
Unraveling the role of superalkalis in modulating the static and dynamic hyperpolarizabilities of emerging calix[4]arenes†
Khalida
Khalil
a,
Shahnaz
b,
Ralf
Ludwig
 cde,
Ammar M.
Tighezza
f,
Khurshid
Ayub
g,
Tariq
Mahmood
gh and
Mazhar Amjad
Gilani
*a
cde,
Ammar M.
Tighezza
f,
Khurshid
Ayub
g,
Tariq
Mahmood
gh and
Mazhar Amjad
Gilani
*a
aDepartment of Chemistry, COMSATS University Islamabad, Lahore Campus, Lahore 54600, Pakistan. E-mail: mazhargilani@cuilahore.edu.pk
bDepartment of Chemistry, Lahore College for Women University, Lahore, Pakistan
cUniversity of Rostock, Institute of Chemistry, Physical and Theoretical Chemistry, Albert-Einstein-Straße 27, Rostock 18059, Germany
dUniversity of Rostock, Faculty of Interdisciplinary Research, Department Science and Technology of Life, Light and Matter, Rostock 18059, Germany
eLeibniz Institute for Catalysis, Rostock 18059, Germany
fDepartment of Chemistry, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia
gDepartment of Chemistry, COMSATS University Islamabad, Abbottabad Campus, Abbottabad 22060, Pakistan
hDepartment of Chemistry, College of Science, University of Bahrain, Sakhir P. O. Box 32038, Bahrain
First published on 7th February 2024
Abstract
Calixarenes, as novel organic materials, can play a pivotal role in the development of high-performance nonlinear optical materials due to the ease of design and fabrication. In this study, DFT simulations were employed to investigate the geometric, electronic, and NLO responses of calix[4]arene doped with Li3O, Na3O, and K3O superalkalis. The computed values of the vertical ionization energies and interaction energies indicate the chemical and thermodynamic stabilities of the targeted M3O@calix[4]arene complexes. The corresponding energy gaps (2.01 to 3.49 eV) are notably reduced, indicating the semiconductor nature of the materials. Surprisingly, the M3O@calix[4]arene complexes exhibit transparency in the UV/visible range as the absorption peaks are shifted in the near infrared (NIR) region. The highest values of 5.9 × 105 a.u. and 2.3 × 108 a.u. for the respective first and second hyperpolarizabilities are observed for Na3O@calix[4]arene. Furthermore, the Na3O@calix[4]arene complex exhibits maximum values of 2.3 × 105 a.u. for second harmonic generation (SHG) and (K3O@calix[4]arene) 2.3 × 106 a.u. for the electro-optical Pockels effect (EOPE) at 1064 nm. Similarly, approximations are made for the dynamic second hyperpolarizability coefficients (EOKE and EFISHG) at different wavelengths. Notably, the Na3O@calix[4]arene complex demonstrates the highest quadratic nonlinear refractive index (n2) of 9.5 × 10−15 cm2 W−1 at 1064 nm. This research paves the way for the development of stable calix[4]arenes doped with superalkalis, leading to an improved nonlinear optical (NLO) response.
1. Introduction
The design of novel nonlinear optical materials with a high NLO response has been the subject of numerous theoretical and practical studies for several decades. This is primarily due to their potential for enhanced technical applications in photonic and optical systems.1,2 Several strategies for improving the NLO response have been described for a variety of organic and inorganic materials. These include the use of donor–acceptor bridges,3 donor π conjugated bridge systems,4 extended π conjugation,5 distorted π electron systems,6 diffuse excess electrons,7,8 metal atom doping, design of metal–ligand frameworks based on charge transfer from the metal to ligand,9etc. The introduction of diffuse excess electrons is a well-known and extensively used strategy in organic NLO materials.10 In many theoretical studies, a significant number of nonlinear optical materials with large hyperpolarizability using the diffuse excess electron strategy have been reported.11 The finding of alkali metals dissolved in gaseous ammonia can be traced back to the earliest investigation of systems containing diffuse excess electrons.12,13 After this, a substantial number of studies emphasized theoretically related systems such as electrides,14,15 trimer of water (H2O)316 and alkalides.17 This revealed that most of these systems exhibit remarkable NLO responses with a noticeably large first hyperpolarizability.16Alkalides are a special type of ionic salts in which alkali metals occupy the anionic sites and have excess electrons associated to them.18 Dye et al. reported the first prepared alkalide (Na+(cryptand-[2.2.2]Na−)).19 Apart from alkalides, another novel class of compounds, superalkalides, was discovered and has an obvious better NLO response as alkalides.20 Superalkalides have very low ionization energies and are even more reactive than alkalides due to their higher electron affinity.21 Li et al. investigated the NLO properties of alkalides (L–M–L–M′) and superalkalides ( ). The results revealed that the highest β0 was observed in the case of the L–K–L–LiO3 complex (3.42 × 106 a.u.).22
). The results revealed that the highest β0 was observed in the case of the L–K–L–LiO3 complex (3.42 × 106 a.u.).22
A variety of methods can be employed to introduce excess electrons into a material. One approach is to use electron-donating groups or dopants (alkali metals or superalkali units), which can introduce extra electrons into the material.23 Superalkalis are a class of “superatoms” that have excess electrons as compared to the constituent alkali metal atoms, giving them unique chemical and electronic properties.24 They have remarkable features (such as diffuse excess electrons) due to their lower IPs (ionization potentials) than alkali metal atoms.25 Doping with superalkalis will lead to a considerable increase in electron transport and a reduction in the HOMO–LUMO energy gap. Recently, significant advances have been made in superalkali doped complexes in the field of optoelectronics.26 It has been reported that superalkali doped dyads (M3O–BC59), which are donor–acceptor (DA) chromophores, have outstanding stability as well as substantial first hyperpolarizability and are good candidates for second-order NLO materials.27 Similarly, Kosar et al. observed that superalkali doping not only affects the electrical characteristics of graphdiyne (GDY), but also has a significant impact on the NLO properties.28 In another DFT investigation, the NLO properties of C2N doped with Li3O have been studied. All the doped structures show a higher NLO response than the undoped C2N nanosheet.29 The effect of superalkali doping on macrocyclic [hexa-]thiophene complexes has been studied by Mehmood et al. Among all the complexes, Na3O@6CT with a hyperpolarizability value of 5 × 104 a.u. has the highest NLO response.30 Similarly, in another study, Ayub et al. have studied Li3O@[12-crown-4]M complexes and all the doped complexes exhibited significant NLO response compared to the undoped moiety.31 Noormohammadbeigi et al. reported the influence of superalkali (Li3O, Na3O, and K3O) doping on the C20 fullerene and the studied Na3O@C20 fullerene showed high first hyperpolarizability (2.3 × 104 a.u.).32 Provided the foregoing discussions, it is evident that superalkali doping is a viable approach for enhancing the nonlinear optical response of the system.
In the realm of nonlinear optics (NLO), organic materials have taken the lead over inorganic counterparts. This shift is attributed to their lower dielectric constants,33 ultrafast response times, enhanced nonlinear optical susceptibilities,34 high laser damage thresholds,35 improved processability36 and wide transparency ranges.37 Furthermore, organic NLO materials can undergo significant chemical modifications, particularly those that enhance specific physical characteristics.38 Despite these advancements, significant efforts are currently underway to further enhance the NLO response of various nanomaterials. Calixarenes are organic compounds having 3D structures. These are cyclic oligomers formed by combining phenols and formaldehyde. The name “calixarenes” is attributed to the hydrophobic cup-like cavity formed by intramolecular hydrogen bonding among phenolic hydroxyls. They possess unique properties such as the ability to serve as excellent host molecules,39 the ability to be adapted to form reversible complexes,40 and applicability in separation chemistry.41 They have a wide range of applications in different areas like in the synthesis of metal nanoparticles, the manufacturing of sensors, nonlinear optics, etc. Different classes of calixarenes such as calix[4]arene, calix[5]arene, calix[6]arene, and calix[8]arene have been reported.42 The structural modifications in the calix[4]arene (CX [4]) structure have been investigated by many groups to check its impact in different applications.43,44 For instance, the CX [4] skeleton was fused with a cycloparaphenylene (CPP) ring to produce a new hybrid host. The optoelectronic and structural properties of the calix–CPP hybrid host were also studied. The results revealed an unexpected selectivity of calix–CPP for Li+ over Na+ due to the more favorable interaction through the CPP bridge and its greater size.13 Paterson et al. studied the ability of CX [4] to bind to a range of small guest molecules. It has been found that coordination to the lower-rim of CX [4] with both Mn3+ (quartet) and Fe3+ (quartet) causes greater binding energies of CX [4] toward these guest molecules. Binding energies were investigated for a range of metal types, spins, and oxidation states.45 The effect of doping of alkali metal atoms (Li, Na, and K) on the structural, electrical, and nonlinear optical properties of CX [4] has also been studied.46 The polarizability and first hyperpolarizability of the CX [4] molecule are dramatically enhanced by alkali metal atoms. The Li@calix[4]arene complex has the greatest increase in NLO response (9.3 × 104 a.u.).47 In a study carried out by Hou et al., it has been shown that the impact of a single alkali metal doping considerably increases the first hyperpolarizability of the t-Bu-calix[4]arene molecule.48
However, despite the abundance of superalkali-based nanomaterial advancements, there is a lack of sufficient exploration in the field of nonlinear optical (NLO) properties, especially concerning CX [4]. To the best of our knowledge, no investigation has been conducted on how superalkali clusters impact the geometrical, electrical, and NLO characteristics of calix[4]arenes. As a result, our keen interest lies in exploring the superalkalide properties and NLO response of superalkali-doped CX [4]. The current study focuses on the doping of selected superalkali (SA) subunits (Li3O, Na3O, and K3O) on CX [4], presenting a novel range of NLO materials with potential applications in nanotechnology.
2. Computational details
The pristine calix[4]arene (CX [4]) molecule (C28H24O4) and superalkali-doped calix[4]arenes have undergone geometry optimization using the ωB97XD functional and the 6-31+G(d,p) basis set in an unrestricted formalism. The ωB97XD long-range corrected hybrid functional is employed for the prediction of non-covalent interactions, particularly in systems that encompass doping.49 Following the geometry optimization, frequency calculations have been conducted to verify that the optimized geometries are real minima on the potential energy surface (PES). In this study, no scale factor to the frequencies has been applied when calculating the zero-point vibrational energy (ZPVE) or enthalpy corrections (Delta H). The convergence criteria for self-consistent field (SCF) calculations are as follows: a requested convergence of 1.00D-08 on the RMS density matrix, a requested convergence of 1.00D-06 on the MAX density matrix, and a requested convergence of 1.00D-06 on the energy. The standard default convergence criteria for optimization were set as follows: 0.00045 for the maximum force, 0.0003 for the root mean square (RMS) force, 0.0018 for maximum displacement, and 0.0012 for the root mean square (RMS) displacement. The ωB97XD/6-31+G(d,p) level of theory has been employed to investigate the interaction energies (Eint), NBO (natural bond orbital) charges, frontier molecular orbital (FMO) energy gaps, and vertical ionization energies (VIEs). The following formula can be utilized to compute the interaction energy: | (1) |
Other studies are also carried out by using the ωB97XD functional in combination with the 6-31+G(d,p) basis set, including density of states (DOS), non-covalent interaction (NCI), and quantum theory of atoms in molecules (QTAIM) analyses, and the Multiwfn50 and VMD51 software packages are employed. The absorption studies are performed at the TD-ωB97XD/6-31+G(d,p) level of theory. The CAM-B3LYP52 functional and the 6-31+G(d,p)53 basis set are used to determine the linear and nonlinear optical parameters including polarizability and second order polarizability (static first hyperpolarizability). The static hyperpolarizability of superalkali doped calix[4]arenes is also computationally tested by using the CAM-B3LYP functional. Based on previously reported results, the aforementioned level of theory is suitable to be employed to analyze the electro-optical characteristics of superalkali-doped systems.27,54 The default integral grid for SCF energies is set to UltraFine, which employs 99 radial and 590 angular points. For the coupled perturbed equations, a grid that is lesser by two steps is utilized. This default grid is SG1, consisting of 50 radial and 194 angular points. The finite electric field is employed to estimate higher derivatives. The Polar keyword is utilized to generate analytic first, second, and third derivatives with respect to the external field. Gamma (which involves the 4th derivative of energy with respect to the electric field) is calculated using differences of third derivatives with a finite electric field. The default value for this field is 0.000333 a.u. This methodology strikes a balance between numerical accuracy and computational efficiency, ensuring robust results while optimizing computational resources. The optical polarizability (α0), dipole moment (μ0), and first hyperpolarizability (β0) can all be calculated using the following formulae:
| μ0 = [μx2 + μy2 + μz2]1/2 | (2) |
 | (3) |
| β0 = [βx2 + βy2 + βz2]1/2 | (4) |
Additionally, calculations are performed to determine the βvec (beta vector) and γ (second hyperpolarizability) of SA-doped calix[4]arenes.
The mathematical expression for βvec is as follows:
 | (5) |
The equation used to calculate the average (isotropic) second hyperpolarizability (〈γ〉) is as follows:55
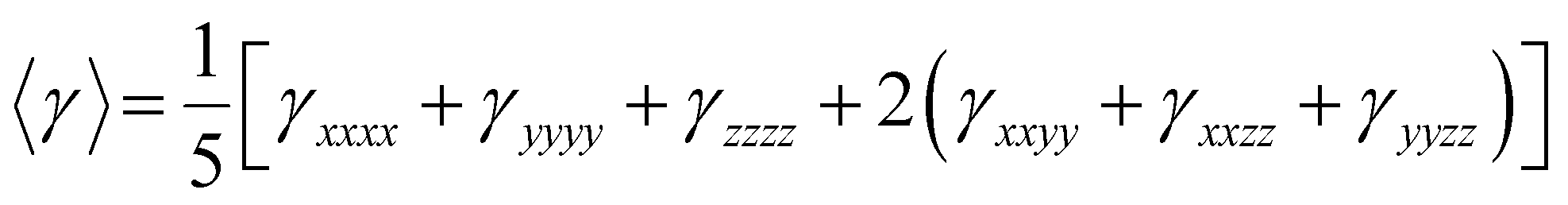 | (6) |
To further evaluate the practical applicability of the superalkali doped CX [4], the dynamic first hyperpolarizability (β(ω)) computations at laser wavelengths (532 nm and 1064 nm) are performed as well. The β(ω) can be obtained using the following equation:56
 | (7) |
At the same level of theory, the γ(ω) (dynamic second hyperpolarizability) of superalkali-doped calix[4]arenes is also estimated. The nonlinear refractive indices of M3O@calix[4]arene complexes are computed as follows:57
| n2 (cm2 W−1) = 8.23 × 10−23γDFWM | (8) |
Furthermore, the degenerate four-wave mixing (DFWM) using the relation provided by Tarazkar et al.58 is determined:
 | (9) |
Gaussian 1659 software is used for all DFT computations, and GaussView 6.1.160 software is utilized for illustration purposes.
3. Results and discussion
3.1 Optimized geometries and stabilities
It has been found that the 3D macrocycle CX [4], which consists of four phenolic oxygen atoms, is a neutral ligand that may bind to metal cations.61 In the CX [4] host, the 1,3-alternate structure is considerably preferred, and pure CX [4] possesses the C2 symmetry. The decreased repulsions between the lone pairs of oxygen atoms are therefore assumed to be a contributing factor for the conformational stability of arene moieties.62 Due to the presence of methylene linkages in CX [4], all isolated benzene rings produce a cavity that has a box-like shape. The optimized CX [4] has O–H, O–C and C–C (methylene linked) bond distances of 0.98 Å, 1.37 Å, and 1.51 Å, respectively, which are consistent with previously reported bond lengths.61 Furthermore, the tailored Li3O superalkali has a Li–O bond length of 1.69 Å, which nearly approaches the experimentally determined Li–O bond distance.63 From these results, it can be observed that the ωB97XD/6-31G+(d,p) level of theory is valid for the current study. The optimized stable geometries of CX [4] along with superalkalis (Li3O, K3O, and Na3O) are displayed in Fig. 1. | ||
| Fig. 1 Optimized structures of pure calix[4]arene (CX [4]) along with Li3O, Na3O & K3O superalkalis. | ||
The Li3O, Na3O, and K3O superalkalis are doped at CX [4] in the present work to investigate the different interaction sites. The various sensitive sites of CX [4] enable several possible interaction orientations. All of these orientations have been studied. There were totally four orientations taken into consideration during superalkali doping on CX [4], including two horizontal patterns (bottom and top) and two vertical patterns (bottom and top). Since most of the structures were distorted such as in the top position and the vertical bottom positions were merged to the horizontal ones, only the horizontal bottom position with the oxygen atoms of the complexant (CX [4]) has been taken into account for further analyses. A small change in the dihedral angle of the O1–O2–O3–O4 cavity of CX [4] indicates that the structural integrity of the system remains unaffected by the doping of superalkalis. The Li–O, Na–O, and K–O bond distances of the superalkalis in the proposed M3O@calix[4]arene (where M3O = Li3O, Na3O and K3O) are also 1.67, 2.01, and 2.40 Å, respectively, which are comparable to the values of the distinct superalkalis depicted in Fig. 1. The M3O subunits retain their structural integrity as a result of being incorporated into CX [4], with barely slight modifications to the geometric features of the superalkali subunits. The positive values of the first frequencies of the M3O@calix[4]arene complexes (Table 1) indicate the presence of true local minima on the PES (potential energy surface). The resulting optimized structures of SA-doped heteroatomic macrocycles are illustrated in Fig. 2.
| Complex | ν 1 | Symmetry | I d(M–CA) | E int | ΔG | VIE |
|---|---|---|---|---|---|---|
| Li3O@calix[4]arene | 48.87 | C 1 | 2.3 | −47.69 | −34.07 | 3.02 |
| Na3O@calix[4]arene | 10.56 | C 1 | 2.9 | −21.83 | −14.31 | 2.59 |
| K3O@calix[4]arene | 13.78 | C 1 | 3.3 | −18.79 | −7.35 | 2.44 |
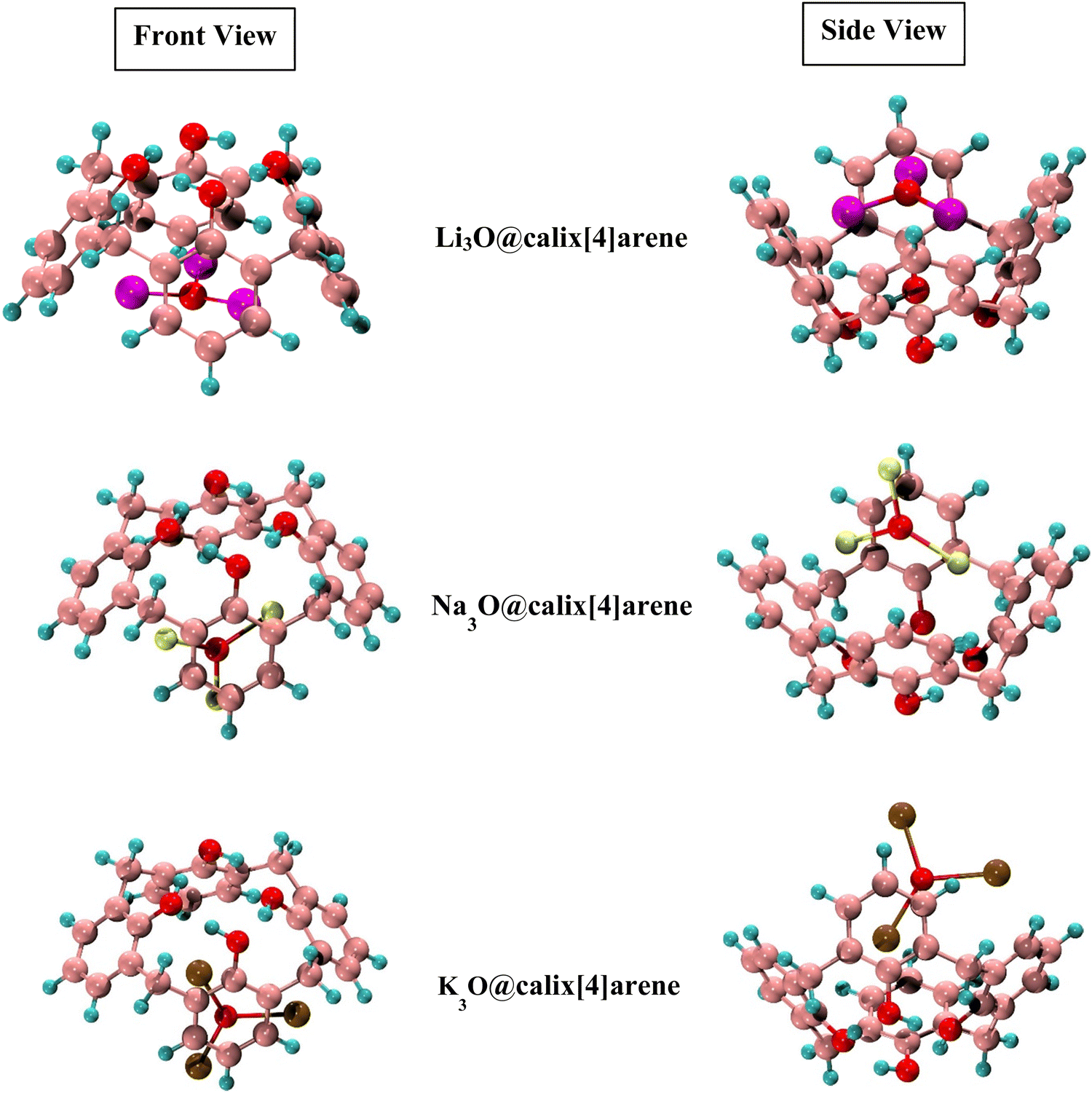 | ||
| Fig. 2 Optimized structures of Li3O, Na3O and K3O doped calix[4]arene (CX [4]) complexes (side and front views). | ||
The symmetry of CX [4] has changed as a result of the presence of superalkali subunits. Complexes of M3O@calix[4]arene now display the C1 point group, contributing to the confirmation that superalkali doping may alter the substrate symmetry which corroborates prior studies.
The practical applications of nonlinear optical materials place a considerable emphasis on the stability of complexes. The key factors that define the thermal and chemical stability of any system are interaction energy (Eint) and vertical ionization energy (VIE). The interaction energy of the system indicates that any framework with particularly exothermic nature exhibits enhanced thermodynamic stability.64 The Eint values of these superalkali-doped M3O@calix[4]arenes are illustrated in Table 1. The stability of Li3O@calix[4]arene, Na3O@calix[4]arene, and K3O@calix[4]arene complexes was evaluated by calculating their respective interaction energy (Eint) values, which were found to be −47.69, −21.83 and −18.79 kcal mol−1. It was observed that Li3O@calix[4]arene is the most stable among the three designed complexes. Lithium is in contact far more closely with CX [4] due to its smaller atomic size. This is supported by the fact that it has the shortest interaction distance (Id), 2.3 Å, compared to all other SA@calix[4]arenes (see Table 1). With an increase in the atomic number (from Li to K), the interaction distance seems to increase monotonically. The M3O@calix[4]arene systems are thermodynamically more stable than the previously reported M@calix[4]arene (M = Li, Na and K) complexes.47 The reason for the high thermal stability of SA-doped systems is the low ionization potential of superalkalis than the alkali metal atoms. To attain a more comprehensive insight into the stability of the complexes, the pertinent Gibbs enthalpies of formation are also computed. These values range from −34.07 to −7.35 kcal mol−1 in all superalkali doped calix[4]arene complexes (Table 1). A negative ΔG indicates that the fabrication of these complexes is spontaneous in nature and the complexes are thermodynamically stable.
The vertical ionization energy (VIE) is another factor that validates the chemical and electronic stability of complexes. A significantly large VIE value implies the chemical stability and rigidity of the systems. The SA@calix[4]arene complexes exhibit VIE values between 3.02 and 2.44 eV. The highest VIE value of 3.02 eV is observed in the case of Li3O@calix[4]arene. The Na3O@calix[4]arene and K3O@calix[4]arene possessed VIEs of 2.59 eV and 2.44 eV, respectively. It is observed that, with an increase in the metal size of superalkalis, the vertical ionization energy decreases: Li3O@calix[4]arene > Na3O@calix[4]arene > K3O@calix[4]arene.
3.2 Electronic properties
Frontier molecular orbital (FMO) analysis is an effective method for examining the change in the conductivity of compounds after complexation.65 This provides comprehensive information related to the energies of the highest occupied molecular orbitals (EHOMO), lowest unoccupied molecular orbitals (ELUMO), and energy gaps (Eg). Pristine CX [4] has a large energy gap, which restricts its application in optoelectronic devices [3]. Doping of CX [4] with superalkalis (Li3O, Na3O, and K3O) can elevate its electronic properties.The results showed that the Eg (EH–L) values of the complexes (Li3O@calix[4]arene to K3O@calix[4]arene) were considerably reduced. The primary factor responsible for the reduction in EH–L is the creation of new HOMOs. The generation of the new HOMO is due to excess electrons, and the previously existing HOMO transforms into HOMO−1. The EHOMO (HOMO energy), ELUMO (LUMO energy), corresponding energy gaps, EH–L percentage reductions, and Fermi level energies (EFL) are indexed in Table 2.
| Complexes | Q M3O | E HOMO | E LUMO | E H–L | % EHL | E FL |
|---|---|---|---|---|---|---|
| Calix[4]arene | — | −8.07 | 0.87 | 8.94 | — | −3.6 |
| Li3O@calix[4]arene | 0.89 | −3.24 | 0.25 | 3.49 | 59.7 | −1.49 |
| Na3O@calix[4]arene | −0.03 | −2.64 | −0.002 | 2.64 | 67.2 | −1.32 |
| K3O@calix[4]arene | −0.02 | −2.25 | −0.24 | 2.01 | 72.1 | −1.24 |
For SA-doped systems, the HOMO–LUMO energy gaps vary from 3.49 to 2.01 eV. K3O@calix[4]arene exhibits the highest reduction in EH–L which is due to the low ionization potential (2.44 eV), whereas Li3O@calix[4]arene has the lowest EH–L reduction value, which can be attributed to the high ionization potential (3.02 eV) of the complex. These findings reveal that superalkali-doped CX [4] systems possess semiconducting properties and probably can be practiced in optoelectronics.
All the newly designed complexes have percentage reduction in EH–L values that vary from 59.7 to 72.1%. Furthermore, the electron densities lie on the superalkalis in complexes, reflecting that the superalkali subunits have an important part in new HOMO formation. Since superalkalis have metal atoms of different sizes, the distribution of electron density on HOMOs and LUMOs and superalkalide properties also vary for the corresponding complexes, as shown in Fig. S1 (ESI†).
3.3 NBO analysis
Natural bond order (NBO) analysis has been employed for calculating NBO charges in order to estimate the direction and magnitude of charge transfer in SA@calix[4]arenes. The overall charge on the superalkali units (QM3O) is negative in the case of Na3O@calix[4]arene and K3O@calix[4]arene, whereas the charge on the calix[4]arene is positive, demonstrating that charge has been transferred from the substrate (calix[4]arene) towards the dopant (superalkalis) (Table 2). In accordance with Fig. S1 (ESI†), the electronic cloud of the HOMOs of Na3O@calix[4]arene and K3O@calix[4]arene is distributed on the entire Na3O and K3O structures. These intriguing complexes may have good NLO characteristics owing to their HOMOs’ electronic cloud distributions and NBO studies. The Li3O@calix[4]arene complex exhibits a peculiar behavior since Li3O has a net positive charge of 0.89 |e|. The charge (positive) on the superalkali (Li3O) and the electronic density which primarily lies over CX [4] (Fig. S1, ESI†) show that the charge is being shifted from the dopant towards the substrate. As demonstrated by Biglari and colleagues, the low ionization energy and smaller size of lithium make it more convenient to release electrons, which account for the greater charge on Li3O.66 It concludes that Li3O@calix[4]arene reflects an excess electron system rather than an alkalide character.3.4 Total and partial densities of states
A density of states study was carried out to gain more insight into the frontier molecular orbitals (FMOs), electronic behavior, and energy changes of doped complexes. It is evident from the PDOS spectra that superalkalis (except Li3O) significantly contribute to the creation of new HOMOs. In the case of Li3O@calix[4]arene, the major contribution is from calix[4]arene as the charge is transferred from the calix[4]arene towards the superalkali (Li3O). In comparison to the energy of pure CX [4] (−8.0 eV), the newly generated HOMOs in all complexes were in the high-energy range (between −2 and −5 eV). The PDOS and TDOS spectra of the undoped CX [4] and SA@calix[4]arene materials are shown in Fig. S2 (ESI†). It is anticipated that the newly formed HOMOs would contribute to the reduction of EH–L (HOMO–LUMO energy gap), as well as to the enhancement of the optoelectronic performance of the designed complexes.3.5 IRI analysis
The interaction region indicator (IRI) analysis is a computational technique that enables us to gain insight into the nature of intra- and intermolecular interactions.67,68 This is achieved by visualizing the electron density overlap between different regions. It includes 3D-isosurfaces and 2D-IRI plots. The IRI can also be written as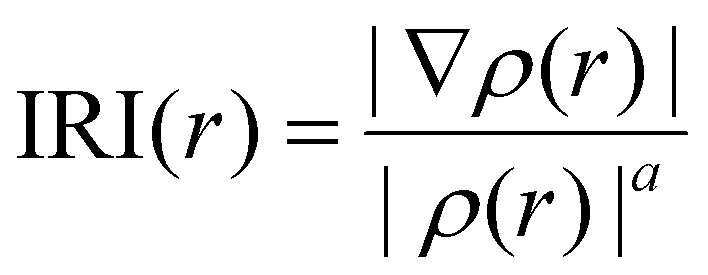 | (10) |
Therefore, interpreting 3D-isosurfaces and 2D-IRI plots gives valuable details regarding covalent and non-covalent interactions of SA@calix[4]arene complexes. In general, 3D isosurfaces portray patches of various colors such as blue, green, and red describing non-covalent, attractive, and repulsive interactions, respectively. The IRI maps with spikes at sign(λ2)ρ < 0 indicate non-bonded interactions (repulsive), while those with sign(λ2)ρ > 0 depict weakly bonded forces (dispersive).67 Consequently, both (isosurfaces and IRI graphs) have been generated to examine the kind of interactions that exist between superalkalis and CX [4] as shown in Fig. 3. In the graphs of SA@calix[4]arene complexes, green patches are indicative of weak dispersive forces, particularly vdW (van der Waals) interactions. The appearance of spikes in the sign(λ2) ρ region (around −0.04 and 0.005 a.u.) of IRI graphs in different complexes further supports the existence of non-covalent interactions (see Fig. 3).
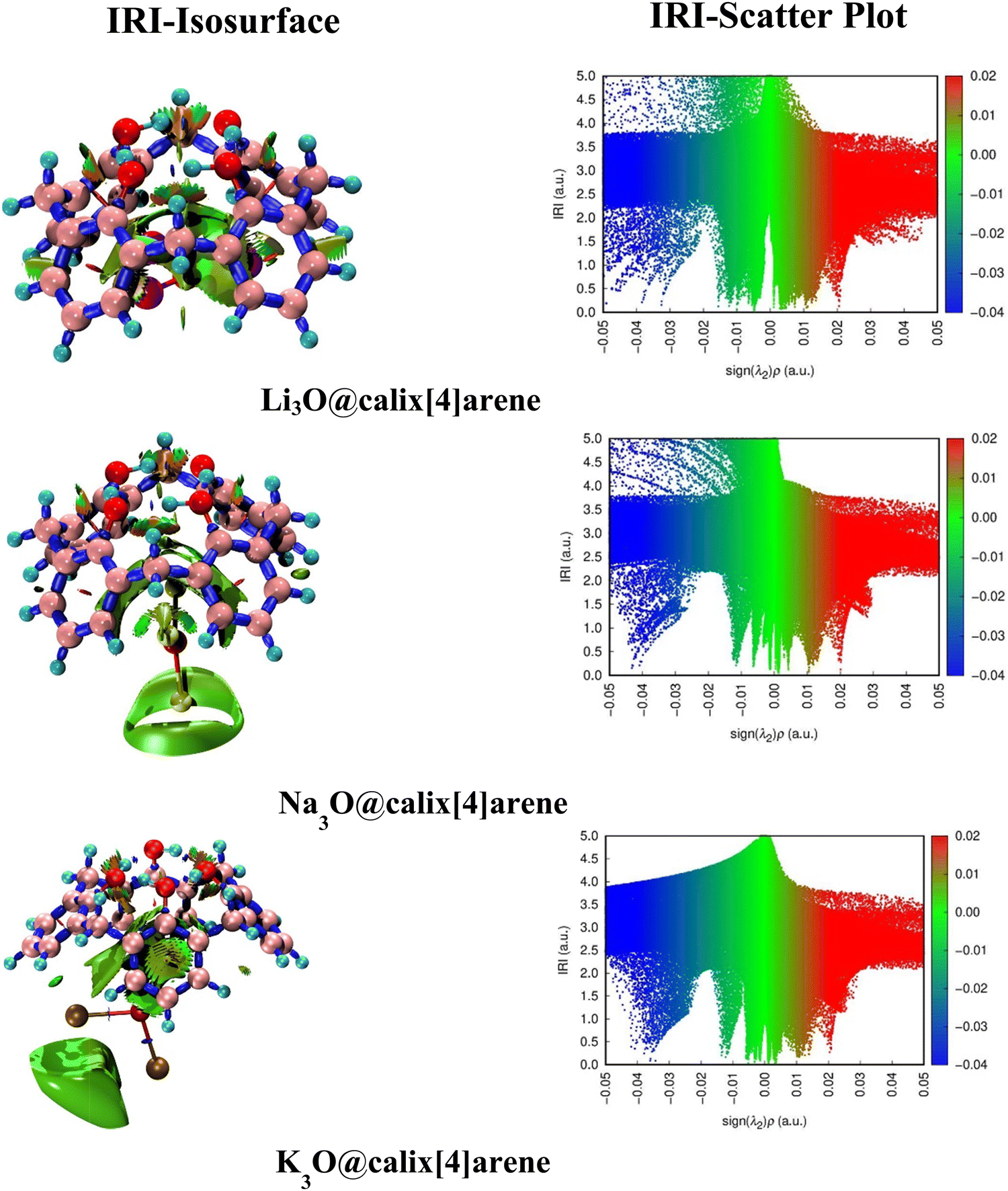 | ||
| Fig. 3 2-isosurfaces and 3D-IRI scatter plots of M3O@calix[4]arene complexes. | ||
3.6 QTAIM analysis
The QTAIM analysis is used for the visualization of different types of interactions, which include hydrogen bonding and van der Waals interactions.69,70 The nature of bond critical points (BCPs) and of different interactions can be examined using topological factors, including electron density and ∇2ρ (Laplacian of electron density). The territory with maximum electron density interconnecting the atoms of a molecule is termed as the bond critical point (BCP).71 The values of different BCPs can be expressed by using the following equations:| Hr = Gr + Vr | (11) |
 | (12) |
 | ||
| Fig. 4 QTAIM topological BCPs (orange spots) of superalkali doped calix[4]arene. | ||
3.7 UV-visible analysis
The UV-visible study has been carried out to investigate the absorbance features of the doped systems. The λmax (maximum absorption) values, transition energy and oscillator strength (f0) can be computed with the help of TD-DFT computations. Nonlinear optical materials must be transparent for commonly used lasers. The maximum absorption of pristine CX [4] takes place at 242 nm. The SA@calix[4]arene exhibits λmax values of 1246–1535 nm in the near infrared (NIR) region (Table 3). This rise in absorption maxima (λmax) is primarily attributed to the smaller Eex (excitation energy) values of the substances (0.81–1.09 eV) in comparison to bare calix[4]arene, which has 5.11 eV of Eex. It is inferred from the results that all SA@calix[4]arene complexes have an intense red shift (Fig. 5). These UV-visible transparent materials are compelling candidates to serve as optical materials since they have a significant role in the reinforcement of SHG (second harmonic generation).72| Complexes | ΔE | f 0 | Excited state | λ max | Major contribution |
|---|---|---|---|---|---|
| Calix[4]arene | 5.11 | 0.08 | (E2) | 242 | HOMO−1 → LUMO |
| Li3O@calix[4]arene | 0.99 | 0.04 | (E1) | 1246 | HOMO → LUMO+6 |
| Na3O@calix[4]arene | 0.81 | 0.22 | (E3) | 1236 | HOMO → LUMO+11 |
| K3O@calix[4]arene | 1.09 | 0.24 | (E3) | 1535 | HOMO → LUMO+18 |
 | ||
| Fig. 5 Absorption spectra of pure and superalkali doped calix[4]arene complexes. | ||
3.8 NLO properties of SA-doped calix[4]arenes
The measured α0 values (between 493 and 733 a.u.) of the complexes are larger than the pristine CX [4] (320 a.u.). The findings reflect that the increase in the dipole moment and polarizability of the complexes is triggered by the excess electrons of superalkalis. These results are in agreement with previously reported studies.81 Meanwhile, the β0 (static hyperpolarizability) of SA-doped complexes ranges from 7.0 × 104 to 5.9 × 105 a.u. The observed enhancement in the hyperpolarizability value is attributed to the surplus electrons introduced by the superalkalis, which perturb the symmetry of CX [4]. The Na3O@calix[4]arene attained the largest β0 (5.9 × 105 a.u.), whereas the K3O@calix[4]arene exhibited the smallest β0 (7.0 × 104 a.u.) (see Table 4). Another fact is that the β0 is also influenced by the excitation energies of the complexes. The extent of the first hyperpolarizability increases as the excitation energies of the complexes decrease. The results indicate that the highest β0 (5.9 × 105 a.u.) was observed for Na3O@calix[4]arene with the lowest excitation energy (0.81 eV). On the other hand, K3O doped calix[4]arene shows the lowest β0 (7.0 × 104) due to the highest excitation energy of 1.09 eV. (Table 3). The β0 of SA@calix[4]arene complexes aligns well when compared to already studied superalkali doped cyclic oligomers.82 As compared to the previously observed hyperpolarizability value of Li3O@B12N12 (3.7 × 104 a.u.), the β0 value of the newly introduced superalkali-doped calix[4]arene (5.9 × 105 a.u.) is substantially higher.83 The β0 value of the newly formed Na3O@calix[4]arene complex is greater than those of the already studied complexes of AM@calix[4]arenes (9.3 × 104 a.u.),84 alkalides (Li+(calix[4]pyrrole)M−) (2.4 × 104 a.u.)85 and superalkalides (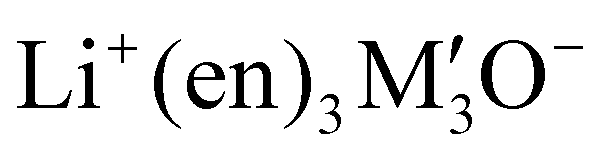 ) (9.1 × 104 a.u.).24 Apart from that, nonlinear optical prototypes involving p-nitroaniline (p-NA) and urea are also used to compare the β0 of the designed complexes. Urea and p-nitroaniline have respective β0 values of 43 a.u.86 and 1.2 × 103 a.u.87 In contrast to urea and p-NA, Na3O@calix[4]arene has a higher observed first hyperpolarizability (β0) value (5.9 × 105 a.u.). Therefore, the doped materials with substantial β0 values are considered to be more desirable NLO candidates.
) (9.1 × 104 a.u.).24 Apart from that, nonlinear optical prototypes involving p-nitroaniline (p-NA) and urea are also used to compare the β0 of the designed complexes. Urea and p-nitroaniline have respective β0 values of 43 a.u.86 and 1.2 × 103 a.u.87 In contrast to urea and p-NA, Na3O@calix[4]arene has a higher observed first hyperpolarizability (β0) value (5.9 × 105 a.u.). Therefore, the doped materials with substantial β0 values are considered to be more desirable NLO candidates.
| Complexes | μ 0 | α 0 | β 0 (a.u.) | β 0 (esu) | β Vec | γ 0 |
|---|---|---|---|---|---|---|
| Calix[4]arene | 2.16 | 320 | 556 | 4.80 × 10−30 | 556 | 7.5 × 104 |
| Li3O | 5.66 | 396 | 2.0 × 105 | 1.73 × 10−27 | — | 3.5 × 107 |
| Na3O | 4.05 | 605 | 2.9 × 105 | 2.51 × 10−27 | — | 3.1 × 107 |
| K3O | 5.03 | 693 | 8.4 × 105 | 7.26 × 10−27 | — | 1.1 × 108 |
| Li3O@calix[4]arene | 6.11 | 493 | 1.3 × 105 | 1.12 × 10−27 | 1.2 × 105 | 2.8 × 107 |
| Na3O@calix[4]arene | 9.44 | 340 | 5.9 × 105 | 5.10 × 10−27 | 5.9 × 105 | 2.3 × 108 |
| K3O@calix[4]arene | 8.98 | 733 | 7.0 × 104 | 6.05 × 10−28 | 5.6 × 104 | 8.5 × 107 |
The obtained results are verified by the computation of static hyperpolarizability projection on the dipole moment vector (βvec). Based on the trends of βvec (that are in perfect agreement with the β0 of the complexes), it is found that Na3O@calix[4]arene is quite NLO sensitive, while K3O@calix[4]arene has the least sensitivity. These results are quite similar to the electronic properties, particularly the NBO (charge transfer) charges and energy gaps.
Moreover, the static second hyperpolarizability for the proposed systems was also computed. The results indicated that the γ0 (static second hyperpolarizability) values show the same pattern as β0 but much larger in number. The SA@calix[4]arene complexes have γ0 values between 2.8 × 107 and 2.3 × 108 a.u. (see Table 4).
| Complexes | EOPE (−ω, ω, 0) | SHG (−2ω, ω, ω) | ||
|---|---|---|---|---|
| λ = 1064 nm | λ = 532 nm | λ = 1064 nm | λ = 532 nm | |
| Li3O@calix[4]arene | 7.1 × 104 | 9.5 × 104 | 2.1 × 104 | 1.9 × 105 |
| Na3O@calix[4]arene | 1.5 × 106 | 5.7 × 104 | 2.3 × 105 | 4.1 × 104 |
| K3O@calix[4]arene | 2.3 × 106 | 4.4 × 104 | 9.7 × 103 | 5.0 × 104 |
The third-order NLO (nonlinear optical) responses of all designed complexes have been predicted at 532 nm and 1064 nm by means of the EOKE (electro-optic dc-Kerr effect) and EFISHG (electric field-induced second harmonic generation). The findings reflect that M3O@calix[4]arene complexes exhibit augmented EFISHG and dc Kerr effects at both (532 nm and 1064 nm) wavelengths. The values of the estimated γ(−2ω; ω, ω, 0) (EFISHG) effect varied between 1.0 × 107 and 2.2 × 108 a.u. at 532 nm, while at 1064 nm they varied from 7.8 × 106 to 1.8 × 109 a.u. The γ(−ω; ω, 0, 0) (EOKE) effect values were in the range of 1.8 × 107 to 4.1 × 109 a.u. at 532 nm and from 4.9 × 107 to 4.0 × 1010 a.u. at 1064 nm. It is evident from the results (Table 6) that the K3O@calix[4]arene complex has the maximum values of EFISHG (1.8 × 109 a.u.) and EOKE (4.0 × 1010 a.u.) coefficients at 1064 nm.
| Complexes | Wavelength (nm) | γ(−ω; ω, 0, 0) EOKE | γ(−2ω; ω, ω, 0) EFISHG | γ DFWM (−ω; ω, −ω, ω) | n 2 (cm2 W−1) |
|---|---|---|---|---|---|
| Li3O@calix[4]arene | 1064 | 4.9 × 107 | 7.8 × 106 | 4.2 × 107 | 3.5 × 10−15 |
| 532 | 3.0 × 108 | 2.2 × 108 | 4.3 × 109 | 3.5 × 10−13 | |
| Na3O@calix[4]arene | 1064 | 8.3 × 107 | 3.3 × 108 | 1.1 × 108 | 9.5 × 10−15 |
| 532 | 4.1 × 109 | 4.5 × 107 | 4.3 × 109 | 3.6 × 10−13 | |
| K3O@calix[4]arene | 1064 | 4.0 × 1010 | 1.8 × 109 | 4.0× 1010 | 3.3 × 10−12 |
| 532 | 1.8 × 107 | 1.0 × 107 | 4.6 × 109 | 3.8 × 10−13 | |
In optoelectronics, NLO materials exhibiting higher quadratic nonlinear refractive indices (n2) are widely used as optical switches, optical pulse modulators and wavelength converters.88 The above equation may be used to calculate the n2 (quadratic nonlinear refractive indices) of SA-doped calix[4]arene systems using DFWM (degenerate four-wave mixing) γDFWM.89
| n2 (cm2 W−1) = 8.28 × 10−23γDFWM(a.u.) |
| γDFWM(−ω; ω,−ω, ω) ≈ (1/3)γ(−2ω; ω, ω, 0) + γ(−ω; ω, 0, 0) − (1/3)γ(0; 0, 0, 0) |
The estimated results are presented in Table 6. The Na3O@calix[4]arene has striking results of n2 (refractive index) (9.5 × 10−15 cm2 W−1) at 1064 nm as well as (3.6 × 10−13 cm2 W−1) at 532 nm. The computed results demonstrate that the newly designed systems possess excellent NLO responses at different wavelengths.
4. Conclusion
DFT simulations are employed to thoroughly investigate the geometric, electrical, and nonlinear responses of SA-doped calix[4]arenes (M3O@calix[4]arenes, where M3O represents Li3O, Na3O, and K3O). The results of the geometrical parameters indicated that both the superalkali subunits and CX [4] maintained their structural integrity. The designed M3O@calix[4]arene complexes exhibited a broad range of interaction energies from −18.79 to −47.69 kcal mol−1 and vertical ionization energies (2.44–3.02 eV), evidencing their chemical and thermodynamic stability. The Na3O@calix[4]arene complex exhibits the highest observed negative charge transfer of −0.03 |e|, as determined through NBO analysis. The reduced HOMO–LUMO energy gaps of the M3O@calix[4]arene complexes (ranging from 2.01 to 3.49 eV) compared to pure calix[4]arene (8.94 eV) indicate their semiconducting nature. Comprehensive IRI and QTAIM analyses offer a clear illustration of the non-bonding interactions present between the superalkalis and calix[4]arene. Additionally, the M3O@calix[4]arene complexes exhibit maximum absorption in the near infrared (NIR) region, specifically between 1236 nm and 1535 nm, rendering them UV-visible transparent. The first and second hyperpolarizabilities of the M3O@calix[4]arene complexes are determined to be 5.9 × 105 a.u. and 2.3 × 108 a.u., respectively, highlighting their pronounced nonlinear responses. The Na3O@calix[4]arene complex demonstrates significant values in second harmonic generation (SHG) (2.3 × 105 a.u.) and the electro-optical Pockels effect (5.0 × 104 a.u.) in the case of K3O@calix[4]arene at a wavelength of 1064 nm. Furthermore, an improvement in the dynamic second hyperpolarizability (third-order nonlinear optical response) coefficients, including the EFISHG and dc-Kerr effect, is observed. The nonlinear refractive indices (n2) of the M3O@calix[4]arene systems also show a noticeable increase, with the Na3O@calix[4]arene complex achieving a maximum n2 value of 9.5 × 10−15 cm2 W−1. These outcomes suggest that superalkali doped calix[4]arenes hold great potential as candidates for diverse applications, such as optical switching, wavelength conversion, and SHG, making them attractive options for future developments in these fields.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Researchers Supporting Project (no. RSPD2023R765), King Saud University, Riyadh, Saudi Arabia.References
- D. J. P. Arivuoli, Pramana, 2001, 57, 871–883 CrossRef CAS.
- A. Nayak, J. Park, K. De Mey, X. Hu, T. V. Duncan, D. N. Beratan, K. Clays and M. J. Therien, ACS Cent. Sci., 2016, 2, 954–966 CrossRef CAS PubMed.
- Y. Yao, H.-L. Xu and Z.-M. Su, J. Mater. Chem. C, 2022, 10, 886–898 RSC.
- T. Zhang, X. Wei, Y. Zuo and J. Chao, Optik, 2019, 182, 295–302 CrossRef CAS.
- M. Rajeshirke and N. Sekar, Opt. Mater., 2018, 76, 191–209 CrossRef CAS.
- H. Sajid, F. Ullah, M. Yar, K. Ayub and T. Mahmood, New J. Chem., 2020, 44, 16358–16369 RSC.
- N. Kosar, H. Tahir, K. Ayub and T. Mahmood, J. Mol. Graphics Modell., 2021, 105, 107867 CrossRef CAS PubMed.
- F. Ullah, N. Kosar, K. Ayub and T. Mahmood, Appl. Surf. Sci., 2019, 483, 1118–1128 CrossRef CAS.
- M. A. Gilani, S. Tabassum, U. Gul, T. Mahmood, A. I. Alharthi, M. A. Alotaibi, M. Geesi, R. Sheikh and K. Ayub, Appl. Phys. A: Mater. Sci. Process., 2018, 124, 1–9 CrossRef CAS.
- A. Ahsin and K. Ayub, J. Nanostruct. Chem., 2021, 1–17 Search PubMed.
- M. Asif, H. Sajid, M. A. Gilani, K. Ayub and T. Mahmood, Vacuum, 2022, 203, 111301 CrossRef CAS.
- D. Holton and P. Edwards, Chem. Br., 1985, 21, 1007 CAS.
- S. J. M. Thomas, P. P. Edwards and V. L. Kuznetsov, ChemPhysChem, 2008, 9, 59–66 CrossRef PubMed.
- J. L. Dye, Science, 1990, 247, 663–668 CrossRef CAS PubMed.
- J.-J. Wang, Z.-J. Zhou, Y. Bai, Z.-B. Liu, Y. Li, D. Wu, W. Chen, Z.-R. Li and C.-C. Sun, J. Mater. Chem., 2012, 22, 9652–9657 RSC.
- W. Chen, Z.-R. Li, D. Wu, F.-L. Gu, X.-Y. Hao, B.-Q. Wang, R.-J. Li and C.-C. Sun, J. Chem. Phys., 2004, 121, 10489–10494 CrossRef CAS PubMed.
- L.-T. Fan, Y. Li, D. Wu, Z.-R. Li and C.-C. Sun, Aust. J. Chem., 2012, 65, 138–144 CrossRef CAS.
- R. Bano, M. Arshad, T. Mahmood, K. Ayub, A. Sharif, S. Perveen, S. Tabassum, J. Yang and M. A. Gilani, J. Phys. Chem. Solids, 2022, 160, 110361 CrossRef CAS.
- J. L. Dye, J. M. Ceraso, M. Lok, B. Barnett and F. J. Tehan, J. Am. Chem. Soc., 1974, 96, 608–609 CrossRef CAS.
- W.-M. Sun, D. Wu, Y. Li and Z.-R. Li, Dalton Trans., 2014, 43, 486–494 RSC.
- M. Karplus and R. N. Porter, Atoms and molecules; an introduction for students of physical chemistry, 1970 Search PubMed.
- B. Li, D. Peng, F. L. Gu and C. Zhu, ChemistrySelect, 2018, 3, 12782–12790 CrossRef CAS.
- F. Ullah, K. Ayub and T. Mahmood, New J. Chem., 2020, 44, 9822–9829 RSC.
- J. Mai, S. Gong, N. Li, Q. Luo and Z. Li, Phys. Chem. Chem. Phys., 2015, 17, 28754–28764 RSC.
- N. V. Tkachenko, Z. M. Sun and A. I. Boldyrev, ChemPhysChem, 2019, 20, 2060–2062 CrossRef CAS.
- N. Hou, W.-M. Sun, F.-Y. Du and H.-S. Wu, Optik, 2019, 183, 455–462 CrossRef CAS.
- C. Tu, G. Yu, G. Yang, X. Zhao, W. Chen, S. Li and X. Huang, Phys. Chem. Chem. Phys., 2014, 16, 1597–1606 RSC.
- N. Kosar, K. Shehzadi, K. Ayub and T. Mahmood, J. Mol. Graphics Modell., 2020, 97, 107573 CrossRef CAS PubMed.
- T. Ishfaq, R. Ahmad Khera, S. Zahid, U. Yaqoob, R. Aqil Shehzad, K. Ayub and J. Iqbal, Comput. Theor. Chem., 2022, 1211, 113654 CrossRef CAS.
- H. Sajid, F. Ullah, S. Khan, K. Ayub, M. Arshad and T. Mahmood, RSC Adv., 2021, 11, 4118–4128 RSC.
- A. Ahsin and K. Ayub, Mater. Sci. Semicond. Process., 2022, 138, 106254 CrossRef CAS.
- M. Noormohammadbeigi and H. R. Shamlouei, J. Inorg. Organomet. Polym. Mater., 2018, 28, 110–120 CrossRef CAS.
- T. S. Girisun and S. Dhanuskodi, Mater. Res. Bull., 2010, 45, 88–91 CrossRef.
- R. Ittyachan and P. Sagayaraj, J. Cryst. Grow., 2003, 249, 553–556 CrossRef CAS.
- M. Arivanandhan, X. Huang, S. Uda, G. Bhagavannarayana, N. Vijayan, K. Sankaranarayanan and P. Ramasamy, J. Cryst. Growth, 2008, 310, 4587–4592 CrossRef CAS.
- M. J. Cho, D. H. Choi, P. A. Sullivan, A. J. Akelaitis and L. R. Dalton, Prog. Polym. Sci., 2008, 33, 1013–1058 CrossRef CAS.
- S. Kulshrestha and A. Shrivastava, Crystal growth and characterization of organic NLO materials and their application in optical devices, 2020 Search PubMed.
- E. Nestoros and M. C. Stuparu, Chem. Commun., 2018, 54, 6503–6519 RSC.
- S. Shinkai, Pure Appl. Chem., 1986, 58, 1523–1528 CrossRef CAS.
- C. Kahlfuss, E. Metay, M.-C. Duclos, M. Lemaire, M. Oltean, A. Milet, E. Saint-Aman and C. Bucher, C. R. Chim, 2014, 17, 505–511 CrossRef CAS.
- R. Ludwig, Microchim. Acta, 2005, 152, 1–19 CrossRef CAS.
- C. D. Gutsche, Calixarenes: an introduction, Royal Society of Chemistry, 2008 Search PubMed.
- J. H. Hymel, J. Townsend and K. D. Vogiatzis, J. Phys. Chem. A, 2019, 123, 10116–10122 CrossRef CAS PubMed.
- R. E. Sardjono and R. Rachmawati, Green Chemical Processing and Synthesis, IntechOpen, 2017 Search PubMed.
- P. Murphy, S. J. Dalgarno and M. J. Paterson, J. Phys. Chem. A, 2016, 120, 824–839 CrossRef CAS PubMed.
- A. Azadi and H. R. Shamlouei, Comput. Theor. Chem., 2021, 1202, 113332 CrossRef CAS.
- H. R. Shamlouei and F. Parvinzadeh, Phys. E, 2021, 127, 114539 CrossRef CAS.
- N. Hou, Y. Li, D. Wu and Z.-R. Li, Acta Phys.-Chim. Sin., 2014, 30, 1223–1229 CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- E. R. Davidson and D. Feller, Chem. Rev., 1986, 86, 681–696 CrossRef CAS.
- X. Li, Q. Han, X. Yang, R. Song and L. Song, Chem. Phys. Lett., 2016, 659, 93–99 CrossRef CAS.
- H. A. Kurtz, J. J. Stewart and K. M. Dieter, J. Comput. Chem., 1990, 11, 82–87 CrossRef CAS.
- J. E. Rice and N. C. Handy, Int. J. Quantum Chem., 1992, 43, 91–118 CrossRef CAS.
- C. Bree, A. Demircan and G. Steinmeyer, IEEE J. Quantum Electron., 2010, 46, 433–437 CAS.
- D. P. Shelton and J. E. Rice, Chem. Rev., 1994, 94, 3–29 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. B.01., Wallingford, CT, 2016 Search PubMed.
- R. Dennington., T. A. Keith and J. M. Millam, Gauss View, Version 5., Semichem Inc., Shawnee Mission, KS, 2019 Search PubMed.
- D.-F. Wang and Y.-D. Wu, J. Theor. Comput. Chem., 2004, 3, 51–68 CrossRef CAS.
- C. S. Zuo, O. Wiest and Y. D. Wu, J. Phys. Org. Chem., 2011, 24, 1157–1165 CrossRef CAS.
- D. Bellert and W. Breckenridge, J. Chem. Phys., 2001, 114, 2871–2874 CrossRef CAS.
- N. Kosar, T. Mahmood, K. Ayub, S. Tabassum, M. Arshad and M. A. Gilani, Opt. Laser Technol., 2019, 120, 105753 CrossRef CAS.
- A. M. Smith and S. Nie, Acc. Chem. Res., 2010, 43, 190–200 CrossRef CAS PubMed.
- E. Tahmasebi, E. Shakerzadeh and Z. Biglari, Appl. Surf. Sci., 2016, 363, 197–208 CrossRef CAS.
- T. Lu and Q. Chen, Chem.: Methods, 2021, 1, 231–239 CAS.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- R. F. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- R. F. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- F. Huang, S. Zhuang, W. Liu, L. Lin and L. Sun, Spectrochim. Acta, Part A, 2021, 248, 119277 CrossRef CAS PubMed.
- Y. Shen, Y. Yang, S. Zhao, B. Zhao, Z. Lin, C. Ji, L. Li, P. Fu, M. Hong and J. Luo, Chem. Mater., 2016, 28, 7110–7116 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- G. Khanum, A. Ali, S. Shabbir, A. Fatima, N. Alsaiari, Y. Fatima, M. Ahmad, N. Siddiqui, S. Javed and M. Gupta, Crystals, 2022, 12, 337 CrossRef CAS.
- S. Liu, F.-W. Gao, H.-L. Xu and Z.-M. Su, Mol. Phys., 2019, 117, 705–711 CrossRef CAS.
- Maria, J. Iqbal, R. Ludwig and K. Ayub, Mater. Res. Bull., 2017, 92, 113–122 CrossRef CAS.
- F. Ullah, N. Kosar, A. Ali, Maria, T. Mahmood and K. Ayub, Phys. E, 2020, 118, 113906 CrossRef CAS.
- A. Hanif, R. Kiran, R. A. Khera, A. Ayoub, K. Ayub and J. Iqbal, J. Mol. Graphics Modell., 2021, 107, 107973 CrossRef CAS PubMed.
- S. Sajjad, A. Ali, T. Mahmood and K. Ayub, J. Mol. Graphics Modell., 2020, 100, 107668 CrossRef CAS PubMed.
- R. S. Roy, S. Ghosh, K. Hatua and P. K. Nandi, J. Mol. Model., 2021, 27, 74 CrossRef CAS PubMed.
- G. Y. Guo, K. C. Chu, D.-S. Wang and C.-G. Duan, Comput. Mater. Sci., 2004, 30, 269–273 CrossRef CAS.
- H. Sajid, F. Ullah, S. Khan, K. Ayub, M. Arshad and T. Mahmood, RSC Adv., 2021, 11, 4118–4128 RSC.
- W.-M. Sun, X.-H. Li, D. Wu, Y. Li, H.-M. He, Z.-R. Li, J.-H. Chen and C.-Y. Li, Dalton Trans., 2016, 45, 7500–7509 RSC.
- H. R. Shamlouei and F. Parvinzadeh, Phys. E, 2021, 127, 114539 CrossRef CAS.
- W. Chen, Z.-R. Li, D. Wu, Y. Li, C.-C. Sun, F. L. Gu and Y. Aoki, J. Am. Chem. Soc., 2006, 128, 1072–1073 CrossRef CAS PubMed.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195–242 CrossRef CAS.
- C. Teng and A. Garito, Phys. Rev. Lett., 1983, 50, 350 CrossRef CAS.
- S. Lu, C. Zhao, Y. Zou, S. Chen, Y. Chen, Y. Li, H. Zhang, S. Wen and D. Tang, Opt. Express, 2013, 21, 2072–2082 CrossRef CAS PubMed.
- M. Tarazkar, D. A. Romanov and R. J. Levis, J. Chem. Phys., 2014, 140, 214316 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04825j |
| This journal is © the Owner Societies 2024 |