
Holey penta-hexagonal graphene: a promising anode material for Li-ion batteries†
Linguo
Lu
a,
Raven
Gallenstein
b,
Xinghui
Liu
 cd,
Yi
Lin
e,
Shiru
Lin
*b and
Zhongfang
Chen
*f
cd,
Yi
Lin
e,
Shiru
Lin
*b and
Zhongfang
Chen
*f
aDepartment of Physics, University of Puerto Rico, Rio Piedras, San Juan, PR 00931, USA
bDivision of Chemistry and Biochemistry, Texas Woman's University, Denton, TX 76204, USA. E-mail: slin6@twu.edu
cCentre for Integrated Nanostructure Physics (CINAP), Institute of Basic Science (IBS), 2066 Seoburo, Jangan-Gu, Suwon 16419, Republic of Korea
dDepartment of Chemistry, Sungkyunkwan University (SKKU), 2066 Seoburo, Jangan-Gu, Suwon 16419, Republic of Korea
eAdvanced Materials and Processing Branch, NASA Langley Research Center, Hampton, Virginia 23681, USA
fDepartment of Chemistry, University of Puerto Rico, Rio Piedras, San Juan, PR 00931, USA. E-mail: zhongfang.chen1@upr.edu
First published on 8th February 2024
Abstract
Carbon allotropes are widely used as anode materials in Li batteries, with graphite being commercially successful. However, the limited capacity and cycling stability of graphite impede further advancement and hinder the development of electric vehicles. Herein, through density functional theory (DFT) computations and ab initio molecular dynamics (AIMD) simulations, we proposed holey penta-hexagonal graphene (HPhG) as a potential anode material, achieved through active site designing. Due to the internal electron accumulation from the π-bond, HPhG follows a single-layer adsorption mechanism on each side of the nanosheet, enabling a high theoretical capacity of 1094 mA h g−1 without the risk of vertical dendrite growth. HPhG also exhibits a low open circuit voltage of 0.29 V and a low ion migration barrier of 0.32 eV. Notably, during the charge/discharge process, the lattice only expands slightly by 1.1%, indicating excellent structural stability. This work provides valuable insights into anode material design and presents HPhG as a promising two-dimensional material for energy storage applications.
1. Introduction
Rechargeable Li-ion batteries (LIBs) are widely used in electric vehicles, meeting the surging requirement for clean and sustainable energy resources in recent years.1,2 In the pursuit of enhancing the performance of LIBs, carbon allotropes have emerged as promising candidates for anode materials due to their abundant source and cost-effectiveness.3,4 Notably, with excellent conductivity and cycling stability, graphite has become the dominant commercial anode material, but the limited capacity hinders its broader applications.5,6 Integration of two-dimensional (2D) materials may further improve the performance of LIBs and thus have been explored as an important strategy in next-generation anode innovation.7–10Graphene,11 essentially a single layer of graphite, holds immense potential with an impressive theoretical capacity up to 744 mA h g−1, since it follows a two-sided single-layer adsorption mechanism, instead of the staging intercalation reaction mechanism observed in graphite.12–14 However, the weak van der Waals interactions between monolayers may cause undesirable agglomeration, resulting in rapid performance degradation and compromised cycle stability.
To mitigate the restacking issue, holey graphene and its derivatives have emerged as a promising solution, introducing a porous structure that serves multiple purposes.15,16 Firstly, it effectively diminishes the weak interactions between neighboring layers, thus preventing agglomeration and maintaining structural integrity, and also provides additional cross-plane ion transport channels, facilitating a rapid charge/discharge process.17–20 Secondly, the specific porous framework enables accommodation of the local volume variation during Li-ion intercalation/extraction, thereby enhancing cycling.21 Great progress has been achieved in developing holey graphene-based anode materials for LIBs. In 2016, Alsharaeh et al. successfully synthesized holey reduced graphene oxides (HRGO) by utilizing an etching method involving Ag nanoparticles. This method yielded HRGO with specific porous structures, featuring pores ranging from 2 nm to 5 nm. The resulting material exhibited a remarkable capacity, reaching 2.5 times that of reduced graphene oxides, and demonstrated an impressive retention of 94.6% reversible capacity after 100 charging/discharging cycles.22 To further improve the cycling performance, Bulusheva et al. proposed that the presence of various decorated O-containing (O–X) groups within HRGO could potentially influence capacity fading.23 However, despite these advancements, the achieved capacity (around 430 mA h g−1) of holey graphene-based anode materials falls short when compared to the theoretical capacity of graphene, thus presenting a new challenge in the design of anode materials. Notably, by means of first principles computations, Yu showed that modifying graphene's vacancy defects with nitrogen can transform hole sites into additional Li adsorption sites, providing up to 124.5 mA h g−1 capacity per nitrogen decoration.24 Other strategies have also been proposed to increase specific capacities, such as optimization of the synthesis process,25 heteroatom doping modification,26–28 and compositional integration with transitional metal oxides.29,30
Recently, penta-graphene has been proposed as a promising carbon allotrope for high-capacity LIB anodes. Penta-graphene was proposed theoretically in 2015 as a unique carbon allotrope composed entirely of carbon pentagon rings (C5), instead of the hexagon rings (C6) found in graphene.31 This novel structure adopts a buckling multi-decker arrangement with both sp3 and sp2 hybridized carbon atoms. Due to the slightly distorted structure, the sp3-hybridized C atoms would split the pz orbitals of the sp2-hybridized ones and partially localize the electrons, generating an attractive adsorption potential for Li atoms at the sp2 sites. Recognizing this potential, Xiao et al. investigated the performance of penta-graphene as LIB anode materials, and found that the presence of abundant active sites and its lightweight nature contribute to a specific theoretical capacity of up to 1489 mA h g−1, while the isotropic furrow paths of the surface facilitate rapid metal ion diffusion.32 However, the sp2 restructuring process, which occurs in a stepwise exothermic manner, reveals a transformation tendency from penta-graphene to graphene, thereby highlighting the synthesis challenges associated with penta-graphene.33 Nevertheless, one theoretically derived synthesis route has been proposed, starting from the pentalene molecule and progressing to bispentalene and penta-graphene nanoribbons, offering a promising prospect for potential experimental production.34
Inspired by the dibenzocyclooctadiyne molecule first synthesized by Sondheimer and co-workers in 1974,35 Liu et al. successfully synthesized a new 2D carbon allotrope, namely holey graphyne (HGY),36 with a periodic structure. In dibenzocyclooctadiyne, two aromatic benzene rings are connected by two bent acetylenic linkages, resulting in a highly strained octagon ring (C8), and the antiaromatic paratropicity of the central C8 ring is considerably attenuated. In the case of HGY, the carbon framework consists of interlinked C6 rings connected by C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds, forming a distinct pattern of C6 rings alongside highly strained C8 rings. The fusion of aromatic rings and antiaromatic rings in HGY effectively impedes non-planar deformations and the disruption of π interactions within the C8 rings, leading to the stability improvement. This achievement not only introduced a fresh perspective on the design of 2D carbon allotropes, but also serves as an inspiration for exploring the application of this concept in the development of novel LIB anode materials.
C bonds, forming a distinct pattern of C6 rings alongside highly strained C8 rings. The fusion of aromatic rings and antiaromatic rings in HGY effectively impedes non-planar deformations and the disruption of π interactions within the C8 rings, leading to the stability improvement. This achievement not only introduced a fresh perspective on the design of 2D carbon allotropes, but also serves as an inspiration for exploring the application of this concept in the development of novel LIB anode materials.
Dibenzo[a,e]pentalene,37 a molecule comprising one antiaromatic pentalene and two aromatic benzene rings, caught our attention. The two fused benzene rings mitigate the antiaromatic character of pentalene, and thus enhance the stability of the whole molecule. This principle inspired us to design holey penta-hexagonal graphene (HPhG), a planar 2D nanomaterial (Fig. 1). The benzene rings preserve the planarity of neighboring C5 rings, maintaining their π-interactions and creating active sites for Li adsorption on the C5 rings. It is highly possible that the electron characteristic transformation from antiaromaticity to aromaticity for the C5 rings enables strong Li ion adsorption with a single-layer mechanism, preventing dendrite formation in anode applications.
 | ||
| Fig. 1 Top (a) and side (b) views of the optimized unit cell of HPhG, where the gray and white atoms represent C and H atoms, respectively. (c) Phonon dispersion of HPhG. The carbon and hydrogen atoms are denoted by grey and white, respectively. | ||
Herein, we report a new carbon allotrope, HPhG, by DFT computations. We verify its stabilities by binding energies, phonon spectrum, and ab initio molecular dynamics (AIMD) simulations. Our findings confirm that the HPhG monolayer exhibits robust stability as a planar semiconductor with a direct bandgap of 0.58 eV. Furthermore, we have explored the feasibility and mechanisms of HPhG as an anode material. Our computations demonstrate its high theoretical capacity of 1094 mA h g−1, moderate open circuit voltage of 0.29 V, and low ion diffusion barrier of 0.32 eV. Remarkably, the electronic conductivity of HPhG can be substantially enhanced upon Li ions adsorption, transforming it into a conductor. Moreover, the charge/discharge events follow a single-layer adsorption mechanism, effectively preventing the formation of dendrites, which can help improve safety and cycle stability in practical applications. Our work not only introduces HPhG as a new 2D high-capacity anode material for LIBs, but also provides valuable insights into anode material design.
2. Computational methods
In our DFT computations, the CASTEP code38 was employed for the majority of the computations, such as structure optimizations, thermodynamic and dynamic stability assessments of the material, and evaluation of HPhG's performance as LIB anode material, while the Vienna ab initio simulation package (VASP)39 was used for calculating the band structure of HPhG monolayer and evaluating its thermal stability.The electron exchange–correlation function was treated using the generalized gradient approximation (GGA), specifically the Perdew, Burke, and Ernzerhof (PBE) functional,40 and the ultrasoft pseudopotentials were used to describe the ion-electron interactions.41 The energy cutoff was set to 500 eV, and the convergence tolerance was 10−5 eV per atom. The Grimme's DFT-D method was employed to describe the van der Waals interactions,42 and the zero-point energy was neglected due to the marginal effect on the Li adsorption.43 To avoid spurious interactions between adjacent layers, a vacuum space of more than 15 Å was implemented in our computations. The Monkhorst–Pack k points were set as 8 × 8 × 1 for geometry optimization and self-consistent calculations to achieve precision requirements in actual spacing (at least 0.015 1 Å−1). We carried out both spin-polarized and spin-unpolarized computations and ensured the non-magnetic nature of HPhG monolayer.
Since the PBE functional typically underestimates the band gap in semiconductors and insulators, we employed the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional,44,45 which is known to provide a more accurate estimation of the band gap in many materials, to calculate the band structure of HPhG. We also performed AIMD simulations to evaluate the thermal stabilities. During AIMD simulations, the PBE functional and the canonical ensemble (NVT) with a Nose–Hoover thermostat46–48 were employed, and a 2 × 1 × 1 supercell was annealed at various temperatures. Each AIMD simulation was carried out for 10 ps with a time step of 1.0 fs.
3. Results and discussion
3.1. Structures, stability, and electronic properties
The HPhG monolayer exhibits a unique planar and porous structure, characterized by pore openings with a diameter of 5.31 Å. Its fundamental unit cell consists of 24 carbon atoms and six hydrogen atoms. As shown in Fig. 1a and b, among the carbon atoms in the five-membered rings (denoted as C5 atoms), the one at the pore edge shares a single C–C bond with its neighboring atom and is saturated by a hydrogen atom, similar to the antiaromatic molecular pentalene combining sp2- and sp3-hybridization. Notably, the carbon atoms in the six-membered rings (denoted as C6 atoms) in HPhG have two distinct bond types, with bond lengths of 1.38 Å and 1.46 Å, respectively. On the other hand, the C5 atoms display a range of bond lengths, varying between 1.38 Å and 1.44–1.48 Å. Compared with the antiaromatic pentalene (with bond lengths of 1.36 Å and 1.46–1.49 Å),49 the bond lengths in the C5 rings of the HPhG monolayer are more equalized, indicating its increased aromaticity, thus increasing the stability of the whole HPhG system.To investigate the stability of HPhG, we first evaluated its cohesive energies via the formula:
| Ecoh = (EHPhG − nCEC − nHEH)/(nC + nH) |
We then verified the dynamic stability of the HPhG monolayer by phonon dispersions. The absence of imaginary modes (Fig. 1c) in the first Brillouin zone confirms that it is dynamically stable. Given the importance of thermal stability as a critical metric for evaluating the performance across thermal ranges, as indicated in ref. 57, we further investigated this aspect through ab initio molecular dynamics (AIMD) simulations. These simulations were conducted over a duration of 10 ps at three distinct temperatures: 600 K, 800 K, and 1000 K, to comprehensively assess the material's thermal stability. HPhG monolayer maintained structural integrity with little ripples up to 1000 K (Fig. 2 and Fig. S1, ESI†). Note that the thermal stability of the HPhG monolayer is comparable to that of other related 2D materials. For instance, carbon allotropes like graphyne,58 N/W doped BP monolayer,59 and indium bismide,60 also exhibit thermal stability at 1000 K. This similarity underscores the HPhG monolayer's significant temperature tolerance, reinforcing its suitability for advanced battery applications.
 | ||
| Fig. 2 Initial and final structures of HPhG, accompanied by the energy evolution diagram, following a 10 ps AIMD simulation at 1000 K. The carbon and hydrogen atoms are denoted by grey and white, respectively. | ||
To study the electronic properties of HPhG, we calculated the band structure and Electron Localization Function (ELF) diagrams (shown in Fig. 3a and b). HPhG is a direct semiconductor with a narrow band gap of 0.58 eV (by HSE06 functional, 0.24 by PBE functional) as shown in Fig. 3a, which provides an acceptable conduction property. The ELF plot can be visualized as a contour plot in real space, displaying values ranging from 0 to 1. In this representation, a region with a value of 1 indicates perfect electron localization, suggesting the presence of localized electron pairs or strong covalent bonding. Conversely, a value of 0 signifies a region with low electron density, while a value of 0.5 implies the probability of a free electron-gas state. In the ELF plot, the presence of diffused low electron density surrounding carbon rings indicates the formation of π bonds within the HPhG monolayer. Conversely, the relatively high electron localization highlights the residual antiaromaticity of the C5 rings, resulting in a strong affinity for Li ions. Therefore, the excellent thermodynamic, dynamic, and thermal stabilities, as well as electronic properties, strongly indicate its high synthetic feasibility and application as electrode material for LIBs.
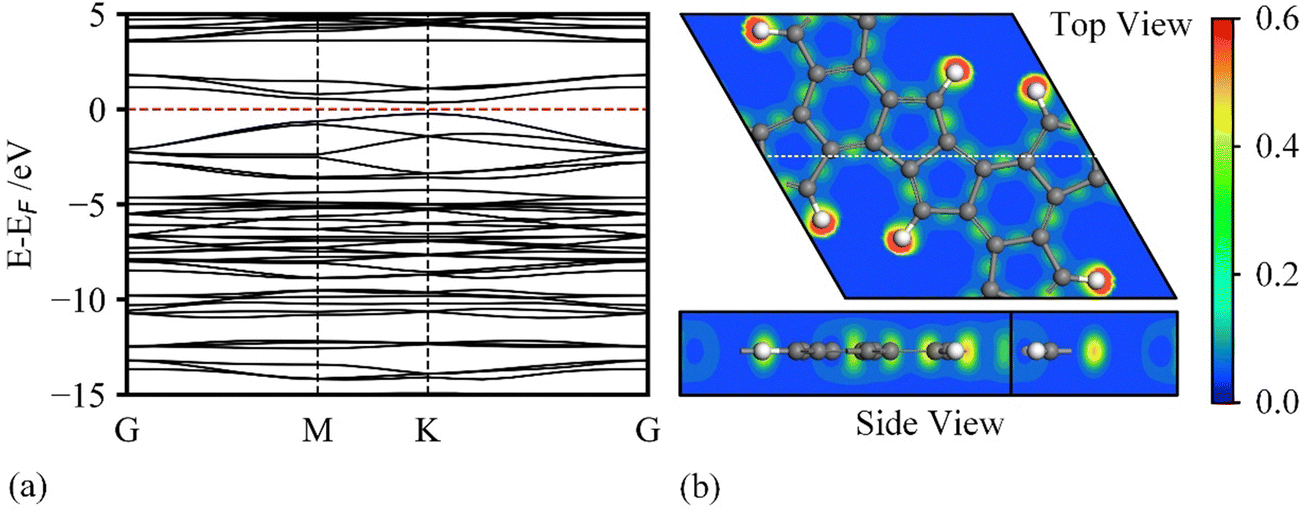 | ||
| Fig. 3 (a) Band structure of holey penta-hexagonal graphene (HPhG) with 0.58 eV band gap (by HSE06 functional). (b) Electron location function of the unit cell. The white dotted line in the top view is the slice section for the side view. The carbon and hydrogen atoms are denoted by grey and white, respectively. | ||
3.2. Application as an anode in LIBs
We first examined the adsorption structures of a single Li atom to evaluate the different possible adsorption sites and calculated the adsorption energies by:| Eads = Esub+nLi − Esub+(n−1)Li − ELi |
Based on symmetry considerations, we categorized the 15 potential sites into four distinct types: (1) polyatomic ring center positions, including site A above the C5 ring and site B above the C6 ring; (2) the bridge site between two bonded carbon atoms (sites C–sites H); (3) the top side of carbon atoms (the site I–site M); (4) side positions around the hollow surface, as the site N in the center and side site O close to the edge. All the proposed adsorption sites (from A to O) are schematically shown in Fig. S2 (ESI†). Among these 15 possible adsorption sites, only sites A, B, and O are thermodynamically stable with negative adsorption energy, which are illustrated in Fig. 4a. Li atoms in the whole bridge and top sites are transferred into A and B sites after geometry optimizations, and the hollow center site N is thermodynamically unstable. These unstable structures will not be considered.
 | ||
| Fig. 4 (a) Three adsorption sites with negative adsorption energy. (b) Top view and (c) side view of the configuration after maximum adsorption. (d) Top view and (e) side view of the charge density differences after maximum adsorption. The cyan and yellow regions represent the charge accumulation and dissipation, respectively. (f) Top and side views of ELF plots of the HPhG with the maximum Li adsorption, where six Li atoms are adsorbed on each side. The carbon, hydrogen, and Lithium atoms are denoted by grey, white, and purple, respectively. | ||
The calculated adsorption energy of Li at the A-, B-, and O-sites are −0.54, −0.53, and −0.40 eV, respectively. Since a more negative value of Eads corresponds to a higher binding strength between Li and the adsorption site, the A site is the most favorable adsorption position to store Li atoms during the charging process.
Theoretical storage capacity (TSC) and open-circuit voltage (OCV) are crucial parameters for evaluating the performance of electrode materials in LIBs from the thermodynamic point of view. In our study, we estimated these parameters by taking into account the Li adsorption on both sides of HPhG. In this way, we can assess the potential capacity for storing Li ions and determine the OCV, providing valuable insights into the electrochemical performance of HPhG as an anode material for LIBs. The OCV is calculated by the equation below:
| OCV = [Esub+n1Li − Esub+n2Li − (n1 − n2)ELi]/(n2 − n1)e |
Our calculations indicate that the adsorption of the first two layers of Li atoms (12 Li atoms, as depicted in Fig. 4b and c) is an exothermic process, characterized by negative adsorption energy. On the other hand, the adsorption of the third layer of Li atoms is an endothermic process, meaning it is not favorable energetically. When the maximum concentration of Li atoms is used (with two layers of adsorbed Li atoms), the theoretical storage capacity approaches 1094 mA hg−1, which is much higher than various carbon-based materials and 2D materials, as shown in Table 1. A higher OCV is beneficial for enhancing lithium storage capability but can hinder charge–discharge. To achieve rapid charge and discharge it is essential to maintain an optimal OCV range. Anodes generally prefer a low OCV, while cathodes perform better with a moderately high OCV. In the case of anode materials, typically retaining an OCV range of 0.1–1 V is ideal to prevent the formation of alkali metal dendrites during discharge/charge.61 For the systems under consideration, the OCVs associated with one and two layers of adsorption are 0.35 V and 0.23 V, respectively, and the average OCV is 0.29 V, falling within the ideal potential range. Both the theoretical specific capacity (TSC) and the OCV values indicate that HPhG serves as a high-performance anode material for lithium-ion batteries.
| Materials | Capacity | Diffusion barrier | OCV | Ref. |
|---|---|---|---|---|
| C2N | 2939 | 0.80 | 0.45 | 62 |
| Holey-graphite-46 | 1689 | 0.38 | 0.86 | 63 |
| B-doped graphene | 1564 | 0.24 | 0.37 | 64 |
| Penta-graphene | 1489 | 0.17 | 0.55 | 32 |
| HfS2 | 1378 | 0.13 | 2.37 | 65 |
| Graphenylene | 1116 | 0.25 | 0.46 | 66 |
| HPhG | 1049 | 0.32 | 0.29 | This work |
| C6H4O2-2D-MnO2 | 588 | 0.28 | 2.21 | 67 |
| Borophane | 504 | 0.21 | 0.43 | 68 |
| VS2 | 466 | 0.22 | 0.93 | 69 |
| Ti3C2 | 447.8 | 0.07 | 0.41 | 70 |
| Phosphorene | 433 | 0.76 | 1.63 | 71 |
| ψ-graphene | 372 | 0.31 | 0.01 | 72 |
To investigate the microscopic mechanism of Li adsorption, we conducted several analyses. First, we examined the charge density differences in the maximum adsorption structure (Fig. 4d and e), and found that electrons are accumulated between the Li atoms layer and carbon rings. Notably, there is a continuous charge distribution above the carbon rings (indicated by the blue color), forming a π-bond-like ring configuration, which enhances the delocalization of the charge spatial distribution. Then, we plotted the ELFs (Fig. 4f) to understand the adsorption behavior further. Compared with the ELF diagram before Li adsorption (in Fig. 3b), the HPhG with 12 Li atoms showed higher electron density at the π bond region and lower electron localization with dimmer spots. The feasible Li adsorption facilitated the transition of the C5 rings on the HPhG to a less localized state, which significantly improved the conductivity, transforming it from a semiconductor to a conductor (shown in Fig. S3, ESI†). The enhanced π-bonds preserved the maximum p orbital overlap, maintaining the characteristic planar structure. Furthermore, considering the adsorbed Li atoms, the electron cloud dissipation (the yellow part in Fig. 4d and e) and the absence of electron arrangement (in Fig. S4, ESI†) chemisorption appeared to exclusively occur between the adsorbates and the substrate, effectively preventing lateral dendrite growth between Li atoms. The lack of electron cloud arrangement also helped maintain the single-layer adsorption mechanism in HPhG and mitigates the risk of vertical dendrite growth. Moreover, there was no electron distribution outside the first Li atoms layer. It increased the transfer barrier of electron channels between the first Li layer and potential extra layers, which served to uphold the single-layer adsorption mechanism in HPhG and effectively prevented the formation of vertical dendrites.
The formation of Li clusters is undesirable and will affect cycle performance. After the adsorption on both sides, the nearest distance between two adsorbed Li atoms is 2.82 Å. It is larger than the bond length in Li2 dimers (2.67 Å),73 which further implies the prevention of Li clusters. From the adsorption and growth mechanism of Li storage mode, the one-layer adsorption mechanism on both sides of HPhG prevents disordered growth and avoids the Li vertical dendrite, which significantly improves its safety and feasibility in future applications. During the Li loading, the HPhG monolayer with 12 adsorbed Li atoms maintains its planar structure while the lattice constant expands slightly by 1.1%, indicating its high structural stability. Therefore, the anode material potential of HPhG shows a suitable capacity and good cycling stability.
To examine the Li transport kinetics of HPhG, we calculated the migration of Li atoms between adjacent A sites using the Linear Synchronous Transit/Quadratic Synchronous Transit (LST/QST) method.74 From the perspective of the phase space, the stability of each geometry structure can be considered as one point in the energy surface, and the stable structures are at the local (B- and O-site) or global minimum (A-site). When a Li atom from one adsorption site transfers into a new one, it corresponds to the energy points’ movement on the 3D surface. Typically, this transition occurs through a specific state known as the transition state. The presence of a transition state serves as a valuable means to assess the validity of proposed diffusion paths, providing an effective method for evaluating the accuracy of such pathways.
Thus, we examined three potential Li diffusion paths, as illustrated in Fig. 5a. Path-I involves the transfer of Li atoms between two interconnected A–A sites. In path-II, Li atoms move along the A–B–A sites on the surface, path-III entails crossing the surface hollow via A–O–A sites. However, there are no viable transition states identified between A- and O-sites, suggesting that path-III is unlikely. As shown in Fig. 5b and c, the computed diffusion barriers for path-I and path-II are 0.21 eV and 0.32 eV, respectively, with Path-II being the step that limits the overall rate of the diffusion process. The diffusion barrier of HPhG is lower than that of commercial graphite (0.4–0.6 eV) and compares favorably with various 2D electrode materials, as shown in Table 1. Moreover, the energy gap vanishes upon Li adsorption (Fig. S3, ESI†), resulting in a distinctive metallic characteristic and good electronic conductivity. Consequently, the combination of fast ion diffusion and exceptional electronic conductivity in HPhG further indicates its good performance as an anode material for LIBs.
 | ||
| Fig. 5 (a) Three proposed ion transition pathways. (b) and (c) are the diagrams of transition structures and energy barriers. The carbon, hydrogen, and lithium atoms are denoted by grey, white, and purple, respectively. | ||
4. Conclusion
By means of DFT computations and ab initio molecular dynamic simulations, we identified HPhG as a promising 2D holey carbon material for application as an anode material for LIBs. HPhG is thermodynamically, dynamically, and thermally stable. Moreover, upon Li adsorption, HPhG undergoes a semiconductor-to-conductor transformation, showing improved electrical conductivity. The low diffusion barrier (up to 0.32 eV) indicates the fast charge/discharge rate for LIBs. The maximum Li capacity reaches an impressive 1094 mA h g−1, accompanied by a competitively low OCV of 0.29 V. By using charge density difference and ELF analyses, we have identified the single-layer adsorption mechanism and elucidated the reasons for the prevention of dendrite growth in HPhG. The concept of HPhG represents a remarkable exploration into harnessing the distinctive properties of pentalene and graphene-like structures. This investigation offers fundamental insights that can guide the design of future anode candidates through comprehensive bond analysis. With exceptional properties, we strongly believe that HPhG will emerge as a new platform for the development of high-performance anode materials for advanced lithium-based energy storage systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF Centre for the Advancement of Wearable Technologies (grant 1849243) and by NASA (grant number 80NSSC19M0236). S. L. acknowledges the startup fund from Texas Woman's University.References
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- A. Rajkamal and R. Thapa, Adv. Mater. Technol., 2019, 4, 1900307 CrossRef CAS.
- M. M. Obeid and Q. Sun, Carbon, 2022, 188, 95–103 CrossRef CAS.
- H.-H. Lee, C.-C. Wan and Y.-Y. Wang, J. Power Sources, 2003, 114, 285–291 CrossRef CAS.
- K. Persson, Y. Hinuma, Y. S. Meng, A. Van der Ven and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125416 CrossRef.
- Y. Jing, Z. Zhou, C. R. Cabrera and Z. Chen, J. Mater. Chem. A, 2014, 2, 12104–12122 RSC.
- C. Zhang, A. Wang, J. Zhang, X. Guan, W. Tang and J. Luo, Adv. Energy Mater., 2018, 8, 1802833 CrossRef.
- Z. Xiao, R. Wang, D. Jiang, Z. Qian, Y. Li, K. Yang, Y. Sun, Z. Zeng and F. Wu, ACS Appl. Energy Mater., 2021, 4, 7440–7461 CrossRef CAS.
- A. Hayat, M. Sohail, A. El Jery, K. M. Al-Zaydi, S. Raza, H. Ali, Z. Ajmal, A. Zada, T. A. Taha, I. U. Din, M. A. Khan, M. A. Amin, Y. Al-Hadeethi, A. Z. Barasheed, Y. Orooji, J. Khan and M. Z. Ansari, Energy Storage Mater., 2023, 59, 102780 CrossRef.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- G. Wang, X. Shen, J. Yao and J. Park, Carbon, 2009, 47, 2049–2053 CrossRef CAS.
- Y. Liu, Y. M. Wang, B. I. Yakobson and B. C. Wood, Phys. Rev. Lett., 2014, 113, 028304–028308 CrossRef CAS PubMed.
- Y. Jing, Z. Zhou, C. R. Cabrera and Z. Chen, J. Mater. Chem. A, 2014, 2, 12104–12122 RSC.
- X. Han, M. R. Funk, F. Shen, Y. C. Chen, Y. Li, C. J. Campbell, J. Dai, X. Yang, J. W. Kim, Y. Liao, J. W. Connell, V. Barone, Z. Chen, Y. Lin and L. Hu, ACS Nano, 2014, 8, 8255–8265 CrossRef CAS PubMed.
- Y. Lin, X. Han, C. J. Campbell, J. W. Kim, B. Zhao, W. Luo, J. Dai, L. Hu and J. W. Connell, Adv. Funct. Mater., 2015, 25, 2920–2927 CrossRef CAS.
- T. Liu, L. Zhang, B. Cheng, X. Hu and J. Yu, Cell Rep. Phys. Sci., 2020, 1, 100215 CrossRef CAS.
- A. C. Lokhande, I. A. Qattan, C. D. Lokhande and S. P. Patole, J. Mater. Chem. A, 2020, 8, 918–977 RSC.
- Y. Lin, Y. Liao, Z. Chen and J. W. Connell, Mater. Res. Lett., 2017, 5, 209–234 CrossRef CAS.
- Z. Jiang, B. Pei and A. Manthiram, J. Mater. Chem. A, 2013, 1, 7775–7781 RSC.
- J. Xu, Y. Lin, J. W. Connell and L. Dai, Small, 2015, 11, 6179–6185 CrossRef CAS PubMed.
- E. Alsharaeh, F. Ahmed, Y. Aldawsari, M. Khasawneh, H. Abuhimd and M. Alshahrani, Sci. Rep., 2016, 6, 29854–29866 CrossRef CAS PubMed.
- L. G. Bulusheva, S. G. Stolyarova, A. L. Chuvilin, Y. V. Shubin, I. P. Asanov, A. M. Sorokin, M. S. Mel’Gunov, S. Zhang, Y. Dong, X. Chen, H. Song and A. V. Okotrub, Nanotechnol., 2018, 29, 134001 CrossRef CAS PubMed.
- Y. X. Yu, Phys. Chem. Chem. Phys., 2013, 15, 16819–16827 RSC.
- Y. Lin, C. O. Plaza-Rivera, L. Hu and J. W. Connell, Acc. Chem. Res., 2022, 55, 3020–3031 CrossRef CAS PubMed.
- X. Wang, L. Lv, Z. Cheng, J. Gao, L. Dong, C. Hu and L. Qu, Adv. Energy Mater., 2016, 6, 1502100 CrossRef.
- D. Wu, Y. Niu, C. Wang, H. Wu, Q. Li, Z. Chen, B. Xu, H. Li and L. Y. Zhang, J. Colloid Interface Sci., 2019, 552, 633–638 CrossRef CAS PubMed.
- R. Zhang, Y. Hou, X. Guo, X. Li, W. Li, X. Tao and Y. Huang, Phys. Chem. Chem. Phys., 2023, 26, 455–462 RSC.
- C. Zhu, Z. Hui, H. Pan, S. Zhu, Q. Zhang, J. Mao, Z. Guo, Y. Li, M. Imtiaz and Z. Chen, J. Mater. Chem. A, 2019, 7, 4788–4796 RSC.
- D. Wu, W. Zhao, H. Wu, Z. Chen, H. Li and L. Y. Zhang, Scr. Mater., 2020, 178, 187–192 CrossRef CAS.
- S. Zhang, J. Zhou, Q. Wang, X. Chen, Y. Kawazoe and P. Jena, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2372–2377 CrossRef CAS PubMed.
- B. Xiao, Y. Li, X. Yu and J. Cheng, ACS Appl. Mater. Interfaces, 2016, 8, 35342–35352 CrossRef CAS PubMed.
- C. P. Ewels, X. Rocquefelte, H. W. Kroto, M. J. Rayson, P. R. Briddon and M. I. Heggie, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15609–15612 CrossRef CAS PubMed.
- S. Li, Y. Shen, D. Ni and Q. Wang, J. Mater. Chem. A, 2021, 9, 23214–23222 RSC.
- H. N. C. Wong, P. J. Garratt and F. Sondheimer, J. Am. Chem. Soc., 1974, 96, 5604–5605 CrossRef CAS.
- X. Liu, S. M. Cho, S. Lin, Z. Chen, W. Choi, Y.-M. Kim, E. Yun, E. H. Baek, D. H. Ryu and H. Lee, Matter, 2022, 5, 1–13 CrossRef.
- M. Saito, M. Nakamura, T. Tajima and M. Yoshioka, Angew. Chem., Int. Ed., 2007, 46, 1504–1507 CrossRef CAS PubMed.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045–1097 CrossRef CAS.
- G. Kresse and J. Furthmü, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. M. Smith, S. P. Jones and L. D. White, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- Y. X. Yu, Phys. Chem. Chem. Phys., 2024, 26, 323–335 RSC.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Paier, M. Marsman, K. Hummer, G. Kresse, I. C. Gerber and J. G. Angyán, J. Chem. Phys., 2006, 124, 154709 CrossRef CAS PubMed.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- S. Nose, Prog. Theor. Phys. Suppl., 1991, 103, 1–46 CrossRef CAS.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- T. Bally, S. Chai, M. Neuenschwander and Z. Zhu, J. Am. Chem. Soc., 1997, 119, 1869–1875 CrossRef CAS.
- X.-L. Sheng, Q.-B. Yan, F. Ye, Q.-R. Zheng and G. Su, Phys. Rev. Lett., 2011, 106, 155703 CrossRef PubMed.
- A. R. Puigdollers, G. Alonso and P. Gamallo, Carbon, 2016, 96, 879–887 CrossRef CAS.
- Y. Yuan, J. Ma, S. Wu, J. Y. Lee and B. Kang, Chem. Phys., 2021, 548, 111257 CrossRef CAS.
- M. Naseri, J. Jalilian, D. R. Salahub, M. P. Lourenço and G. Rezaei, Computation, 2022, 10, 19–27 CrossRef CAS PubMed.
- M. Wang, Z. Zhang, Y. Gong, S. Zhou, J. Wang, Z. Wang, S. Wei, W. Guo and X. Lu, Appl. Surf. Sci., 2020, 502, 144067 CrossRef CAS.
- C. Liu, Z. Liu, X. Ye, P. Cheng and Y. Li, RSC Adv., 2020, 10, 35349–35355 RSC.
- P. Álvarez-Zapatero, A. Herrero, A. Lebon, L. J. Gallego and A. Vega, Int. J. Hydrogen Energy, 2021, 46, 15724–15737 CrossRef.
- D. G. Sangiovanni, R. Faccio, G. K. Gueorguiev and A. Kakanakova-Georgieva, Phys. Chem. Chem. Phys., 2022, 25, 829–837 RSC.
- D. Yang, R. Jia, X. Wang and S. Bai, ACS Appl. Nano Mater., 2023, 6, 16684–16693 CrossRef CAS.
- J. Wu, J. H. Li and Y. X. Yu, ACS Appl. Mater. Interfaces, 2021, 13, 10026–10036 CrossRef CAS PubMed.
- C. Lundgren, A. Kakanakova-Georgieva and G. K. Gueorguiev, Nanotechnol., 2022, 33, 335706 CrossRef PubMed.
- D. Çakir, C. Sevik, O. Gülseren and F. M. Peeters, J. Mater. Chem. A, 2016, 4, 6029–6035 RSC.
- D. Wu, B. Yang, H. Chen and E. Ruckenstein, Energy Storage Mater., 2019, 16, 574–580 CrossRef.
- C. Yang, X. Zhang, J. Li, J. Ma, L. Xu, J. Yang, S. Liu, S. Fang, Y. Li, X. Sun, X. Yang, F. Pan, J. Lu and D. Yu, Electrochim. Acta, 2020, 346, 136244 CrossRef CAS.
- X. Guo, Y. Hou, X. Chen, R. Zhang, W. Li, X. Tao and Y. Huang, Phys. Chem. Chem. Phys., 2022, 24, 21452–21460 RSC.
- Y. Kadioglu and Phys Chem, Chem. Phys., 2022, 25, 1114–1122 Search PubMed.
- Y. X. Yu, J. Mater. Chem. A, 2013, 1, 13559–13566 RSC.
- J. H. Li, J. Wu and Y. X. Yu, J. Phys. Chem. C, 2021, 125, 3725–3732 CrossRef CAS.
- N. K. Jena, R. B. Araujo, V. Shukla and R. Ahuja, ACS Appl. Mater. Interfaces, 2017, 9, 16148–16158 CrossRef CAS PubMed.
- Y. Jing, Z. Zhou, C. R. Cabrera and Z. Chen, J. Phys. Chem. C, 2013, 117, 25409–25413 CrossRef CAS.
- D. Er, J. Li, M. Naguib, Y. Gogotsi and V. B. Shenoy, ACS Appl. Mater. Interfaces, 2014, 6, 11173–11179 CrossRef CAS PubMed.
- S. Zhao, W. Kang and J. Xue, J. Mater. Chem. A, 2014, 2, 19046–19052 RSC.
- X. Li, Q. Wang and P. Jena, J. Phys. Chem. Lett., 2017, 8, 3234–3241 CrossRef CAS PubMed.
- H. L. Xu, Z. R. Li, D. Wu, B. Q. Wang, Y. Li, F. L. Gu and Y. Aoki, J. Am. Chem. Soc., 2007, 129, 2967–2970 CrossRef CAS PubMed.
- N. Govind, M. Petersen, G. Fitzgerald, D. King-Smith and J. Andzelm, Comput. Mater. Sci., 2003, 28, 250–258 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp06146a |
| This journal is © the Owner Societies 2024 |