
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModeling of Cu(II)-based protein spin labels using rotamer libraries†
Zikri
Hasanbasri‡
a,
Maxx H.
Tessmer‡
 b,
Stefan
Stoll
*b and
Sunil
Saxena
*a
b,
Stefan
Stoll
*b and
Sunil
Saxena
*a
aDepartment of Chemistry, University of Pittsburgh, PA 15260, USA. E-mail: sksaxena@pitt.edu; Tel: 412-624-8680
bDepartment of Chemistry, University of Washington, WA 98195, USA. E-mail: stst@uw.edu; Tel: 206-543-2906
First published on 1st February 2024
Abstract
The bifunctional spin label double-histidine copper-(II) capped with nitrilotriacetate [dHis-Cu(II)-NTA], used in conjunction with electron paramagnetic resonance (EPR) methods can provide high-resolution distance data for investigating protein structure and backbone conformational diversity. Quantitative utilization of this data is limited due to a lack of rapid and accurate dHis-Cu(II)-NTA modeling methods that can be used to translate experimental data into modeling restraints. Here, we develop two dHis-Cu(II)-NTA rotamer libraries using a set of recently published molecular dynamics simulations and a semi-empirical meta-dynamics-based conformational ensemble sampling tool for use with the recently developed chiLife bifunctional spin label modeling method. The accuracy of both the libraries and the modeling method are tested by comparing model predictions to experimentally determined distance distributions. We show that this method is accurate with absolute deviation between the predicted and experimental modes between 0.0–1.2 Å with an average of 0.6 Å over the test data used. In doing so, we also validate the generality of the chiLife bifunctional label modeling method. Taken together, the increased structural resolution and modeling accuracy of dHis-Cu(II)-NTA over other spin labels promise improvements in the accuracy and resolution of protein models by EPR.
Introduction
Pulsed dipolar EPR spectroscopy coupled with site-directed spin labeling (SDSL) is a powerful tool for obtaining sparse distance restraints1–7 that relate to the structure and conformational plasticity of proteins. When a protein is engineered to have two spin labels, distance measurements between the labels provide information about conformational changes,8–18 ligand binding site location,19–24 and tertiary and quaternary structures of large proteins and protein assemblies.25–32 In these applications, the prediction of spin label conformations is invaluable. Specifically, pulse EPR measurements primarily report on the distance between the unpaired electrons, which are usually localized at the end of a side chain, several Ångströms away from the protein backbone. Hence, understanding label conformations is essential for extracting information about backbone conformations and dynamics from the spin–spin distances.32–34 Predicting the distance distributions can also determine whether the experimental measurements correspond to a known PDB structure or generated protein model. Additionally, the conformational space of the spin label can shed light on whether the breadth of the distance distribution is due to the flexibility of the protein or the spin label itself. An understanding of the spin label rotamers is also crucial for assessing the interactions between the label and the surrounding environment in different protein conformations.17,35–40 Additionally, effective experimental designs requires rapid spin label modeling methods, such as in silico screening to identify site pairs that will provide a high distance contrast between two or more states of interest.37,41 Overall, accurate and fast spin label modeling methods are essential for experimental design and the interpretation of SDSL EPR data for the investigation of protein structure, allostery, and conformational heterogeneity.For monofunctional labels, such as the widely used S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate (MTSL), many in silico prediction strategies are available.38,42,43 However, methods to calculate the preferred rotamers for bifunctional labels, such as RX44,45 and dHis-Cu(II),46–48 are more limited. These bifunctional labels are rigidly attached to two nearby residues, significantly restraining their conformational flexibility. The restrained conformations make bifunctional labels highly desirable since they minimize the influence of spin label dynamics on the experimental data and, by extension, improve the resolution of the protein backbone structure and dynamics. Recently, a general rotamer library approach was developed for bifunctional spin label in the modeling package chiLife and applied to the RX label to predict distance distributions given a protein structure model.49 While this method showed promising results, it has not been applied to bifunctional spin labels beyond RX, as has been done with monofunctional label modeling methods.43 Furthermore, utilization of the RX spin label requires the introduction of 4 non-native cysteines, which can make purification and labeling cumbersome, prone to intra- and intermolecular cross-linking artifacts, and suffer from low yield. Therefore, developing an accurate and accessible rotamer library and validating the chiLife bifunctional label modeling method is desirable for other bifunctional labels, such as dHis-Cu(II), to expand the toolkit of convenient labels that can be accurately modeled.
The dHis-Cu(II) label consists of a Cu(II) coordinated to a chelator, such as iminodiacetic acid or nitrilotriacetic acid (NTA),46,48 and two strategically-placed histidine residues, referred to as the dHis motif.47Fig. 1 shows the structure of dHis-Cu(II)-NTA. Because the labeling strategy uses histidine residues, the label can be used in proteins without removing native cysteine residues that may be critical for function or protein folding. Furthermore, the highly restrained Cu(II) provides distance distributions that are up to five times narrower than those obtained from the common monofunctional nitroxide labels.47 Such narrow distributions are particularly well-suited for discerning small conformational changes.50 The narrow distributions also enable trilateration of a native metal binding site with the fewest distance measurements reported.20 In addition to distances, the rigidity of dHis-Cu(II)-NTA can resolve the relative orientations of the two labeled sites,51 providing additional structural information. Furthermore, dHis labeling with Cu(II)-NTA is efficient due to the sub-micromolar affinity.52 The labeling strategy is also robust in various buffers53 and in the presence of different competitive metals.54 Importantly, dHis-Cu(II) labeling does not require the removal of native histidine residues, further simplifying the labeling process.55 When combined with other labels, the measurements can be performed at protein concentrations as low as 45 to 500 nM, depending on the label.52,56,57 Consequently, the measurements can provide protein dimerization affinities at concentrations that are hard to measure by isothermal titration calorimetry.58
 | ||
| Fig. 1 The dHis-Cu(II)-NTA bifunctional spin label. (A) 3D representation of the dHis-Cu(II)-NTA spin label. Carbon atoms are shown in green, nitrogen atoms in blue, oxygen atoms in red, and the copper(II) atom is shown as a small copper sphere. Flexible dihedral angles are indicated. (B) Lewis structure of the dHis-Cu(II)-NTA label. | ||
Currently, two approaches are available for predicting dHis-Cu(II) distance distributions, each with drawbacks. First, a method using a pre-computed rotamer library for dHis-Cu(II) was developed and implemented in the software MMM.59 This method is fast and straightforward but has an error ranging from 2.5 Å to 5 Å compared to the experimental distance.59,60 This error is significant, considering the standard deviation of distributions from rigid dHis-Cu(II)-NTA on a small globular protein is on the order of 1 Å. Second, force field parameters were developed, enabling molecular dynamics (MD) simulations that yielded good predictions within 2 Å of the experimental data.60 The drawback of the MD simulations is that they require extensive computational resources and time and do not integrate well with other analyses and modeling pipelines like routine in-silico label site screening, or iterative structural sampling methods like Markov chain Monte Carlo sampling. Therefore, there is still a need for fast, extensible, and accurate modeling methods for dHis-Cu(II) spin labels. The recent development of chiLife's41 general approach to bifunctional label modeling49 offers a potential solution.
In this work, we computationally explore the conformational space of the dHis-Cu(II) motif and develop bifunctional rotamer libraries for the chiLife bifunctional label modeling method using in silico molecular modeling methods. We first analyze the conformational preferences of dHis-Cu(II)-NTA in the context of α-helix and β-sheet sites. We then construct rotamer libraries from the resulting structural ensembles and test the efficacy of the chiLife bifunctional label modeling method and the rotamer libraries by comparing predicted and experimental distance distributions collected from three proteins. We show that the modeling method and the rotamer libraries provide accurate predictions of the experimental distance distributions. These results and the rigidity of the dHis-Cu(II) spin label allow for more stringent SDSL EPR distance restraints for protein modeling with less label ambiguity, which in turn should lead to higher-resolution protein structure and conformational ensemble models.
Experimental
Generation of MD-based rotamer ensembles of dHis-Cu(II)
Ten previously published MD simulations of dHis-Cu(II)-NTA-labeled GB1 labeled at different sites61 were analyzed using the MDAnalysis Python module.62 In summary, PDB:2QMT for the structure of GB1 was simulated using the ff14SB Amber force fields.63 On the other hand, the dHis-Cu(II)-NTA was simulated based on the previously published force field parameters.60 The solvent water was treated with the TIP3P water model.64 Sodium and chloride ions were added to neutralize the charge and maintain the salt concentration of about 50 mM. The MD simulation steps are performed using the pmemd program in the AMBER18 package. The system was minimized by applying a harmonic restraint force constant on the bonds, released from 20 to 0 kcal mol−1 Å−2 over 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps. Minimization was followed by gradual heating from 0 K to 298.15 K, which was then maintained for the production phase of the MD simulation for a total of 200 ns simulation time. The temperature control used a Langevin thermostat with a collision frequency of 5/ps. The pressure of the system was maintained at 1 atm with a relaxation time of 1 ps. The MD simulations were performed for 10 different sites of GB1.61
000 steps. Minimization was followed by gradual heating from 0 K to 298.15 K, which was then maintained for the production phase of the MD simulation for a total of 200 ns simulation time. The temperature control used a Langevin thermostat with a collision frequency of 5/ps. The pressure of the system was maintained at 1 atm with a relaxation time of 1 ps. The MD simulations were performed for 10 different sites of GB1.61
Each site was classified as either α-helical or β-sheet based on the backbone environment. Of the ten trajectories, seven were in the β-sheet context and three were in the α-helical context. The dHis motif and the Cu(II)-NTA atoms were extracted from each frame of each trajectory and pooled into either an α-helical or β-sheet aggregate ensemble.
Generation of CREST-based rotamer ensembles
The conformational space of the dHis-Cu(II)-NTA label was also explored using the conformer-rotamer ensemble sampling tool (CREST).65 CREST-based rotamer ensembles were constructed as previously described.49 For both the α-helical and β-sheet contexts, a minimal system was constructed manually using PyMol. For each backbone context, four different starting structures were used to ensure sampling of four different possible coordination geometries (Fig. S3, ESI†). Two octahedral six-coordinate geometries were used where the ε-nitrogen (Nε) coordinating the copper atom coaxially with the nitrogen of the NTA capping ligand belonged to either the N-terminal or C-terminal histidine. Two square pyramidal five-coordinate geometries were used analogous to the six-coordinate geometries where the carboxyl group of NTA coordinating coaxially with the other histidine is instead no longer coordinating, as observed in a related crystal structure66 and in previous quantum calculations.59 Several other geometries were attempted but were energetically unstable when minimized using the GFN2-xTB semiempirical tight binding method67,68 and resulted in dissociation of one of the histidines. Each minimal system was capped with an N-terminal acetyl cap and a C-terminal amide cap. All non-dHis-Cu(II) residues were glycine to minimize bias caused by side chain atoms. Conformations were sampled with CREST65 using the default sampling settings, the generalize born/surface area implicit water model and the GFN2-xTB68 semiempirical method to evaluate energy. To maintain α-helical or β-sheet contexts, backbone torsion angles were restrained to (ϕ, ψ) = (−64°, −41°) and (−135°, 135°) respectively, using a restraining cartesian force constant of 0.01 Hartree per bohr2 (11.88 kcal mol−1 Å−2). Additionally, bifunctional coordination was enforced by adding 2.03 Å distance restraints between the histidine Nε atoms and the Cu2+ ion and a 2.36 Å restraint between the NTA nitrogen and the Cu2+ ion, using the same force constant. These distances are comparable to those observed both computationally and experimentally for five- and six-coordinated geometries.60Ensemble clustering and rotamer library generation
For each ensemble, the hundreds to tens of thousands of structures were clustered to eliminate redundancy for construction of the rotamer libraries. Each ensemble was clustered in dihedral space using only the dihedral angles of the bifunctional label heavy atoms including the angles illustrated in Fig. 1 as well as the dihedral angles of the NTA cap, but ignoring backbone, non-label, and hydrogen dihedrals. Clustering was performed using the DBSCAN algorithm69 with a distance cutoffs of 20° for any one dihedral and a minimum cluster size of 200 for the 7 β-sheet MD ensembles, 20 for the 3 α-helical MD ensembles, and 1 for the CREST ensembles. Cluster centers were defined as the structure closest to all neighbors in a cluster for MD and the lowest-energy structure for CREST ensembles. Cluster centers were then used for rotamer library construction.The CREST-based cluster center structures were pruned in two additional steps. In the first step, chemically non-viable structures were eliminated. This includes structures that did not coordinate the copper atom with both histidines, the nitrogen of the NTA and at least two carboxyl oxygens of the NTA cap. Additionally, structures where the NTA cap was hydrogen bonding with the backbone were also discarded. These structures are likely highly unfavorable and arise as artifacts from using an implicit-solvent model. In the second pruning step, redundant structures that differ only in atom numbering, but not in geometric structure, were eliminated. Due to the threefold symmetry of the NTA capping ligand, a specific NTA geometry can have three different atom numberings and therefore different dihedral angles. To identify such subsets of equivalent structures, structures were compared pairwise using iterative closest-point alignment of the side chain heavy atoms where the numbering of non-NTA atoms was fixed and the numbering of the NTA atoms was varied. Two structures were deemed equivalent if the pairwise root-mean-square deviation (RMSDs) was less than 0.3 Å. From each subset, all but the lowest-energy structure were eliminated.
Cluster centers were used to generate bifunctional rotamer libraries using chiLife.41 Each rotamer was weighted based on the size of the cluster (MD libraries) or the expected Boltzmann equilibrium populations based on the calculated rotamer energies (CREST libraries). For the CREST libraries, the bottom 1% were discarded due to a large number of high-energy structures. Mobile dihedral angles were defined to include all rotatable side chain dihedrals between the α-carbons (Cαs) of the coordinating histidines as illustrated in Fig. 1A.
Analysis of experimental data
All data from previous publications were reanalyzed using a custom Python script to ensure consistent error estimation across different systems. X-band data (all GB1 sites) were modeled using a separable non-linear least squares approach70 whereby the DEER background and foreground were fit simultaneously. The foreground was modeled using Tikhonov regularization with the second derivative operator and the background using a homogeneous 3D spin distribution model. To regularize background fitting against long-distance artifacts in the distribution, an additional restraint against the magnitude of the modulation depth was used, like that used in LongDistances.71 The regularization parameter was selected using generalized cross-validation. All data were analyzed using the isotropic dHis-Cu(II)-NTA g-value of 2.13046 = (g‖ + 2g⊥)/3.For Q-band data (all YopO data and hGSTA1), we summed DEER traces at three field positions55,72 to eliminate orientational selection and performed background correction by subtracting a 3rd-order polynomial which was fit to the last ¾ of the time-domain signal. The summed and background corrected data were fit using model-free Tikhonov regularization with only the modulation depth as a nonlinear parameter, i.e. no background model.
Confidence intervals for all fitted distance distributions were obtained using bootstrap sampling with 100 samples. For each sample, the Tikhonov regularization parameter was fixed at the fit value and all other parameters were free.
Results and discussion
Development of MD-based dHis-Cu(II)-NTA rotamer libraries
The development of dHis-Cu(II)-NTA rotamer libraries requires accurate and efficient sampling of the conformational diversity of the label. We first sought to investigate this conformational diversity by analyzing 10 recently published MD simulations of dHis-Cu(II)-NTA.61 These simulations consist of dHis-Cu(II)-NTA attached to 7 different β-sheet sites and 3 different α-helix sites on a globular protein, GB1, providing insight into the conformational diversity of dHis-Cu(II)-NTA in different environments. This conformational diversity primarily originates from the six side chain dihedrals described in Fig. 1 (three on each histidine). Fig. 2 shows histograms of the six dihedral angles for all frames of all MD simulations of both the α-helical and β-strand sites. Correlation plots between additional dihedral pairs are shown in Fig. S1 and S2 (ESI†). More details of the context of each dihedral angle are discussed in the ESI.† These data reveal relatively little conformational diversity compared to monofunctional labels such as R173 and the more flexible bifunctional label RX.45 These findings are consistent with the observations that distance distributions between dHis-Cu(II)-NTA spin labels are often considerably sharper than those of other common labels.47 Furthermore, these data reveal distinct rotameric preferences for dHis-Cu(II)-NTA that can be used as the basis of a rotamer library. Notably, the conformations of dHis-Cu(II)-NTA on β-sheet sites are more diverse than the conformations on α-helix sites, suggesting that dHis-Cu(II)-NTA rotamers in the context of α-helices are more restrained than the rotamers in the β-sheet context. | ||
| Fig. 2 MD-based dHis-Cu(II)-NTA rotamer libraries. Left: Blue and red structural ensembles depict α-helix (top) and β-strand libraries (bottom). Right: Histograms of the six side chain dihedral angles over the corresponding MD trajectories, in grayscale, with rotamer library members indicated as blue (α-helix) and red (β-strand) circles. | ||
Next, we developed rotamer libraries from the MD trajectories by pooling all frames with similar backbone contexts (i.e., α-helix or β-sheet) into aggregate trajectories. Then, we clustered the dHis-Cu(II)-NTA rotamers in dihedral space as described in the Methods section. The clustering provided 2 and 5 representative rotamers for α and β contexts, respectively (Fig. 2). These representative rotamers were then compiled into a multi-state PDB and assigned weights based on the number of represented frames in the MD trajectory, i.e., cluster size. These rotamers are plotted as blue (α-helical, top) and red (β-strand, bottom) circles on Fig. 2. The plot shows that the obtained rotamers correspond to the dominant conformations of the MD trajectories.
Note that the conformational variations observed in the MD simulations relies on the force-field parameterization of dHis-Cu(II)-NTA as a unit.60 Hence, the MD simulations do not contain conformational diversity that arises from the heterogeneity of the coordination between Cu(II) and NTA. Notably, the coordination geometry of the NTA cap has been modeled both as octahedral60 and as square pyramidal59 and conclusive experimental evidence for the prevalence of either state has not yet been published. Even within a given coordination geometry, NTA coordination can have slightly different isomeric structures, as illustrated in Fig. S3 (ESI†). Therefore, we explored an additional rotamer sampling method to obtain different coordination geometries and isomeric arrangements.
Development of CREST-based rotamer libraries of dHis-Cu(II)
We consider alternative coordination geometries and isomeric structures on the conformational landscape of dHis-Cu(II)-NTA by using the conformer-rotamer ensemble search tool (CREST),65 as described in the Methods section. To allow for alternative coordination geometries, we used the GFN-xTB2 semi-empirical method for energy evaluation and to maximize the sampling of alternative coordination geometries. Furthermore, we used four different starting structures (Fig. S3, ESI†) with different coordination geometries (see Methods section) for both α-helical and β-strand contexts.The resulting rotamer libraries are shown in Fig. 3, with 17 rotamer in the α-helical context and 40 in the β-strand context. Like the MD rotamer libraries, the β-strand context exhibits more conformational diversity than the α-helical context. Comparison of Fig. 2 and 3 reveals similar patterns in both rotamer libraries for α and β contexts; however, the CREST-based libraries show considerably more conformational diversity. Interestingly, the vast majority of rotamers exhibit an octahedral six-site coordination geometry with 11 out of 40 rotamers of the β-sheet library having square pyramidal, five-site coordination geometries and none in the α-helix library, despite half of the starting structures having five-site coordination geometries. Both contexts exhibited a significant amount of isomeric heterogeneity, where the nitrogen of the NTA cap coordinated coaxially with either the N-terminal or C-terminal histidine. While this conformational diversity may not affect the placement of the Cu2+, it may reflect alternate conformations that arise to accommodate clashes from neighboring atoms when labeling more complex environments.
 | ||
| Fig. 3 CREST-based dHis-Cu(II)-NTA rotamer libraries. Blue and red structural ensembles depict α-helix (top) and β-strand libraries (bottom). Dihedral angles of library rotamers are plotted as blue (α-helix) and red (β-strand) circles. | ||
Bifunctional modeling of dHis-Cu(II) with chiLife
We incorporated the clustered rotamers and the associated weights into the software package chiLife41 as bifunctional rotamer libraries.49 chiLife performs bifunctional rotamer modeling by splitting bifunctional labels into two monofunctional labels with a subset of redundant atoms (henceforth referred to as the “cap”) and then optimizing the cap alignment by fitting the mobile dihedrals of the label. The cap region is made up of all atoms beyond the terminal mobile dihedrals of each monofunctional subunit. This includes the Cu2+ ion and the NTA capping ligand (Fig. 4A). Fig. 4B illustrates the fitting procedure. A movie of this procedure for dHis-Cu(II)-NTA is available in the ESI.† First, the two monofunctional labels are individually aligned to the protein backbone sites of interest. Minor differences between the labeling site backbone structures and the backbone structure the rotamer was derived from causes slight mismatches in the position of the two caps, including the two copies of Cu2+. Next, the mobile dihedral angles of each rotamer in the library are varied until the two cap copies overlap as previously described49 and the monofunctional subunits are merged back into a bifunctional label. After achieving optimal overlap, the final rotamers are evaluated for clashes with other molecules or sidechains from the protein. High-energy rotamers, caused by either poor cap alignment or external clashes, are discarded. | ||
| Fig. 4 Cap definition and bifunctional modeling procedure of dHis-Cu(II)-NTA. (A) Cap definition and construction of monofuntional subunits of dHis-Cu(II)-NTA. (B) ChiLife bifunctional label modeling procedure for a single rotamer. | ||
Rotamer libraries provide accurate predictions of Cu(II)–Cu(II) distance distributions
To evaluate the accuracy of the dHis-Cu(II)-NTA rotamer libraries as well as the chiLife bifunctional label modeling method, we modeled dHis-Cu(II)-NTA on 7 sites on 3 proteins and compared the predicted distance distributions to previously published experimentally derived distributions between site pairs on the GB1 domain of protein G, human glutathione S-transferase A1 (hGSTA1), and Yersinia outer protein O (YopO).55,60,72 This analysis is shown in Fig. 5. Both rotamer libraries predicted the experimental distributions with high accuracy, with an absolute mode deviation between 0.0–1.0 Å with an average of 0.46 Å for the MD-based libraries and between 0.0–1.2 Å with an average of 0.56 Å for the CREST-based libraries. We note that the deviations are comparable to the resolutions of 1–1.5 Å of the PDB structures used in Fig. 5. Therefore, given the Å resolution of pulsed-EPR distance measurements and the rigidity of dHis-Cu(II), the small deviations in the modeling strategy enable meaningful interpretation of the relative positions of the protein backbones.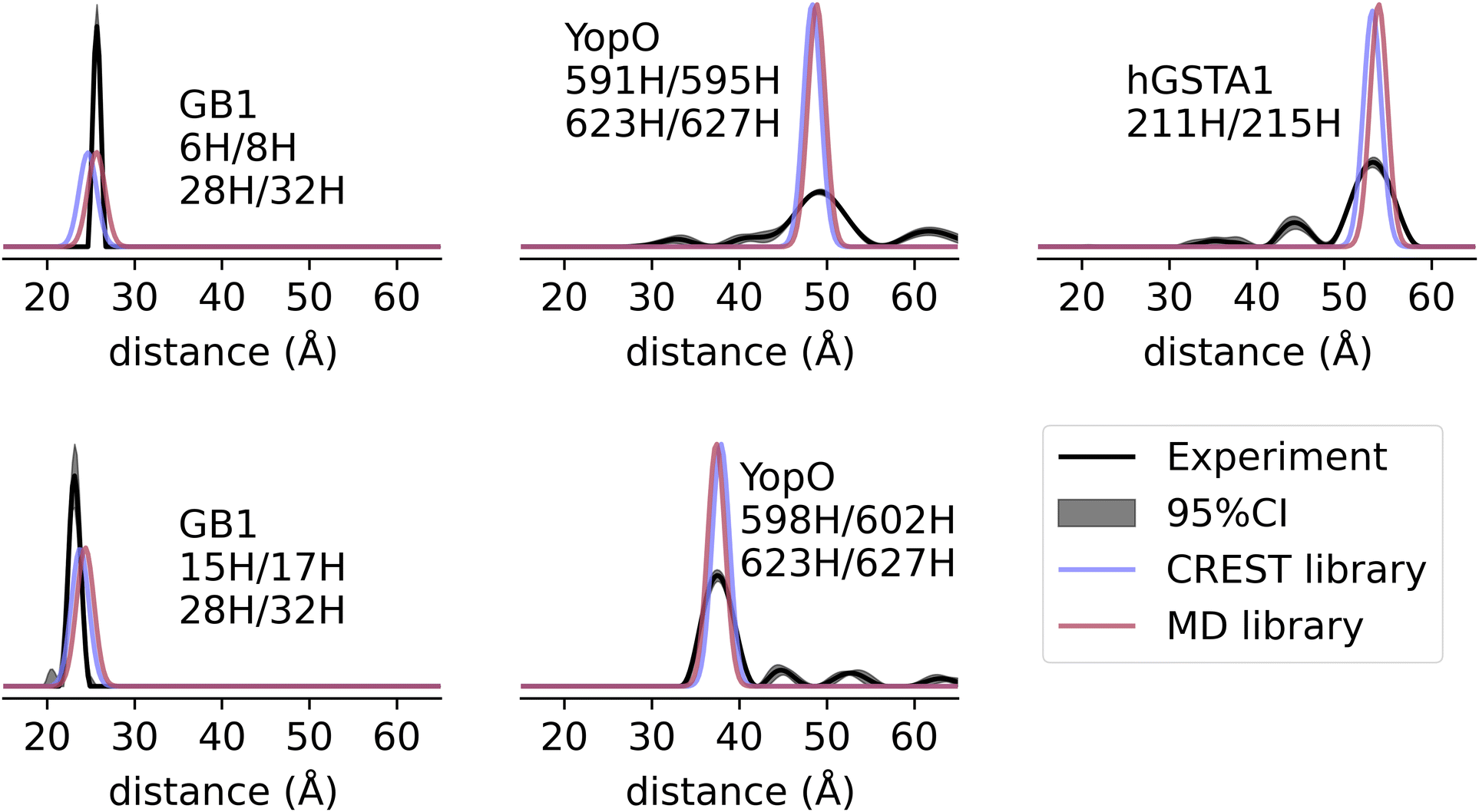 | ||
| Fig. 5 Prediction of experimental Cu–Cu distance distributions. Comparison of predicted and experimental distance distributions for two sites on GB1 (PDB: 2QMT), human GSTA1 (PDB: 1K3L), and YopO (PDB: 2H7O). | ||
It is also instructive to compare the predicted distribution widths with experiments.55,60,72 For GB1, the predicted widths are close to the experimental results. GB1 has limited backbone fluctuations61,74–76 and therefore this protein serves as an important test case to establish the robustness of the analysis. On the other hand, the experimental distributions for hGSTA-1 and YopO are broader than predicted. While over-smoothing due to low signal-to-noise ratio (SNR) can result in such broadenings, this does not apply to the original signal for hGSTA1-1 and YopO, which have high SNR.55,72 Therefore, the broader distances are likely from the flexible protein backbones that we cannot model from a single PDB structure. In the case of Fig. 5, the labeled sites on hGSTA1-1 and YopO are likely more flexible than the labeled sites on GB1. These results highlight the utility of dHis-Cu(II) to resolve protein backbone fluctuations and conformations that are obfuscated when using more flexible spin labels.
With the caveat of only using a small sample size, the accuracy of our dHis-Cu(II) modeling method outperforms monofunctional label modeling methods which generally have an absolute mode error on the order of 2–4 Å.43 Furthermore, the rotamer libraries in this work have ∼4 times better accuracy than previously published rotamer library method for modeling dHis-Cu(II),59 which exhibited an average absolute mode deviation of 2.1 Å over this same data set (Fig. S4, ESI†). Finally, the distance predictions of GB1 in Fig. 5 have similar accuracy as MD simulations of GB1 that have deviations up to 2 Å.60
Notably, the smaller, less conformationally diverse MD-based rotamer libraries performed equally as well as the CREST-based libraries, reinforcing the observations that the dHis-Cu(II) motif is highly restrained. This result also demonstrates the effectiveness of the chiLife bifunctional ensemble modeling method, and it establishes the generality of the method to bifunctional labels other than RX, for which it was first developed and validated. The modeling method was also relatively fast at an average of 82 ms per rotamer, resulting in average ensemble modeling times of 2.0 s and 0.2 s for the CREST and MD libraries, respectively, using an AMD Ryzen 5980HX laptop processor. The speed of the modeling provides an accessible method to perform high-throughput in silico screening of site pairs on a given PDB structure.
Prediction of distance distribution is robust to protein structural diversity in PDB structures
One of the major advantages of SDSL EPR spectroscopic methods is their ability to probe protein conformational elasticity. GB1 has been a well-studied molecule with several models deposited in the protein data bank using both X-ray diffraction77 and nuclear magnetic resonance methods,78–83 each with modest structural differences resulting from both conformational heterogeneity and differences in model construction. To assess the tolerance of the rotamer libraries and the bifunctional label modeling method to model structural variability, we modeled dHis-Cu(II)-NTA using several available PDB structures of GB1 and compared the predicted distance distributions with the experimentally derived distance distributions. The results are shown in Fig. 6 and illustrate that the dHis-Cu(II)-NTA models make accurate and consistent predictions despite the minor differences in protein structure used. Again, both rotamer libraries performed similarly better with a mean absolute deviation of the mode of 0.6 Å for the CREST library and 0.7 Å for the MD-based library. Both predictions are considerably more accurate than those of monofunctional labels and RX.43,49 | ||
| Fig. 6 Prediction of distance distributions on different GB1 PDB structures. Comparison of predicted and experimental distance distributions for two sites on GB1 with PDB IDs shown next to corresponding distributions. | ||
Model selection using dHis-Cu(II)-NTA
Next, we assess the dHis-Cu(II) rotamer libraries as a tool for distinguishing two different protein conformations, using YopO. Previously, dHis-Cu(II) measurements were performed on two YopO constructs, 598H602H/620H624H and 591H595H/620H624H.55 The two constructs have both dHis motifs on α-helix 14. Fig. 7A shows the α-helix 14 of YopO and the dHis-Cu(II) sites based on the two available crystal structures of YopO, PDB:2H7O84 and PDB:4CI6,85 color-coded as blue and red, respectively. In the PDB:2H7O structure the α-helix 14 is straight, while in PDB:4CI6 it is slightly bent as a result of a minor allosteric change induced by the interaction of YopO with actin.85 We used the two PDB structures to model the distance distributions and compared them to the previously measured experimental distance distribution. | ||
| Fig. 7 Prediction of distance distributions on two different YopO structures. (A) Structures of YopO with a straight (PDB:2H7O, blue) and a bent (PDB:4CI6, purple) α-helix 14. The teal sticks and balls represent the dHis-Cu(II)-NTA sites. (B) Comparison of the predicted and experimental distance distributions of two YopO constructs using the MD-based rotamer libraries. (C) Comparison of the predicted and experimental distance distributions of two YopO constructs using the CREST-based rotamer libraries. | ||
Fig. 7B shows the expected distance distributions for the two YopO constructs using the MD-based libraries and Fig. 7C show the same for the CREST-based libraries. The predicted distance distributions are color-coded with blue and red corresponding to PDB:2H7O and PDB:4CI6, respectively. For comparison, the experimental distance distributions are shown in black. For the short-distance YopO construct on the bottom panels of Fig. 7B and C, the experimental distances agree well with the prediction from PDB:2H7O using both rotamer libraries. On the other hand, prediction of the long-distance YopO construct is less conclusive. Particularly, the measured distance distribution has a maximum between the prediction from the two crystal structures. Furthermore, the distribution is wide enough to have significant overlap with the predictions from either crystal structures. These observations are consistent with previous work.55 Particularly, previous predictions using MMM and MD also provided the same ambiguous interpretation of the long YopO construct and a straightforward agreement between the PDB:2H7O model and the short YopO experimental data. Nonetheless, the predictions using the CREST and MD-based rotamer libraries best support that YopO adopts a straight α-helix 14 conformation, a similar conclusion with previous study.55 Therefore, the dHis-Cu(II)-NTA rotamer libraries and chiLife bifunctional modeling method are effective, or at least as effective as previous methods, for selecting an appropriate model that matches the experimental data. Furthermore, Fig. 7 highlights that this modeling strategy and distances from dHis-Cu(II) can distinguish structures that differ by a backbone RMSD as little as 2.0 Å, as is the case between helix 14 of 2H7O and 4CI6, resulting in distance distribution difference of ∼4 Å. Overall, the dHis-Cu(II) MD-rotamer library approach is an accessible method for calculating distance distributions.
The dHis-Cu(II)NTA rotamer libraries and the chiLife bifunctional modeling method benefit from the relatively narrow experimental data. In the presence of flexible protein backbones, the approach needs to be combined with other methods to completely model distance distributions. Protein backbone diversity can be generated using elastic network modeling, as implemented in MMM and MMMx38,86 or using MD simulations, especially with the use of enhanced sampling methods.72,87,88 As a scriptable Python-based API, chiLife can easily be integrated with an implementation of the same, or an equivalent elastic network modelling protocol, utilize full MD trajectories of native proteins as inputs to model Cu–Cu distance distributions, or integrate with other modelling software like Rosetta17,89 and Xplor-NIH,90 allowing dHis-Cu(II)-NTA distance constraints to be used in a wide variety of existing and future analysis and protein modelling pipelines.
Conclusions
This work establishes an accessible, robust, and accurate method for modeling the dHis-Cu(II)-NTA label on a protein structure using rotamer libraries generated using two different approaches. The method is validated against experimental data from three proteins, and we hope that future work will extend this validation to a broader range of proteins. Fast calculation times compared to full MD provide an efficient tool for designing dHis variants for experiments and for applying restraints in iterative protein modeling pipelines. Additionally, the narrow widths of dHis-Cu(II) distance distributions coupled with the improved accuracy of distance distribution predictions provides a basis for better model selection and modeling accuracy when using sparse EPR distance restraints. Of particular interest is the enhanced resolution of backbone conformational heterogeneity, making minor backbone fluctuations resolvable both experimentally and in silico. This capability arises from limited rotameric preferences of the dHis-Cu(II) that are easier to calculate and sample than more flexible mono-functional labels.91–93 With a thorough understanding of dHis-Cu(II) conformations, we can start to explore methods for generating accurate protein models by using experimentally measured dHis-Cu(II) distance restraints, as demonstrated for nitroxide labels.17,35–37,39Author contributions
The study and methods were conceived by Z. H., M. H. T., S. Stoll, and S. Saxena. Research was performed by Z. H. and M. H. T. Software was implemented by M. H. T. The manuscript was written by Z. H. and M. H. T. and edited by Z. H., M. H. T., S. Stoll, and S. Saxena.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Institutes of Health (NIH) grants R01 GM125753 (S. Stoll) and National Science Foundation (NSF) – Binational Science Foundation (BSF) grant MCB 2006154 (S. Saxena). The authors would also like to thank Eric Evans, Xiaowei Bogetti and Prof. Junmei Wang for their helpful discussion regarding the coordination geometry of dHis-Cu(II)-NTA, and the Olav Schiemann Group at University of Bonn for collaborative work on the YopO EPR data.Notes and references
- P. P. Borbat and J. H. Freed, Chem. Phys. Lett., 1999, 313, 145–154 CrossRef CAS.
- L. V. Kulik, S. A. Dzuba, I. A. Grigoryev and Y. D. Tsvetkov, Chem. Phys. Lett., 2001, 343, 315–324 CrossRef CAS.
- S. Milikisyants, F. Scarpelli, M. G. Finiguerra, M. Ubbink and M. Huber, J. Magn. Reson., 2009, 201, 48–56 CrossRef CAS PubMed.
- A. D. Milov, A. G. Maryasov and Y. D. Tsvetkov, Appl. Magn. Reson., 1998, 15, 107–143 CrossRef CAS.
- M. Pannier, S. Veit, A. Godt, G. Jeschke and H. W. Spiess, J. Magn. Reson., 2000, 142, 331–340 CrossRef CAS PubMed.
- G. Jeschke, M. Pannier, A. Godt and H. W. Spiess, Chem. Phys. Lett., 2000, 331, 243–252 CrossRef CAS.
- M. Bonora, J. Becker and S. Saxena, J. Magn. Reson., 2004, 170, 278 CrossRef CAS PubMed.
- T. E. Assafa, K. Anders, U. Linne, L.-O. Essen and E. Bordignon, Structure, 2018, 26, 1534–1545 CrossRef CAS PubMed.
- Z. Liu, T. M. Casey, M. E. Blackburn, X. Huang, L. Pham, I. M. S. de Vera, J. D. Carter, J. L. Kear-Scott, A. M. Veloro, L. Galiano and G. E. Fanucci, Phys. Chem. Chem. Phys., 2016, 18, 5819–5831 RSC.
- I. Hänelt, D. Wunnicke, E. Bordignon, H.-J. Steinhoff and D. J. Slotboom, Nat. Struct. Mol. Biol., 2013, 20, 210–214 CrossRef PubMed.
- O. Dalmas, P. Sompornpisut, F. Bezanilla and E. Perozo, Nat. Commun., 2014, 5, 3590 CrossRef PubMed.
- B. Verhalen, R. Dastvan, S. Thangapandian, Y. Peskova, H. A. Koteiche, R. K. Nakamoto, E. Tajkhorshid and H. S. Mchaourab, Nature, 2017, 543, 738–741 CrossRef CAS PubMed.
- B. Joseph, A. Sikora and D. S. Cafiso, J. Am. Chem. Soc., 2016, 138, 1844–1847 CrossRef CAS PubMed.
- K. Stone, J. Townsend, J. Sarver, P. Sapienza, S. Saxena and L. Jen-Jacobson, Angew. Chem., Int. Ed., 2008, 120, 10346–10348 CrossRef.
- T. Hett, T. Zbik, S. Mukherjee, H. Matsuoka, W. Bönigk, D. Klose, C. Rouillon, N. Brenner, S. Peuker, R. Klement, H.-J. Steinhoff, H. Grubmüller, R. Seifert, O. Schiemann and U. B. Kaupp, J. Am. Chem. Soc., 2021, 143, 6981–6989 CrossRef CAS PubMed.
- E. G. B. Evans, J. L. W. Morgan, F. DiMaio, W. N. Zagotta and S. Stoll, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 10839–10847 CrossRef CAS PubMed.
- D. Sala, D. Del Alamo, H. S. Mchaourab and J. Meiler, Structure, 2022, 30, 1157–1168 CrossRef CAS PubMed.
- G. Jeschke, J. Chem. Theory Comput., 2012, 8, 3854–3863 CrossRef CAS PubMed.
- A. K. Upadhyay, P. P. Borbat, J. Wang, J. H. Freed and D. E. Edmondson, Biochemistry, 2008, 47, 1554–1566 CrossRef CAS PubMed.
- A. Gamble Jarvi, T. F. Cunningham and S. Saxena, Phys. Chem. Chem. Phys., 2019, 21, 10238–10243 RSC.
- D. Abdullin, N. Florin, G. Hagelueken and O. Schiemann, Angew. Chem., Int. Ed., 2015, 54, 1827–1831 CrossRef CAS PubMed.
- B. J. Gaffney, M. D. Bradshaw, S. D. Frausto, F. Wu, J. H. Freed and P. Borbat, Biophys. J., 2012, 103, 2134–2144 CrossRef CAS PubMed.
- D. M. Yin, J. S. Hannam, A. Schmitz, O. Schiemann, G. Hagelueken and M. Famulok, Angew. Chem., Int. Ed., 2017, 56, 8417–8421 CrossRef CAS PubMed.
- D. Nguyen, D. Abdullin, C. A. Heubach, T. Pfaffeneder, A. Nguyen, A. Heine, K. Reuter, F. Diederich, O. Schiemann and G. Klebe, Angew. Chem., Int. Ed., 2021, 60, 23419–23426 CrossRef CAS PubMed.
- S. Milikisiyants, S. Wang, R. A. Munro, M. Donohue, M. E. Ward, D. Bolton, L. S. Brown, T. I. Smirnova, V. Ladizhansky and A. I. Smirnov, J. Mol. Biol., 2017, 429, 1903–1920 CrossRef CAS PubMed.
- S.-Y. Park, P. P. Borbat, G. Gonzalez-Bonet, J. Bhatnagar, A. M. Pollard, J. H. Freed, A. M. Bilwes and B. R. Crane, Nat. Struct. Mol. Biol., 2006, 13, 400–407 CrossRef CAS PubMed.
- S. Valera, K. Ackermann, C. Pliotas, H. Huang, J. H. Naismith and B. E. Bode, Chem. – Eur. J., 2016, 22, 4700–4703 CrossRef CAS PubMed.
- H. A. DeBerg, J. R. Bankston, J. C. Rosenbaum, P. S. Brzovic, W. N. Zagotta and S. Stoll, Structure, 2015, 23, 734–744 CrossRef CAS PubMed.
- M. H. Tessmer, D. M. Anderson, A. M. Pickrum, M. O. Riegert, R. Moretti, J. Meiler, J. B. Feix and D. W. Frank, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 525–530 CrossRef CAS PubMed.
- D. Z. Herrick, W. Kuo, H. Huang, C. D. Schwieters, J. F. Ellena and D. S. Cafiso, J. Mol. Biol., 2009, 390, 913–923 CrossRef CAS PubMed.
- S. M. Hanson, E. S. Dawson, D. J. Francis, N. Van Eps, C. S. Klug, W. L. Hubbell, J. Meiler and V. V. Gurevich, Structure, 2008, 16, 924–934 CrossRef CAS PubMed.
- M. H. Tessmer, S. A. DeCero, D. Del Alamo, M. O. Riegert, J. Meiler, D. W. Frank and J. B. Feix, Sci. Rep., 2020, 10, 19700 CrossRef CAS PubMed.
- A. W. Fischer, D. M. Anderson, M. H. Tessmer, D. W. Frank, J. B. Feix and J. Meiler, ACS Omega, 2017, 2, 2977–2984 CrossRef CAS PubMed.
- T. Schmidt, D. Wang, J. Jeon, C. D. Schwieters and G. M. Clore, J. Am. Chem. Soc., 2022, 144, 12043–12051 CrossRef CAS PubMed.
- D. Del Alamo, M. H. Tessmer, R. A. Stein, J. B. Feix, H. S. Mchaourab and J. Meiler, Biophys. J., 2020, 118, 366–375 CrossRef CAS PubMed.
- D. Del Alamo, K. L. Jagessar, J. Meiler and H. S. Mchaourab, PLoS Comput. Biol., 2021, 17, e1009107 CrossRef CAS PubMed.
- G. Jeschke, Protein Sci., 2018, 27, 76–85 CrossRef CAS PubMed.
- G. Jeschke, Protein Sci., 2021, 30, 125–135 CrossRef CAS PubMed.
- N. S. Alexander, R. A. Stein, H. A. Koteiche, K. W. Kaufmann, H. S. McHaourab and J. Meiler, PLoS One, 2013, 8, e72851 CrossRef CAS PubMed.
- A. W. Fischer, N. S. Alexander, N. Woetzel, M. Karakas, B. E. Weiner and J. Meiler, Proteins, 2015, 83, 1947–1962 CrossRef CAS PubMed.
- M. H. Tessmer and S. Stoll, PLoS Comput. Biol., 2023, 19, e1010834 CrossRef CAS PubMed.
- G. Hagelueken, R. Ward, J. H. Naismith and O. Schiemann, Appl. Magn. Reson., 2012, 42, 377–391 CrossRef CAS PubMed.
- M. H. Tessmer, E. R. Canarie and S. Stoll, Biophys. J., 2022, 121, 3508–3519 CrossRef CAS PubMed.
- M. R. Fleissner, M. D. Bridges, E. K. Brooks, D. Cascio, T. Kálai, K. Hideg and W. L. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16241–16246 CrossRef CAS PubMed.
- M. A. Stevens, J. E. McKay, J. L. S. Robinson, H. El Mkami, G. M. Smith and D. G. Norman, Phys. Chem. Chem. Phys., 2016, 18, 5799–5806 RSC.
- M. J. Lawless, S. Ghosh, T. F. Cunningham, A. Shimshi and S. Saxena, Phys. Chem. Chem. Phys., 2017, 19, 20959–20967 RSC.
- T. F. Cunningham, M. R. Putterman, A. Desai, W. S. Horne and S. Saxena, Angew. Chem., Int. Ed., 2015, 54, 6330–6334 CrossRef CAS PubMed.
- S. Ghosh, M. J. Lawless, G. S. Rule and S. Saxena, J. Magn. Reson., 2018, 286, 163–171 CrossRef CAS PubMed.
- M. H. Tessmer and S. Stoll, Appl. Magn. Reson., 2023 DOI:10.1007/s00723-023-01576-1.
- H. Sameach, S. Ghosh, L. Gevorkyan-Airapetov, S. Saxena and S. Ruthstein, Angew. Chem., Int. Ed., 2019, 131, 3085–3088 CrossRef.
- A. Gamble Jarvi, K. Ranguelova, S. Ghosh, R. T. Weber and S. Saxena, J. Phys. Chem. B, 2018, 122, 10669–10677 CrossRef CAS PubMed.
- J. L. Wort, K. Ackermann, A. Giannoulis, A. J. Stewart, D. G. Norman and B. E. Bode, Angew. Chem., Int. Ed., 2019, 58, 11681–11685 CrossRef CAS PubMed.
- A. Gamble Jarvi, J. Casto and S. Saxena, J. Magn. Reson., 2020, 320, 106848 CrossRef CAS PubMed.
- J. L. Wort, S. Arya, K. Ackermann, A. J. Stewart and B. E. Bode, J. Phys. Chem. Lett., 2021, 12, 2815–2819 CrossRef CAS PubMed.
- C. A. Heubach, Z. Hasanbasri, D. Abdullin, A. Reuter, B. Korzekwa, S. Saxena and O. Schiemann, Chem. – Eur. J., 2023, e202302541 CrossRef CAS PubMed.
- S. Kucher, C. Elsner, M. Safonova, S. Maffini and E. Bordignon, J. Phys. Chem. Lett., 2021, 12, 3679–3684 CrossRef CAS PubMed.
- N. Fleck, C. Heubach, T. Hett, S. Spicher, S. Grimme and O. Schiemann, Chem. – Eur. J., 2021, 27, 5292–5297 CrossRef CAS PubMed.
- M. Oranges, J. L. Wort, M. Fukushima, E. Fusco, K. Ackermann and B. E. Bode, J. Phys. Chem. Lett., 2022, 13, 7847–7852 CrossRef CAS PubMed.
- S. Ghosh, S. Saxena and G. Jeschke, Appl. Magn. Reson., 2018, 49, 1281–1298 CrossRef CAS.
- X. Bogetti, S. Ghosh, A. Gamble Jarvi, J. Wang and S. Saxena, J. Phys. Chem. B, 2020, 124, 2788–2797 CrossRef CAS PubMed.
- K. Singewald, J. A. Wilkinson, Z. Hasanbasri and S. Saxena, Protein Sci., 2022, 31, e4359 CrossRef CAS PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilsonet al., AMBER 2018, University of California, San Francisco, 2018.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- C. J. Burns, L. D. Field, T. W. Hambley, T. Lin, D. D. Ridley, P. Turner and M. P. Wilkinson, ARKIVOC, 2001, 2001, 157–165 Search PubMed.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, WIREs Comput. Mol. Sci., 2020, 11, e1493 Search PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- M. Ester, H.-P. Kregel, J. Sander and X. Xu, AAAI Press, 1996, 226–231 Search PubMed.
- L. Fábregas Ibáñez, G. Jeschke and S. Stoll, Magn. Reson., 2020, 1, 209–224 CrossRef PubMed.
- C. J. López, Z. Yang, C. Altenbach and W. L. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E4306 Search PubMed.
- X. Bogetti, A. Bogetti, J. Casto, G. Rule, L. Chong and S. Saxena, Protein Sci., 2023, e4770 CrossRef CAS PubMed.
- G. Jeschke, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 72, 42–60 CrossRef CAS PubMed.
- D. Idiyatullin, V. A. Daragan and K. H. Mayo, J. Magn. Reson., 2003, 161, 118–125 CrossRef CAS PubMed.
- X. Shi and C. M. Rienstra, J. Am. Chem. Soc., 2016, 138, 4105–4119 CrossRef CAS PubMed.
- D. Idiyatullin, V. A. Daragan and K. H. Mayo, J. Phys. Chem. B, 2003, 107, 2602–2609 CrossRef CAS.
- H. L. F. Schmidt, L. J. Sperling, Y. G. Gao, B. J. Wylie, J. M. Boettcher, S. R. Wilson and C. M. Rienstra, J. Phys. Chem. B, 2007, 111, 14362–14369 CrossRef PubMed.
- B. J. Wylie, L. J. Sperling, A. J. Nieuwkoop, W. T. Franks, E. Oldfield and C. M. Rienstra, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16974–16979 CrossRef CAS PubMed.
- L. B. Andreas, K. Jaudzems, J. Stanek, D. Lalli, A. Bertarello, T. Le Marchand, D. Cala-De Paepe, S. Kotelovica, I. Akopjana, B. Knott, S. Wegner, F. Engelke, A. Lesage, L. Emsley, K. Tars, T. Herrmann and G. Pintacuda, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 9187–9192 CrossRef CAS PubMed.
- W. T. Franks, B. J. Wylie, H. L. F. Schmidt, A. J. Nieuwkoop, R.-M. Mayrhofer, G. J. Shah, D. T. Graesser and C. M. Rienstra, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4621–4626 CrossRef CAS PubMed.
- P. Robustelli, A. Cavalli and M. Vendruscolo, Structure, 2008, 16, 1764–1769 CrossRef CAS PubMed.
- A. J. Nieuwkoop, B. J. Wylie, W. T. Franks, G. J. Shah and C. M. Rienstra, J. Chem. Phys., 2009, 131, 095101 CrossRef PubMed.
- D. J. Wilton, R. B. Tunnicliffe, Y. O. Kamatari, K. Akasaka and M. P. Williamson, Proteins, 2008, 71, 1432–1440 CrossRef CAS PubMed.
- G. Prehna, M. I. Ivanov, J. B. Bliska and C. E. Stebbins, Cell, 2006, 126, 869–880 CrossRef CAS PubMed.
- W. L. Lee, J. M. Grimes and R. C. Robinson, Nat. Struct. Mol. Biol., 2015, 22, 248–255 CrossRef CAS PubMed.
- G. Jeschke and L. Esteban-Hofer, Methods Enzymol., 2022, 666, 145–169 CAS.
- Y. I. Yang, Q. Shao, J. Zhang, L. Yang and Y. Q. Gao, J. Chem. Phys., 2019, 151, 070902 CrossRef PubMed.
- X. Bogetti and S. Saxena, ChemPlusChem, 2024, 89, e202300506 CrossRef CAS PubMed.
- S. J. Hirst, N. Alexander, H. S. McHaourab and J. Meiler, J. Struct. Biol., 2011, 173, 506–514 CrossRef CAS PubMed.
- C. M. Hammond, T. Owen-Hughes and D. G. Norman, Methods, 2014, 70, 139–153 CrossRef CAS PubMed.
- M. I. Fajer, H. Li, W. Yang and P. G. Fajer, J. Am. Chem. Soc., 2007, 129, 13840–13846 CrossRef CAS PubMed.
- K. Sale, L. Song, Y.-S. Liu, E. Perozo and P. Fajer, J. Am. Chem. Soc., 2005, 127, 9334–9335 CrossRef CAS PubMed.
- J. L. Sarver, J. E. Townsend, G. Rajapakse, L. Jen-Jacobson and S. Saxena, J. Phys. Chem. B, 2012, 116, 4024–4033 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05951k |
| ‡ These authors contributed equally. |
| This journal is © the Owner Societies 2024 |