−1), which are consistent with the values for the oxidized forms of common flavin compounds14,15 and 7,8-dimethyl-10-[4′-(methoxycarbonyl)phenyl]-isoalloxazine (abbreviated to FlH–MB, flavin–methyl benzoate) as a reference compound for 1.16 In contrast to the solution, the spectrum of a solid sample (KBr pellet) of 1 exhibited absorption maxima at approximately 480 nm and 415 nm, which could be assigned to intermolecular flavin–Trp charge transfer (CT) bands supported by the crystal structure described above. In other words, in solutions of 1 at concentrations below 100 μM, intramolecular interaction is likely to predominate, with little contribution from intermolecular interaction (Fig. S10, ESI†). Excitation of 1 at 450 nm resulted in fluorescence emission at 515 nm, which also corresponds well with the emission for the oxidized forms of flavins (Fig. S11, ESI†).13,15 Significant fluorescence quenching by Trp resulted in only 25% emission compared to FlH–MB. Based on a previous investigation, FlH–MB exhibits solvent-dependent aggregation: FlH–MB forms a self-assembled structure and self-quenches much more effectively in DMSO than in acetonitrile (MeCN).16 To investigate the solvent dependence of 1, the fluorescence quantum yield was evaluated, as well as that for FlH–MB.16 Contrary to expectations, 1 exhibited a smaller solvent effect, in that the fluorescence quantum yield for a 0.1 μM solution in DMSO was as low as 0.011, whereas that for a MeCN solution was 0.059 (Table S1, ESI†). The results are interpreted as the effects of quenching by intramolecular Trp, and only slightly by solvent-dependent intermolecular Trp.
To distinguish the contribution from collisional and static quenching,17 the fluorescence lifetime (τ1) for flavin in 1 was measured for the two solvents, and was found to be 0.64 ns for DMSO and 0.88 ns for MeCN at room temperature ∼293 K (Fig. S12, ESI†). As a control experiment, the fluorescence lifetime for a MeCN solution of FlH–MB was estimated to be 8.8 ns by fitting the data using a single exponential curve (Fig. S13, ESI†), which is probably indicative of decay without quenchers, defined as τ0(MeCN). The resulting ratio τ0(MeCN)/τ1(MeCN) was calculated to be 10, indicating that only collisional quenching contributes. A comparison of F0/F1 (= 9.9) and τ0/τ1 (= 10) indicates that there is little static contribution, i.e., the collisional effect is dominant for quenching by intramolecular Trp for flavin in MeCN solution. On the other hand, the fluorescence lifetimes τ1 for FlH–MB and 1 in DMSO were estimated to be 1.2 and 0.64 ns, respectively. These results can be interpreted as being complicated by aggregation, especially for 1, where there are mainly intramolecular and possibly intermolecular flavin–Trp interactions. To confirm the mechanism, the fluorescence quenching at several temperatures was compared (Fig. S15, ESI†). In most cases, except when intermolecular interactions are particularly strong, both fluorescence intensity (F) and lifetime (τ) should decrease with increasing temperature. Indeed, these temperature-dependent trends were observed in both solutions of 1, indicating that the intermolecular interactions may be weak. This interpretation of the results is also supported by little CT absorption being observed even in 100 μM DMSO solution. From the fluorescence quenching analysis of flavin, the photoinduced ET from (mainly intramolecular) Trp to flavin proceeds predominantly via a singlet precursor in both solutions of 1 (see ESI†).
TREPR studies were performed to gain insight into the photochemical reaction dynamics between flavin and Trp in 1. TREPR measurements for a 100 μM MeCN solution of 1 were recorded at room temperature (298 K) in three dimensions as a function of the magnetic field B0, and the time t, after pulsed laser excitation at 450 nm (Fig. S16, ESI†). Upon photoexcitation, 1 forms a spin-polarized paramagnetic species with a g-value centered at 2.0023, which can be assigned to RP intermediates generated from oxidized flavin, based on the spectral shape and narrow linewidth (full width at half maximum ∼1.5 mT).7,10 The polarized EPR spectrum appeared with an enhanced microwave emission (E) pattern around the resonant field region for the flavin anion radical (gFl ≈ 2.0034),18 and for the Trp cation radical (gTrp ≈ 2.0025),19 as shown in Fig. 2. Under identical experimental conditions, FlH–MB did not exhibit any TREPR signal above the noise level. Therefore, the Trp in the MeCN solution of 1 was confirmed to be essential as an electron donor for ET to the flavin, which results in the generation of flavin anion radicals (Fl˙−) and Trp cation radicals (Trp˙+). Simulations can be performed using the reported g-tensors, (gx, gy, gz) = (2.0043, 2.0039, 2.0022) for Fl˙−18 and (gx, gy, gz) = (2.0035, 2.0025, 2.0023) for Trp˙+,19 and the resulting exchange interaction parameter |J| ∼ 1–2 mT (which could not be precisely determined) and the dipolar interaction parameter between two electron spins of |d| ∼ 0.7 mT (which dominates the spectral shape), as shown by the red line in Fig. 2a.20 The time evolution at B0 = 344.8 mT revealed that the spectral intensity increased and then decreased while maintaining the spectral shape, and could be fitted by a single exponential function with a time constant of ∼5 μs (the red line in Fig. 2b). The features of the MeCN solution of 1 are similar to those reported for poly(3-hexylthiophene)–fulleropyrrolidine dyads without a bridge spacer, and the net E pattern is explained by a triplet mechanism (TM)10 by which the spin polarization of the precursor-excited triplet state is transferred to the sublevel populations.21 It has been reported that the linewidth becomes larger for the dyad without a spacer due to enhancement of electron spin dipolar coupling of d in the CS state.21 This situation is also most likely applicable to the MeCN solution of 1, as supported by the simulation. Based on the point-dipole approximation: DRP/mT = −2.782 × 103/(RD−A/Å)3, the dipole interaction d between Fl˙− and Trp˙+ of –0.7 mT corresponds to a distance of 15.8 Å, which is longer than the centers of the intramolecular flavin and Trp rings from the crystal structure analysis (i.e., 11.4 Å). This difference may indicate that the intramolecular structure is elongated in the solution. Even though the simulation is a rough approximation, this elongation seems reasonable, given the lack of intermolecular interaction between the dyad units in the crystal structure in Fig. 1. Most remarkably, TREPR data suggest that the photoinduced ET proceeds via a triplet precursor, which leads to a long-lived RP with a lifetime in the order of microseconds. According to the fluorescence quenching of flavin by Trp in the MeCN solution of 1, the ET reaction probably occurs almost exclusively via a singlet precursor. This discrepancy could be explained by the singlet precursor not contributing to the long-lived RP detected by TREPR,22 and only the minor amount of triplet precursor contributing to it. The lower generation of triplet-born RP in 1 than that reported for poly(3-hexylthiophene)–fulleropyrrolidine without a bridge spacer may be due to the absence of an S atom in the photoresponsive moiety and the resulting slower intersystem crossing rate (3.5 × 107 s−1 for 1vs. 1.3 × 109 s−1 for poly(3-hexylthiophene)–fulleropyrrolidine21).
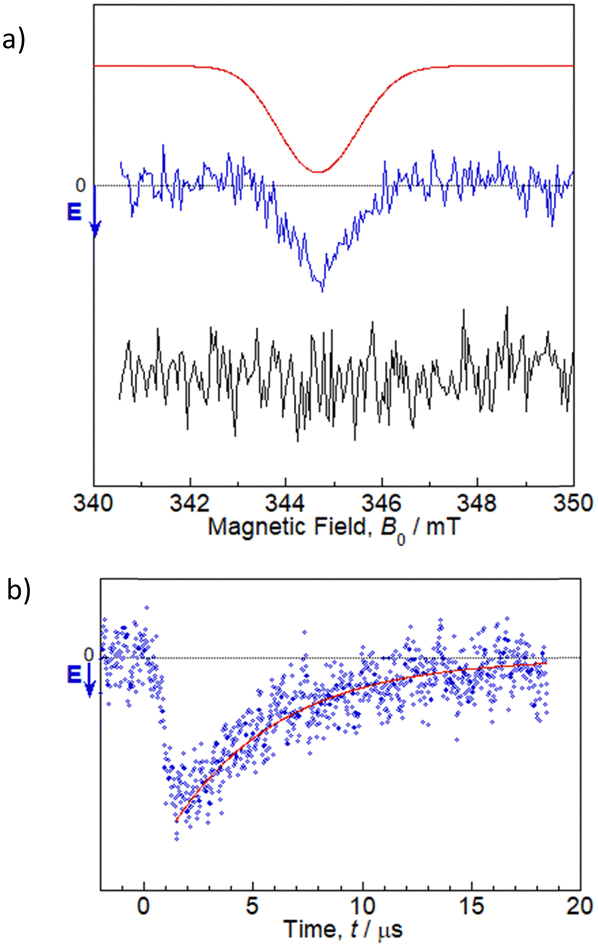 | ||
| Fig. 2 TREPR spectra for a 100 μM MeCN solution of 1 at 298 K. Microwave power: 2.1 mW. (a) Spectrum at a delay of 2 μs after the laser pulse (blue), simulation spectrum using the parameters described in the text (red), and a comparison with FlH–MB (black) as a reference. (b) Transient signal at B = 344.8 mT (blue) and exponential fitting (red). Details of the fitting are described in the text. | ||
On the other hand, the TREPR signal for a 100 μM DMSO solution of 1 recorded at room temperature (298 K) was not polarized (Fig. S18, ESI†). This may be attributed to reduced generation of long-lived RP in intramolecular flavin–Trp due to additional intermolecular interaction. To check the possibility of contribution by intermolecular interaction, FlH–MB was titrated by Trp(OMe) in both solutions (Fig. S19–S21, ESI†). The results showed little significant change during the titration, supporting the idea that intermolecular flavin–Trp may not contribute to RP generation in both solutions of 1. In other words, intramolecular flavin–Trp exclusively generates RP, regardless of whether the precursor is singlet or triplet in the solution state of 1.
To better understand the photochemical reaction dynamics, we employed density functional theory (DFT) and time-dependent (TD) DFT calculations using B3LYP/6-31+G(d,p)//B3LYP/6-31G(d) level with a polarizable continuum model (PCM)23 under the solvent (MeCN) field (see ESI†). Fig. 3 shows the frontier orbitals of the optimized singlet state and the spin density map of the optimized triplet state, which is characterized as a CS state 3(Fl˙−–Trp˙+) with an energy barrier of 2.15 eV above the singlet ground state. After the generation of the 3(Fl˙−–Trp˙+) state (Scheme 1b and c, intramolecular ET), charge recombination should occur to return to the singlet ground state via the 1(Fl˙−–Trp˙+) state (Scheme 1c and d, intramolecular ET). The energy gap between 3(Fl˙−–Trp˙+) and 1(Fl˙−–Trp˙+) using optimized triplet geometry may need a higher level of basis function especially during geometric optimization to evaluate charge recombination and exchange interaction.
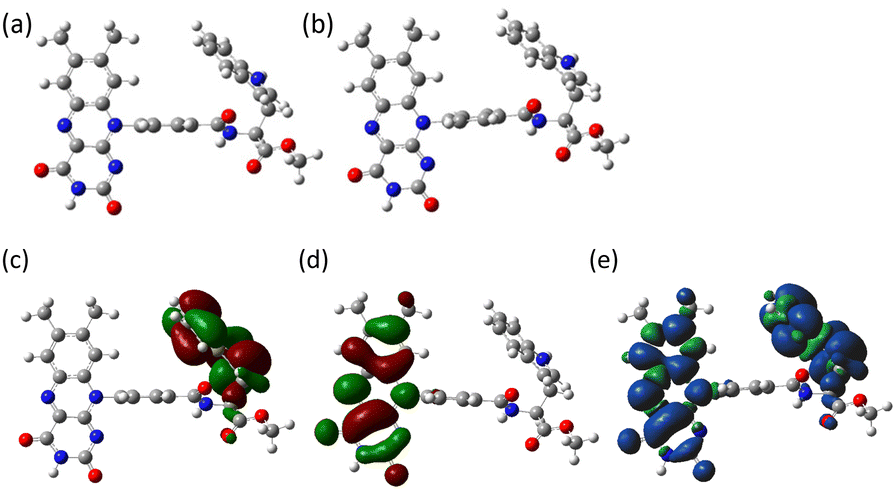 | ||
| Fig. 3 (a) Optimized ground state (S0 state) and (b) optimized triplet state (T1 state) geometries of 1 in MeCN (PCM model). The center-to-center distances of Fl–Trp rings are 9.4 Å in both (a) and (b). Frontier molecular orbitals, (c) HOMO and (d) LUMO of 1 in MeCN (PCM model), isovalue = 0.02 at the ground state geometry of S0. (e) Electron spin density surface of the triplet state of 1 in MeCN (PCM model), isovalue = 0.0004 at the optimized triplet state geometry of T1. Blue and green colors in (e) show α and β spins, respectively. | ||
In summary, a dyad of flavin and Trp bridged by a p-phenylamide linker, 1, was synthesized and characterized against the background that flavin–Trp-linked molecules, as model systems of flavoprotein magnetic-field sensors, were still largely unexplored. The center-to-center distance between intramolecular flavin and Trp rings of 1 (from crystal structure analysis and point-dipole approximation analysis of TREPR) was found to be in a similar range to that reported for RP detected in flavoproteins (10–20 Å), and 1 demonstrated the dynamics of blue-light-induced CS reaction between flavin (acting as A) and Trp (acting as D) by fluorescence quenching and TREPR measurements. Our findings may highlight a unique mechanism on the structure–RP generation mediated by the following competitions: (1) between intramolecular and intermolecular interaction, and especially (2) between singlet and triplet precursors. Although the crystal structure of 1 exhibited not only intramolecular but also intermolecular interactions between flavin and Trp, titration experiments clarified that the intramolecular interaction of flavin–Trp is almost exclusive in solutions of 1 at concentrations below 100 μM. In MeCN solution, the flavin–Trp dyad 1 was shown to have a much shorter fluorescence lifetime (0.88 ns at room temperature) than FlH–MB (8.8 ns at room temperature), suggesting that the singlet quenching process occurs efficiently to generate the singlet CS state 1(Fl˙−–Trp˙+) in 1; that is, the photoinduced ET from (mainly intramolecular) Trp to flavin proceeds predominantly via a singlet precursor in 1 and only faint triplet exciton of 3Fl*–Trp may be generated due to the slow ISC. However, in TREPR spectroscopy, a 100 μM MeCN solution of 1 exhibited a polarized emission (E) pattern, which could be explained by a triplet mechanism, suggesting that the RP is generated via a triplet precursor, and may persist in the order of microseconds. In contrast, RP mainly generated via a singlet precursor in the simple flavin–Trp dyad 1 may not contribute to a long-lived RP. Highly sensitive magnetoreception comparable to that of the photolyase/cryptochrome family could not have been achieved by the synthetic molecule in this study. However, we expect that this molecule will serve as a draft reference for addressing structure–magnetoreception correlation, which may be facilitated by artificial systems, especially through the precise design and synthesis of linker structures. The selection of rigid linkers, such as phenyl, ethynyl and their combinations, and possibly the use of reaction fields such as crystals or membranes, may allow for a certain orientation and fixation between intramolecular D and A units.
Y.O. would like to thank Prof. H. Masu for crystal structure analysis in Instrument and Research Technology Center, Chiba University. We are grateful to M. Fujiwara, T. Ueda and S. Iki for their support in TREPR measurements at Instrument Center (IC), Institute for Molecular Science (IMS). The fluorescence lifetime measurements were performed at IC, IMS and Instrumental Analysis Center, Gifu University. The computation was carried out using Research Center for Computational Science, Okazaki, and the computer resource offered under the category of Comprehensive Projects between Hiroshima University and Kyushu University by Research Institute for Information Technology, Kyushu University. This work was supported by Sekisui Chemical Grant Program for Research on Manufacturing Based on Innovations Inspired by Nature, Iketani Science and Technology Foundation, JSPS KAKENHI Grant Numbers JP16K13980, JP25870261 and JP22H02053, and MEXT Nanotechnology Platform Program “Molecule and Material Synthesis”.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Deisenhofer and J. R. Norris, The Photosynthetic Reaction Center, Academic Press, San Diego, 1993 Search PubMed; R. E. Blankenship, M. T. Madigan and C. E. Bauer, Anoxygenic Photosynthetic Bacteria, Kluwer Academic, Dordrecht, 1995 Search PubMed.
- H. Imahori, D. M. Guldi, K. Tamaki, Y. Yoshida, C. Luo, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 6617 CrossRef CAS PubMed; K. Ohkubo, H. Kotani, J. Shao, Z. Ou, K. M. Kadish, G. Li, R. K. Pandey, M. Fujitsuka, O. Ito, H. Imahori and S. Fukuzumi, Angew. Chem., Int. Ed., 2004, 116, 871 CrossRef; J. W. Verhoeven, J. Photochem. Photobiol., C, 2006, 7, 40 CrossRef.
- L. E. Foley, R. J. Gegear and S. M. Reppert, Nat. Commun., 2011, 2, 356 CrossRef CAS PubMed; T. Ritz, S. Adem and K. Schulten, Biophys. J., 2000, 78, 707 CrossRef PubMed; W. Wiltschko, J. Traudt, O. Güntürkün, H. Prior and R. Wiltschko, Nature, 2002, 419, 467 CrossRef PubMed; T. Ritz, P. Thalau, J. B. Phillips, R. Wiltschko and W. Wiltschko, Nature, 2004, 429, 177 CrossRef PubMed; R. Wiltschko, D. Gehring, S. Denzau, C. Nießner and W. Wiltschko, J. Exp. Biol., 2014, 217, 4225 CrossRef PubMed; K. B. Henbest, P. Kukura, C. T. Rodgers, P. J. Hore and C. R. Timmel, J. Am. Chem. Soc., 2004, 126, 8102 CrossRef PubMed; M. J. Golesworthy, T. Zollitsch, J. Luo, D. Selby, L. E. Jarocha, K. B. Henbest, O. Paré-Labrosse, R. Bartölke, J. Schmidt, J. Xu, H. Mouritsen, P. J. Hore, C. R. Timmel and S. R. Mackenzie, J. Chem. Phys., 2023, 159, 105102 CrossRef PubMed; L. Gerhards, C. Nielsen, D. R. Kattnig, P. J. Hore and I. A. Solovyov, J. Comput. Chem., 2023, 44, 1704 CrossRef PubMed.
- K. Maeda, K. B. Henbest, F. Cintolesi, I. Kuprov, C. T. Rodgers, P. A. Liddell, D. Gust, C. R. Timmel and P. J. Hore, Nature, 2008, 453, 387 CrossRef CAS PubMed.
- K. Maeda, A. J. Robinson, K. B. Henbest, H. J. Hogben, T. Biskup, M. Ahmad, E. Schleicher, S. Weber, C. R. Timmel and P. J. Hore, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4774 CrossRef CAS PubMed.
- T. Biskup, E. Schleicher, A. Okafuji, G. Link, K. Hitomi, E. D. Getzoff and S. Weber, Angew. Chem., Int. Ed., 2009, 48, 404 CrossRef CAS PubMed; D. Nohr, S. Weber and E. Schleicher, Methods Enzymol., 2019, 621, 251 Search PubMed.
- B. Giovani, M. Byrdin, M. Ahmad and K. Brettel, Nat. Struct. Biol., 2003, 10, 489 CrossRef CAS PubMed.
- C. A. Brautigam, B. S. Smith, Z. Ma, M. Palnitkar, D. R. Tomchick, M. Machius and J. Deisenhofer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12142 CrossRef CAS PubMed.
- D. Nohr, B. Paulus, R. Rodriguez, A. Okafuji, R. Bittl, E. Schleicher and S. Weber, Angew. Chem., Int. Ed., 2017, 56, 8550 CrossRef CAS PubMed; T. Hochstoeger, T. Said, D. Maestre, F. Walter, A. Vilceanu, M. Pedron, T. Cushion, W. Snider, S. Nimpf, G. Nordmann, L. Landler, N. Edelman, L. Kruppa, G. Dürnberger, K. Mechtler, S. Schuechner, E. Ogris, P. Malkemper, S. Weber, E. Schleicher and D. Keays, Sci. Adv., 2020, 6, eabb9110 CrossRef PubMed.
- A. J. Hoff, Advanced EPR: Applications in Biology and Biochemistry, Elsevier, Amsterdam, 1989 Search PubMed.
- S. Paul, L. Meng, S. Berger, G. Grampp, J. Matysik and X. Wang, ChemPhotoChem, 2017, 1, 12 CrossRef CAS; S. Wörner, J. Leier, N. C. Michenfelder, A.-N. Unterreiner and H.-A. Wagenknecht, ChemistryOpen, 2020, 9, e202000199 CrossRef PubMed; C. Bialas, L. E. Jarocha, K. B. Henbest, T. M. Zollitsch, G. Kodali, C. R. Timmel, S. R. Mackenzie, P. L. Dutton, C. C. Moser and P. J. Hore, J. Am. Chem. Soc., 2016, 138, 16584 CrossRef PubMed.
- M. Inoue, Y. Okuda, T. Ishida and M. Nakagaki, Arch. Biochem. Biophys., 1983, 227, 52 CrossRef CAS PubMed.
- R. E. MacKenzie, W. Föry and D. B. McCormick, Biochemistry, 1969, 8, 1839 CrossRef CAS PubMed.
- P. Hemmerich, C. Veeger and H. C. S. Wood, Angew. Chem., Int. Ed. Engl., 1965, 4, 671 CrossRef CAS PubMed.
- A. Kotaki and K. Yagi, J. Biochem., 1970, 68, 509 CrossRef CAS PubMed.
- Y. Oka, ACS Omega, 2020, 5, 21226 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 Search PubMed.
- A. Okafuji, A. Schnegg, E. Schleicher, K. Möbius and S. Weber, J. Phys. Chem. B, 2008, 112, 3568 CrossRef CAS PubMed.
- G. Bleifuss, M. Kolberg, S. Pötsch, W. Hofbauer, R. Bittl, W. Lubitz, A. Gräslund, G. Lassmann and F. Lendzian, Biochemistry, 2001, 40, 15362 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- T. Miura, R. Tao, S. Shibata, T. Umeyama, T. Tachikawa, H. Imahori and Y. Kobori, J. Am. Chem. Soc., 2016, 138, 5879 CrossRef CAS PubMed.
- Y. Oka, T. Miura and T. Ikoma, J. Phys. Chem. B, 2021, 125, 4057 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis, crystal structure, absorption and fluorescence spectra, fluorescence decay curves, TREPR spectra, titration results and DFT calculation results. CCDC 2153389. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cp06219h |
| This journal is © the Owner Societies 2024 |