
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural diversity in the membrane-bound hIAPP dimer correlated with distinct membrane disruption mechanisms†
Qin
Qiao
 *ab,
Guanghong
Wei
c and
Zhijian
Song
ab
*ab,
Guanghong
Wei
c and
Zhijian
Song
ab
aDigital Medical Research Center, School of Basic Medical Sciences, Fudan University, Shanghai 200032, China. E-mail: qinqiao@fudan.edu.cn
bShanghai Key Laboratory of Medical Imaging Computing and Computer Assisted Intervention, Shanghai 200032, China
cDepartment of Physics, State Key Laboratory of Surface Physics, Key Laboratory for Computational Physical Science (Ministry of Education), Fudan University, Shanghai 200438, China
First published on 2nd February 2024
Abstract
Amyloid deposits of the human islet amyloid polypeptide (hIAPP) have been identified in 90% of patients with type II diabetes. Cellular membranes accelerate the hIAPP fibrillation, and the integrity of membranes is also disrupted at the same time, leading to the apoptosis of β cells in pancreas. The molecular mechanism of hIAPP-induced membrane disruption, especially during the initial membrane disruption stage, has not been well understood yet. Herein, we carried out extensive all-atom molecular dynamics simulations investigating the hIAPP dimerization process in the anionic POPG membrane, to provide the detailed molecular mechanisms during the initial hIAPP aggregation stage in the membrane environment. Compared to the hIAPP monomer on the membrane, we observed not only an increase of α-helical structures, but also a substantial increase of β-sheet structures upon spontaneous dimerization. Moreover, the random coiled and α-helical dimer structures insert deep into the membrane interior with a few inter-chain contacts at the C-terminal region, while the β-sheet-rich structures reside on the membrane surface accompanied by strong inter-chain hydrophobic interactions. The coexistence of α and β structures constitutes a diverse structural ensemble of the membrane-bound hIAPP dimer. From α-helical to β-sheet structures, the degree of membrane disruption decreases gradually, and thus the membrane damage induced by random coiled and α-helical structures precedes that induced by β-sheet structures. We speculate that insertion of random coiled and α-helical structures contributes to the initial stage of membrane damage, while β-sheet structures on the membrane surface are more involved in the later stage of fibril-induced membrane disruption.
1. Introduction
The human islet amyloid polypeptide (hIAPP), a 37-residue glucose-regulating hormone co-secreted with insulin, is associated with the development of type II diabetes (T2D), as it deposits as amyloid fibrils in more than 90% of T2D patients.1 Experiments have indicated that the hIAPP exerts cytotoxicity by disrupting the cellular membrane during its amyloid aggregation process.2–4 Therefore, understanding the hIAPP–membrane interactions is essential to reveal the hIAPP pathogenic mechanisms, and thus provide guidance for the therapeutic strategies of T2D.Various hypotheses have been proposed to explain the molecular mechanism of hIAPP-induced membrane damage, including: (1) the “pore formation” mechanism: the formation of hIAPP non-selective ion channels across the membrane breaking the cell osmosis;5–8 (2) the “detergent-like” mechanism: the extraction of lipids by the hIAPP N-terminal region leading to the membrane leakage;9–11 and (3) the “fibril elongation” mechanism: the elongation of hIAPP fibrils distorting the cell by inducing the mechanical pressure on the membrane surface.12,13 Several experimental studies indicated that the hIAPP-induced membrane damage is bi-phasic, with the 1st stage caused by prefibrillar oligomers, and the 2nd stage caused by fibril elongation,13,14 and thus different disruption mechanisms may be involved in different aggregation stages. Meanwhile, the particular mechanism of hIAPP-induced membrane damage also depends on experimental conditions, such as lipid compositions, hIAPP-to-lipid ratios, pH conditions, etc.15–21 Therefore, understanding the detailed molecular interactions during the hIAPP aggregation in a membrane environment is important for unravelling the molecular mechanism of hIAPP-induced membrane disruption.
Understanding the conformational changes during hIAPP aggregation in a membrane is essential for identifying its cytotoxic species. However, due to the transient and heterogeneous nature of the hIAPP oligomerization process, it is still difficult to experimentally characterize the structural distribution of hIAPP intermediates in a membrane environment. On the one hand, although the presence of a membrane accelerates the hIAPP fibrillation,22–27 previous experiments also indicated the stabilization of non-amyloid α-helical structures as a key intermediate in membranes containing both anionic and zwitterionic lipids,28–30 and showed the coil-to-α-to-β structural transitions during the hIAPP fibrillation in the negatively charged DPPG membrane.31 On the other hand, sum frequency generation IR spectroscopy indicated a direct transition from coil to β structures of the hIAPP in the DPPG membrane without involving α-helical intermediates.32 Thus, the role of α-helical intermediates still remains controversial.33,34 Several experiments have utilized nano-discs to capture the hIAPP intermediates on membranes and obtained their residue-based secondary structures, e.g., a NMR spectroscopy experiment revealed a three-stranded β-sheet structure of the hIAPP on the membrane of mixed zwitterionic DMPC and anionic DMPG lipids.35 Recently, an isotope-labelled 2D-IR experiment indicated that the hIAPP oligomer contains α-helices in fragment L12A13 and β-sheets in fragment G24A25 on neutrally charged membranes.36 However, the relations between the hIAPP oligomer structural properties and the corresponding membrane disruption effects still remain unstudied.
Computational studies have shed light on the structural properties of the membrane-bound hIAPP in different aggregation stages.21,37,38 As for monomers, in agreement with the EPR experiment of the hIAPP on anionic POPS vesicles,39 several computational studies reported that the hIAPP monomer forms an amphiphilic α-helix parallel to the membrane surface at its N-terminal region, while its amyloid core and C-terminal regions remain flexible.40–42 During the aggregation stage, a previous simulation study utilized a highly mobile membrane mimetic model to accelerate the hIAPP inter-chain diffusion, and captured the hIAPP α-helix to β-sheet transitions on the membrane surface.43 However, as the lipid mimetic model simplifies the lipid tail groups, this study may not be able to reflect the real hIAPP membrane insertion process.43 The stability of membrane-bound α-helical and β-sheet structures has also been investigated via simulations. On the one hand, starting from the α-helical hIAPP structures on SDS micelles resolved by NMR,44 a previous simulation study investigated the inter-chain interactions of the hIAPP dimer with the N-terminal region pre-inserted into the membrane,45 and coarse-grained simulations revealed that the α-helical hIAPP monomers aggregate and form ion channels, starting from either pre-inserted or surface-bound conformations.46,47 On the other hand, several computational studies focused on revealing the different insertion orientations of β-sheet-rich protofibrils with the change of pH and membrane compositions,48–51 and the structural model of a β barrel for hIAPP ion channels has also been proposed.52 A recent computational study created various structural models for hIAPP ion channels, and concluded that both helical-bundle and β-barrel ion channels are meta-stable after examining their structural stability via all-atom simulations.53 In spite of the continuous efforts, there still lacks an understanding of molecular mechanisms during the hIAPP initial spontaneous aggregation process in the membrane environment.
In this study, we performed extensive (30-μs long) all-atom molecular dynamics simulations to investigate the hIAPP dimerization process in the anionic POPG membrane. Dimer, as the smallest oligomer, provides the first and best opportunity to study the monomer–monomer interface that drives the initial aggregation process and experiments have indicated that the hIAPP dimer is the most probable oligomer at low concentration.54,55 Starting from the membrane-bound α-helical hIAPP monomer structures identified in our previous simulations,41 we observed a substantial increase of β structures and an increase of helical structures of the hIAPP upon its spontaneous dimerization in the POPG membrane, although the random coiled structures are still dominant. The coexistence of α and β structures constitutes a diverse structural ensemble of the membrane-bound hIAPP dimer. Meanwhile, the random coiled and α-helical dimer structures insert deep into the membrane interior, while the β-sheet-rich dimer structures reside on the membrane surface with strong inter-chain hydrophobic interactions. Moreover, from β-sheet to α-helical dimer structures, the degree of hIAPP-induced membrane disruption also increases gradually. In detail, the membrane deformation induced by β-sheet-rich structures on the membrane surface is almost negligible, while the deep insertion of random coiled and α-helical dimers induces global membrane disruption including membrane thinning and stretching. We speculate that the insertion of random coiled and α-helical structures contributes to the initial stage of membrane damage during hIAPP oligomerization, while β-sheet structures on the surface may be more involved in the later membrane disruption stage during hIAPP fibrillation. This study reveals the complexity of the membrane-bound hIAPP dimer structural ensemble, and sheds light on the molecular mechanisms of hIAPP-induced membrane disruption from a structural perspective.
2. Methods
2.1. Simulation details
We constructed a dimerization system from the α-helical structures identified in our previous simulations41 of hIAPP monomers in the POPG membrane. To shorten the free diffusion between monomers, we elevated the hIAPP concentration during the system construction, and then performed extensive simulations (accumulated to 30-μs long) on its dimerization process in the membrane. The details of simulations are as follows. | ||
| Fig. 1 (a) hIAPP amino-acid sequence, which is divided into the N-terminal region (K1–N14), the middle region (F15–S28), and the C-terminal region (S29–Y37). (b) Representative snapshot of the simulation system. (c) Secondary structural comparison between the membrane-bounded hIAPP monomer and dimer. The average number of residues adopting each category of secondary structures is plotted. Random coiled category includes coil, bend, and turn structures. β category includes β-sheet and β-bridge structures. Helix category includes α-helix, 5-helix, and 3-helix structures. The gray dashed bars are the monomer and red solid bars are the dimer, with the error bars indicating the standard deviations. (d) Probability of each residue to adopt each type of secondary structures of the hIAPP dimer in the membrane. | ||
We then equilibrated the reduced simulation system. To avoid periodic crash, we first slightly increased both the x and y box lengths to 5.0 nm, and then performed energy minimization with the steepest method. The minimized system was then further equilibrated with a 200-ps long position restraint followed by a 200-ps long NPT equilibrium. In both the position restraint and NPT equilibrium, we utilized the Berendsen method61 for the thermodynamic coupling. The temperature coupling at 310 K was applied separately on three groups, i.e., hIAPP, POPG, and the rest of water and ions, with the coupling constant at 0.5 ps. The pressure coupling at 1 bar was semi-isotropic, in which the pressures of the xy-plane and the z-direction were coupled separately. The pressure coupling constant was 10 ps and the compressibility coefficient was 4.5 × 10−5 bar−1. The electrostatic interactions were calculated via PME method,62 with real space cut-off at 1.6 nm. vDW interactions were switched to zero from 1.4 nm to 1.5 nm. hIAPP and POPG lipids were constrained with the LINCS algorithm,63 and water molecules with the SETTLE algorithm.64 After the equilibration, the size of the simulation box became around 4.7 × 4.7 × 13.4 nm3.
We then duplicated the monomer conformations in either the x or y direction to create the dimer simulation system, as shown in Fig. S1 (ESI†). The dimer system contains 2 hIAPP, 120 POPG lipids (60 lipids in each leaflet), 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 276 water molecules, 144 Na+, and 30 Cl−. In the 6 initial conformations, the minimal distances between two monomers were larger than 1.8 nm. For each initial state, we performed one 5-μs long NPT production run, and observed the spontaneous dimerization process as shown by the time evolution of minimal distances between two hIAPP chains in Fig. S2 (ESI†). In the production simulations, we utilized the Nose–Hoover temperature coupling65 and the Parrinello–Rahman pressure coupling66 for the thermodynamic couplings.
276 water molecules, 144 Na+, and 30 Cl−. In the 6 initial conformations, the minimal distances between two monomers were larger than 1.8 nm. For each initial state, we performed one 5-μs long NPT production run, and observed the spontaneous dimerization process as shown by the time evolution of minimal distances between two hIAPP chains in Fig. S2 (ESI†). In the production simulations, we utilized the Nose–Hoover temperature coupling65 and the Parrinello–Rahman pressure coupling66 for the thermodynamic couplings.
2.2. Analysis methods
First, we conducted the conformational clustering based on the single-chain RMSD via Daura's method,70,71 with an RMSD cut-off of 0.4 nm (step 1 in Fig. S3, ESI†), i.e., the maximal RMSD to the cluster centre is less than the cut-off 0.4 nm within each cluster. Each frame is labelled by the cluster indices of its two hIAPP chains, and forms a pair of single-chain clusters. Depending on whether there are inter-chain contacts or not, the conformations of single-chain clusters are further divided into the dimerized part and the isolated part. The population distribution of single-chain clustering is shown in Fig. S4 (ESI†). We selected the top 12 clusters of the dimerized part with the population cut-off at 2.0%, as shown in the inset of Fig. S4 (ESI†), and their accumulating population reaches 85.9%. In comparison with the monomeric hIAPP in the membrane, we further calculated the single-chain structural properties in the dimerized part, such as the radius of gyration (Rg) and the number of contacts with POPG tail groups, to investigate the changes in the chain dimension and insertion depth upon the dimerization. Moreover, we also calculated the inter-chain H-bonds and the inter-chain hydrophobic contacts, to examine the contributions of different inter-chain interactions to the corresponding structural changes.
Second, we examined the pairs of single-chain clusters, and divided the dimerized pairs of single-chain clusters by performing additional k-means clustering based on the protein–protein and protein–membrane contact maps (step 2 in Fig. S3, ESI†). We selected the top 13 dimerized pairs with population cut-off at 2.0% for the clustering in the step 2. As shown in Fig. S5 (ESI†), the accumulated population of the top 13 dimerized pairs reaches 80.2%. The details of step 2 clustering are as follows. For each frame, we first concatenated the residue-based inter-chain contact maps (37 × 37 elements) and residue-based contact maps with membrane tail groups (37 × 2 elements), and then performed the k-means clustering based on the concatenated vectors (37 × (37 + 2) elements) describing the inter-molecular contact patterns of the dimerized complex. The optimal number of clusters k* was determined via the elbow method, i.e., the k value at which the 2nd derivative of sum of squared distances (SSD) reaches the maximum. In the case that the SSDs were smaller than 104 and the 2nd derivatives of SSDs were smaller than 103, we chose k* = 1 as the optimal number of clusters, as shown in Fig. S6 (ESI†). This step 2 clustering of dimer pairs results in 22 dimer clusters, and the population distribution of these 22 clusters is shown in Fig. S7 (ESI†). In this way, each resulting dimer cluster corresponds to one specific type of single-chain conformations with one particular inter-molecular contact pattern. For each dimer cluster, we also calculated its structural properties, such as the average number of inter-chain contacts, the average number of tail contacts, the residue-based inter-chain H-bonds, the residue-based inter-chain hydrophobic contacts, and the residue-based tail contacts. To elucidate the hIAPP conformational changes upon dimerization, we tracked the time evolution of the 22 dimer clusters in each trajectory. The transitions among these clusters are shown in Fig. S8 (ESI†). We also labelled the inter-chain contacts of the initially collapsed dimer clusters in each trajectory, and plotted the initially collapsed contact map in Fig. S9 (ESI†).
3. Results and discussion
3.1. Spontaneous hIAPP dimerization in the POPG membrane
We observed the spontaneous dimerization of the hIAPP in the POPG membrane bilayer in our simulations. As shown in Fig. S2 (ESI†), in all the six 5-μs long trajectories, the minimal distances between two hIAPP chains decrease with the increase of inter-chain contacts, and later remain at ∼0.2 nm. Fig. 1(b) shows the representative conformation of the last frame in the 1st trajectory.3.2. Structural properties of the membrane-bound hIAPP dimer
 | ||
| Fig. 2 Structural properties of the hIAPP dimer in the membrane. (a) 2-d free energy projection on the number of inter-chain contacts (x-axis) and the number of contacts with membrane tail groups (y-axis). The black circles correspond to the properties of the top 22 dimer clusters of the hIAPP in the membrane, and the size of each circle is proportional to the corresponding dimer cluster population. The dimer clusters are further grouped into 4 categories: group 1 (G1) in purple, G2 in blue, G3 in green, and G4 in orange. (b) Residue-based inter-chain interactions. The bottom right is the number of inter-chain non-polar, i.e., hydrophobic, contacts and the upper left is the number of inter-chain H-bonds, with the corresponding 1-d distribution shown by side. The contacts of different groups are also labelled with different markers: purple circles for G1, blue hexagons for G2, green pentagons for G3, and orange squares for G4. (c) Residue-based contact number of the hIAPP dimer with membrane tail groups in each group. (d)–(g) Representative conformations and residue-based secondary structural distribution of each group, from G1 in (d) to G4 in (g). In each group, the centre conformations of dimer clusters are aligned. The hIAPP dimers are shown in cartoon representation, with one chain in cyan, and the other chain in light-green. The secondary structure α-helices are in red and β-sheets are in yellow. N-termini (Cα atoms of K1 residues) of chains are in gray sphere. Sidechains of residues forming inter-chain hydrophobic contacts are shown in sphere, and residues forming inter-chain H-bonds are shown in stick with H-bonds shown as orange dashes. Sidechains of deeply inserted residues are shown in gray dots. The population of each group and its cluster IDs (excluding the initially collapsed clusters) are also labelled at the bottom. As for the secondary structure distribution in each group, red circles are the distribution of α-helices and yellow squares are that of β-sheets. Residue numbers adopting α-helices or β-sheets are shown by side as red or yellow bars. | ||
G1: deep insertion with α-helical structures at T4–T9 and F15–V17 fragments. Among the 4 groups, G1 contains the fewest inter-chain contacts and inserts deepest. As shown in the purple circles of Fig. 2(b), some inter-chain contacts are formed in the C-terminal region of the hIAPP in G1, i.e., the H-bond between S29 and S29 and the hydrophobic contacts between V32 and V32. Meanwhile, we observed the insertion of the hIAPP throughout the whole sequence in G1. As shown in Fig. 2(c), not only the N-terminal amphiphilic fragment T4–T9 and the hydrophobic fragment L16V17, but also the residue I26 at the amyloid core region and Y37 at the C-terminus, form a large number of contacts with POPG tail groups. As for the secondary structures (Fig. 2(d)), G1 contains the most abundant α-helical contents and the least β-sheet contents among the 4 groups. In detail, both T4–T9 and F15–V17 fragments have substantial α-helical contents, as shown in Fig. 2(d).
G2: α-helical fragment T5–R11 insertion and β-hairpin structures on the membrane surface. As for G2, its contact patterns with membrane tail groups are similar to those of G1, as its N-terminal α-helical-rich amphiphilic segment T5–R11 also inserts into membrane tail groups. However, the hydrophobic fragment L16V17 is on the membrane surface, as shown in Fig. 2(c). In contrast to G1, G2 contains substantial β-sheet structures, and the β-sheet structures are located at N-terminal (C2N3), middle (S20N21, I26–S28), and C-terminal (V32–S34) regions, as shown in Fig. 2(e). In addition, these β-sheet fragments in G2 are on the surface of the membrane. Both H-bonds and hydrophobic contacts contribute to the inter-chain interactions in G2, as shown in the blue hexagons of Fig. 2(b). Moreover, these inter-chain contacts are adjacent to the β-sheet-rich regions, e.g., the H-bond between K1 and Y37 is close to the β-fragments C2N3 in one chain and V32–S34 in the other chain, while the hydrophobic contacts between F23 and I26 are close to the β-fragments S20N21 in one chain and I26–S28 in the other chain.
G3: hydrophobic fragment F15–V17 insertion and β-hairpin structures on the membrane surface. In G3, the hydrophobic fragment F15–V17 inserts deep with random coiled and α-helical structures. Different from G2, although it contains notable α-helical structures, the N-terminal fragment C7–T10 is located on the membrane surface without deep insertion. Instead, the N-terminal residues (K1, T4, and A5) form inter-chain interactions with the residues (S20, A25, and I26) in the middle region, e.g., there are H-bonds formed by K1/T4 with S20/A25, and also hydrophobic contacts between A5 and I26. Moreover, both chains form intra-chain β-hairpin structures, and these β-hairpins are connecting fragments L12–N14 with T30–V32, S20–N22 with N31–G33, and A25–L27 with T30–V32. Consistent with G2, these β-sheet-rich structures in G3 are on the membrane surface, and also participate in the inter-chain interactions.
G4: β-sheet-rich structures on the membrane surface with strong inter-chain hydrophobic interactions. Opposite to G1, G4 contains the most abundant inter-chain contacts and has the shallowest insertion depth among the 4 groups. In G4, there are multiple inter-chain hydrophobic contacts among several fragments, as shown in the orange squares in Fig. 2(b). In detail, the hydrophobic contacts are between L12A13 and F15–V17, L16V17 and L16V17, L12 and A26I27, F23 and F15–V17, F15 and I26, and F15 and V32, covering all the hydrophobic residues of the hIAPP, except A5 and A8 at the amphiphilic N-terminal region. In addition, there are also H-bonds between A13/S34 and L16/T36. As shown in Fig. 2(c), there are only a small number of tail contacts throughout the hIAPP sequence, indicating that hIAPP dimer structures in G4 are mainly located on the membrane surface. Moreover, G4 contains the highest β-sheet contents among the 4 groups, and its β-sheet structures are located at F15L16, N21–I26, and N31–N35 fragments, as shown in Fig. 2(g). These β-sheet-rich regions coincide with the inter-chain contacts, indicating that the β-sheet structures in the dimerized hIAPP correlate well with the formation of inter-chain interactions. Besides, the N-terminal T4–C7 segment contains notable α-helix contents with a few tail contacts.
Moreover, the β-sheet-rich structures on the membrane surface are more stable than the random coiled and α-helical structures deeply inserted into membrane interior. To compare their structural stability, we estimated the population of the 4 groups. For each group, we summed up the population of its dimer clusters, with the initially collapsed clusters excluded. As shown in Fig. 2(d–g), among the 4 groups, the deeply inserted G1 is the least populated (13.0%) while β-sheet-rich G4 on the membrane surface is the most populated (22.2%), with the G2 (16.3%) and G3 (14.5%) in between. The higher metastability of G4 is probably due to two factors: one is the strong inter-chain hydrophobic interactions in G4, and the other is the less severe membrane deformation it induces due to its shallow insertion. In comparison, the deep insertion of G1 leads to fewer inter-chain contacts and also more severe membrane deformation. The membrane deformation induced by different groups will be discussed in detail in the Section 3.3.
To elucidate the conformational rearrangements upon dimerization, we plotted the time evolution of the dimer clusters along each trajectory as shown in Fig. S8 (ESI†). In short, we observed the deepening of membrane insertion in G1 and also the elongation of β-hairpin structures on the membrane surface in G4. Besides, the initial inter-chain contacts contain both the hydrophobic and polar interactions, mostly involving the hydrophobic residues F23, V32, and the positively charged residues K1, R11, as shown in Fig. S9 (ESI†).
 | ||
| Fig. 3 Single-chain structural properties of the hIAPP dimer in the membrane. (a) 2-d free energy projection on the radius of gyration (Rg) (x-axis) and the number of tail contacts (y-axis) of each chain in the dimer ensemble, with the corresponding 1-d probability distribution shown along each axis (red solid lines), in comparison with the properties of the hIAPP monomer in the membrane (gray dotted lines). (b) 2-d free energy projection on the non-polar contacts (x-axis) and the inter-chain H-bonds (y-axis) formed by each single chain in the dimer ensemble. Similarly, the corresponding 1-d probability distribution is shown along each axis. In both (a) and (b), we labelled the average values of the top 12 single-chain clusters as the black circles in the contour plot. In (b), the groups of clusters are shaded with different colors, i.e., purple for group 1 (G1), blue for G2, green for G3, and orange for G4. The single-chain clusters forming a dimer complex with each other are connected with dashed black lines. The cluster C3 forming a dimer with itself is surrounded by dashed black circles. (c) Center conformations of the top 12 single-chain clusters arranged according to their corresponding structural groups. The population of each cluster is labelled in the bracket. The proteins are shown as cartoon representation, with α-helices in red and β-sheets in yellow. N-termini (Cα atoms of K1 residues) of hIAPP chains are in gray sphere. Sidechains forming inter-chain non-polar interactions are shown in sphere and residues forming inter-chain H-bonds are shown in stick. Sidechains of residues deeply inserted into membrane are shown in gray dot. | ||
Dimerization induces more expanded single-chain conformations. As shown in the bottom right subplot of Fig. 3(a), compared to the hIAPP monomer (gray dotted line), the single-chain Rg distribution of the dimerized hIAPP shifts to larger values (red solid line). In detail, the major peak at Rg ∼ 1.01 nm in the monomer shifts to Rg ∼ 1.07 nm in the dimerized hIAPP, while both the monomer and dimer contain the peaks at Rg ∼ 0.93 nm. In particular, the Rgs of single-chain clusters C4 and C8 are significantly larger than the rest clusters, i.e., the Rg of C4 in the β-sheet-rich G4 is ∼1.15 nm, and the Rg of C8 in the deeply inserted G1 is ∼1.30 nm. The dimension expansions of C4 and C8 are caused by different factors. In detail, the dimension expansion of C4 is due to the strong hydrophobic inter-chain interactions as shown in Fig. 3(b), while the dimension expansion of C8 is due to its deep insertion as reflected by its large number of tail contacts (Fig. 3(a)).
Dimerization induces wider distribution of the single-chain insertion depth. For the insertion depth, the dimerized hIAPP adopts a wider distribution of the number of tail contacts, as shown in the upper left subplot of Fig. 3(a). In contrast to the single high peak at ∼0.62 × 103 in the monomer ensemble, there are several peaks in the dimerized hIAPP, i.e., one peak at ∼0.05 × 103 corresponding to the hIAPP on the membrane surface (single-chain cluster C4 in G4), and two flat peaks at ∼0.54 × 103 and ∼0.90 × 103, containing the majority of the single-chain clusters in the dimer. Moreover, the single-chain cluster C8 in G1 contains an exceptionally large number of tail contacts (∼1.43 × 103), deeply inserted into the membrane interior.
Last but not least, the interactions between single-chain clusters also reflect the feature of each group of dimer clusters. In detail, the single-chain clusters in G1 (C1, C5, and C8, shaded in purple) contain the fewest inter-chain contacts among the 4 groups, while the single-chain clusters of G4 (C2, C4, and C9, shaded in orange) contain a considerable large number of hydrophobic contacts. Besides, the single-chain clusters in both G2 and G3 contain a mixture of inter-chain H-bonds and hydrophobic interactions.
From the analyses above, we conclude that the inter-chain hydrophobic interactions in dimerized hIAPP facilitate the β-sheet formation on the membrane surface, while coiled and α-helical structures of hIAPP are responsible for the membrane deep insertion. Moreover, the inter-chain interactions are mainly formed at the middle and C-terminal regions, while the insertion of α-helical and coiled structures is located at the N-terminal and middle regions. Previous NMR experiment observed that the hIAPP1–19 fragment alone causes membrane disruption to the identical extent as the full-length hIAPP, but does not form amyloid fibrils, and concluded that hIAPP membrane disruption and fibrillation are two separate processes.76 Meanwhile, the rat IAPP (rIAPP), which differs from the hIAPP by 6 residues at middle and C-terminal regions, is able to bind to the membrane, but is non-cytotoxic and does not form fibrils.77,78 As the 3 out of the 6 rIAPP residues different from hIAPP are the secondary structure breaker proline (A25P, S28P, and S29P), the non-fibrillation of rIAPP implies the importance of hIAPP middle and C-terminal regions in forming inter-chain interactions in the membrane environment. Besides, the non-toxic IAPP analogues from cattle and bears also contain at least one or two mutations to proline in the middle and the C-terminal regions and do not form amyloid fibrils either.79,80 Taken together, consistent with our simulation results, these experimental observations not only indicate the N-terminal to middle regions is responsible for hIAPP membrane binding and insertion, but also highlight the important role of middle and C-terminal regions during the hIAPP fibrillation in the membrane environment. Moreover, the correlation between cytotoxicity and amyloidogenicity among different IAPP isoforms supports the membrane disruption mechanism of the “fibril elongation” hypothesis, and suggests that hIAPP-induced membrane disruption may be a multi-step process.
The coexistence of α-helical and β-sheet structures in the membrane-bound hIAPP dimer indicates the complexity of hIAPP aggregation pathways in the membrane. Previous experiments have also indicated the structural diversity during the hIAPP oligomerization process, with some species toxic and some species non-toxic.7,8 To explore the membrane deformation induced by various hIAPP dimer structures, we further calculated the changes of membrane properties, such as the membrane thickness, area per lipid, membrane curvature, tail ordering parameter, and hydrophobic packing defects, induced by each structural group of the hIAPP dimer in the following sections.
3.3. Different degrees of membrane deformation induced by different hIAPP dimer structures
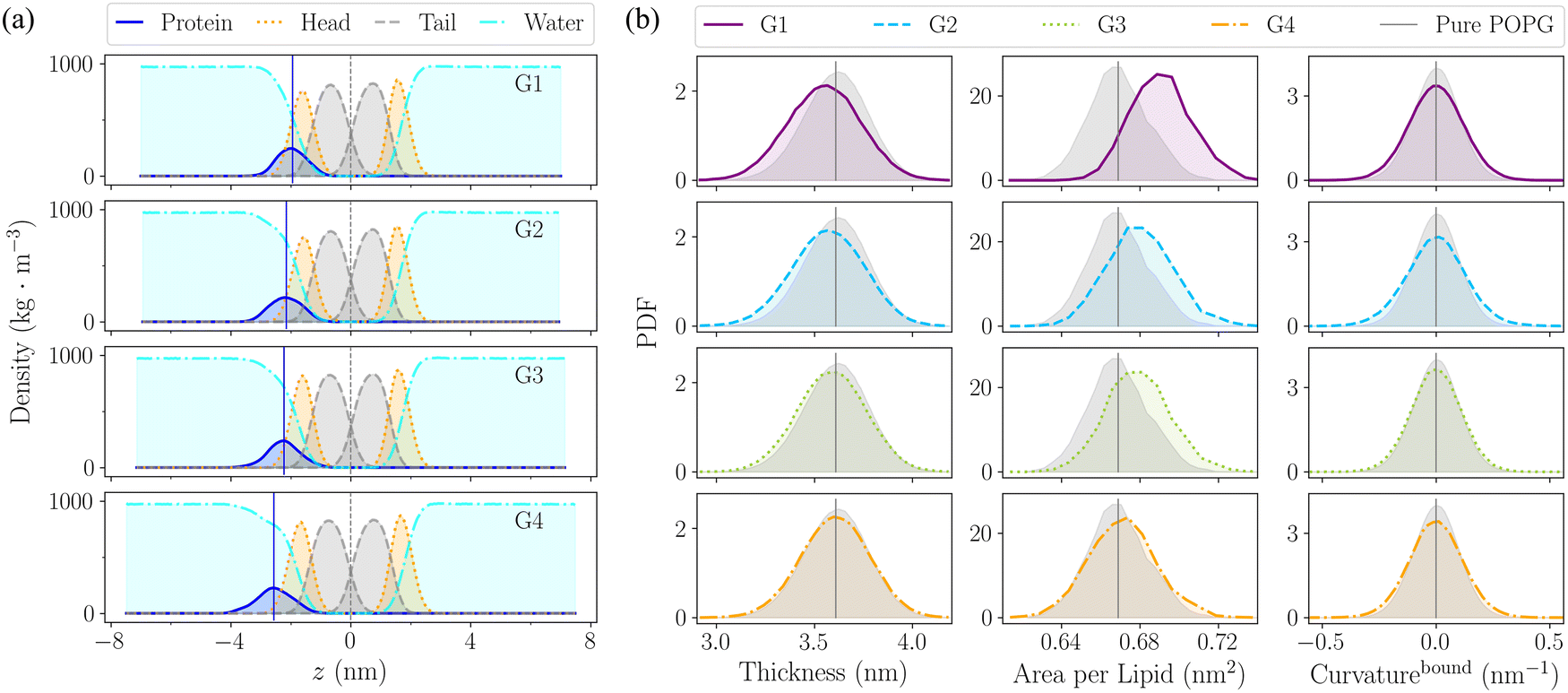 | ||
| Fig. 4 Comparison of (a) hIAPP position and (b) membrane deformation among different groups of the hIAPP dimer in the membrane environment. (a) Density distribution of each component along the z-axis of the simulation box. Subplots from top to bottom correspond to the distributions from group 1 (G1) to G4. In each subplot, the blue solid curve is the hIAPP, the orange dotted curve is POPG head groups, the dashed gray curve is POPG tail groups, and the cyan dash-dotted curve is water solvent. Vertical blue solid lines indicate the density peaks of the hIAPP, and vertical gray dashed lines indicate the COMs of POPG tail groups. (b) Distribution of the membrane thickness (left), area per lipid in the xy-plane (middle), and the mean curvature (right) of the hIAPP-bound membrane leaflet. In each subplot, the distribution in group 1 (G1) is shown in a purple solid line, G2 in a blue dashed line, G3 in a green dotted line, and G4 in an orange dash-dotted line. The vertical gray lines indicate the average values of the pure POPG membrane, with the gray shaded areas indicating its distributions. | ||
 | ||
| Fig. 5 Comparison of (a) order parameters SCDs of membrane tail groups, (b) thickness of each membrane leaflet, and (c) and (d) distribution of sizes of packing defects in the hIAPP-bound membrane leaflet, among different groups of hIAPP dimer in membrane environment. In (a) and (b), the hollow markers/bars are for the hIAPP-bound membrane leaflet, and the solid markers/bars are for the hIAPP-free leaflet. (c) is of the shallow defects and (d) is of the deep defects. In each plot, the properties of group 1 (G1) are in purple, G2 in blue, G3 in green, and G4 in orange, in comparison with the properties of pure POPG membrane in gray. In (a) and (b), the gray shaded areas indicate the corresponding standard deviations of the pure POPG membrane. In (c) and (d), the inset plots are the distribution of defect sizes in two ranges, one for the normal size (within 99% of the pure POPG), and the other for the large size (exceeding 99% of the pure POPG). | ||
The local membrane disturbance in G2 is also reflected by its large-size shallow defects in the hIAPP-bound leaflet. As shown in Fig. 5(c), in the hIAPP-bound leaflet, G2 contains significantly more large-size shallow defects (>0.32 nm2) than the other 3 groups. Meanwhile, G1 contains the most large-size deep defects (>0.29 nm2) among the 4 groups (Fig. 5(d)), which is consistent with its deepest insertion. Besides, as for the size distribution of overall defects (shallow defects plus deep defects) shown in Fig. S11(a) (ESI†), although G1 contains the most large-size overall defects (>0.45 nm2), there is a substantial increase of overall defects larger than 1 nm2 in G2. This increase is mainly due to the large-size shallow defects in G2. Meanwhile, as for the hIAPP-free leaflet, we observed a slight increase of large-size overall defects, and the degrees of size enlargement decrease gradually from G1 to G4, which is consistent with the degree of membrane stretching in each structural group, as shown in Fig. S11(b) (ESI†). In short, both the tail disordering and the size distribution of hydrophobic packing defects indicate that the hIAPP deep insertion in G1 causes more global membrane disruption in the hIAPP-free leaflet, while the shallow insertion in G2 causes more local membrane disruption in the hIAPP-bound leaflet.
From the analyses above, we observe that different hIAPP dimer structures cause different degrees of membrane disruption, and we believe that the structural diversity of the membrane-bound hIAPP dimer may also imply the diversity of hIAPP toxic species. Previous experiments have indicated the structural diversity in the oligomerization stage, although the detailed structures of membrane-bound hIAPP oligomers have still been elusive.7,8 With computational modelling, both helical-bundle and β-barrel structures of oligomeric hIAPP ion channels have been proposed.46,52,53 Here, based on the structural and corresponding membrane disruption properties of the hIAPP dimer ensemble, we speculate that the random coiled and α-helical structures in G1 may be the precursor of helical-bundle ion channels, while the mixed α-and-β G2 may be the precursor for the β-barrel ion channels.
To sum up, the degrees of membrane disruption decrease gradually from the random coiled and α-helical structures in G1 to the β-sheet-rich structures in G4, well correlated with the hIAPP insertion depth in each structural group, as shown in Fig. 6. In particular, the deep insertion of G1 causes the global membrane disruption, inducing the most significant membrane thinning and stretching, and also creating the large-size deep defects. In comparison, the shallow insertion of G2 with a mixture of α-helical and β-sheet structures causes the local membrane disruption and creates large-size shallow defects, resulting in the most severe membrane bending in the hIAPP-bound leaflet. Experiments have indicated that the hIAPP-induced membrane disruption process is bi-phasic, including the 1st phase leakage caused by the prefibrillar oligomer, and the 2nd phase leakage caused by fibril elongation,13,14 and thus both the oligomers in the early nucleation stage and fibrils in the later elongation stage contribute to the hIAPP-induced membrane disruption. Besides, even during the initial oligomer poration stage, previous experiments have observed that the growth of oligomers in the membrane is in a step-wise manner, and there exist two types of oligomers, only one of which is toxic.7,8 Though the detailed structures have still been elusive experimentally, both helical bundles and β barrels of hIAPP ion-channels have been proposed in the computational modelling.46,52,53 In line with these experimental and computational results, we observed a diverse structural ensemble of the membrane-bound hIAPP dimer in our simulations, and different dimer structures induce the membrane disruption to different extents. Based on our simulations, we speculate that the random coiled and α-helical structures of G1 initialize the membrane disruption and are responsible for the membrane poration, while the β-sheet-rich structures in G4 may be the seed for hIAPP fibrillation in the membrane environment and more involved in the later stage of hIAPP-induced membrane disruption.
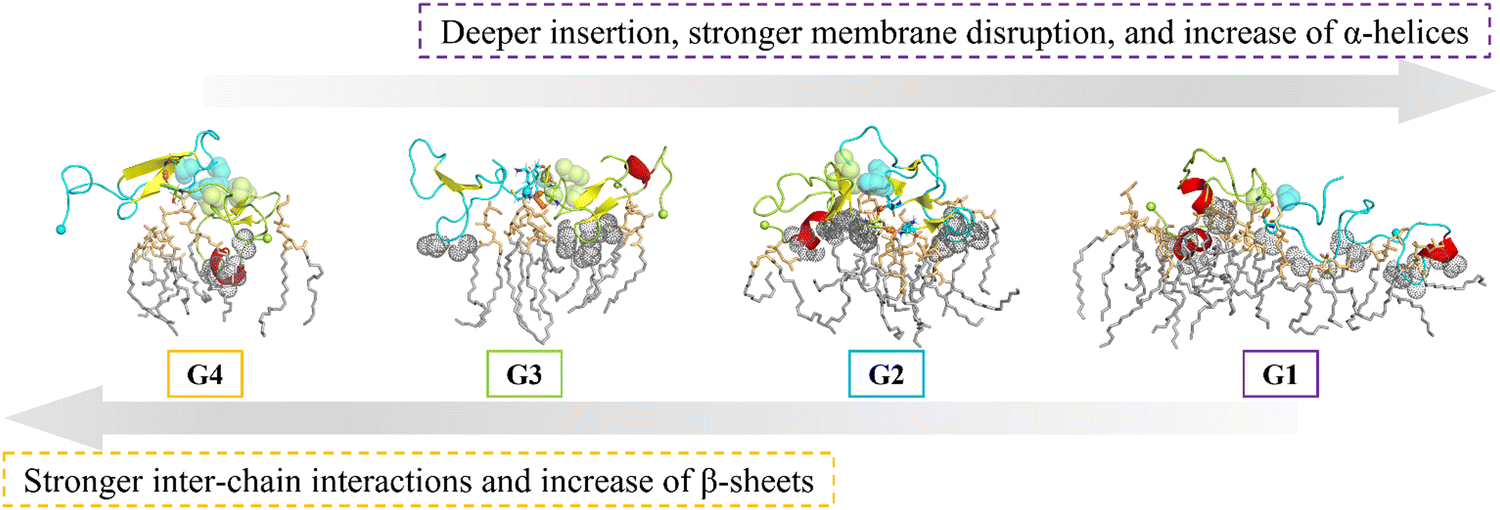 | ||
| Fig. 6 Representative centre conformation of each group with the nearby POPG lipids, whose contact probabilities with the hIAPP dimer are higher than 95%. hIAPP dimers are shown in cartoon representation, with the α-helix in red and β-sheets in yellow. In each conformation, one chain is in cyan and the other chain is in light green. N-termini (the Cα atoms of K1) are shown in a solid sphere. Sidechains forming inter-chain hydrophobic contacts are shown in a sphere, and residues forming inter-chain H-bonds are shown in a stick. Sidechains of deep inserting residues are shown in gray dots. POPG lipids nearby are shown in a stick, with head groups in orange and tail groups in gray. | ||
4. Conclusions
In this study, we performed extensive all-atom molecular dynamics simulations to investigate the hIAPP dimerization process in the anionic POPG membrane environment. From our simulations, we observed a diverse conformational ensemble of the hIAPP dimer in the POPG membrane environment, and different structural groups induce different degrees of membrane deformation. From G4 to G1, the hIAPP dimer shifts from the β-sheet-rich structures on the membrane surface to the random coiled and α-helical structures inserted into the membrane interior. Strong inter-chain hydrophobic interactions facilitate the β-sheet formation in G4, while only a few inter-chain interactions at the C-terminal region are involved in the random coiled and α-helical structures deeply inserted into the membrane in G1. The extent of hIAPP-induced membrane disruption also increases from G4 to G1 gradually. In detail, the deep insertion of random coiled and α-helical structures in G1 induces the strongest membrane deformation among the 4 groups, including membrane thinning, membrane stretching, and global tail disordering. Meanwhile, the mixture of α-helical and β-sheet structures in G2 mainly causes membrane bending and local tail disordering. In contrast, the β-sheet-rich structures on the membrane surface in G4 induce negligible membrane deformation. We believe that the deeply inserted random coiled and α-helical dimer structures identified here contribute to the initial stage of membrane disruption, while the β-sheet-rich dimer structures on the membrane surface may be involved in the later stage during the hIAPP fibril elongation. This study reveals the complexity of the membrane-bound hIAPP dimer structural ensemble, providing a detailed structural explanation on the seemingly conflicting mechanisms of hIAPP-induced membrane disruption.Author contributions
Q. Q. conceived the project, performed the simulations, analyzed the simulation data, and drafted the manuscript. The manuscript was written through contributions of all authors, and all authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (no. 12074079, 11804054), and the Natural Science Foundation of Shanghai Municipality (no. 22ZR1406800). The simulations were performed on local GPU cluster of our group.References
- J. W. M. Höppener, B. Ahrén and C. J. M. Lips, N. Engl. J. Med., 2000, 343, 411–419 CrossRef PubMed.
- S. M. M. Butterfield and H. A. A. Lashuel, Angew. Chem., Int. Ed., 2010, 49, 5628–5654 CrossRef CAS PubMed.
- S. H. Back and R. J. Kaufman, Annu. Rev. Biochem., 2012, 81, 767 CrossRef CAS PubMed.
- L. Haataja, T. Gurlo, C. J. Huang and P. C. Butler, Endocr. Rev., 2008, 29, 303–316 CrossRef CAS PubMed.
- T. A. Mirzabekov, M. Lin and B. L. Kagan, J. Biol. Chem., 1996, 271, 1988–1992 CrossRef CAS PubMed.
- A. Quist, I. Doudevski, H. Lin, R. Azimova, D. Ng, B. Frangione, B. Kagan, J. Ghiso and R. Lal, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10427–10432 CrossRef CAS PubMed.
- N. B. Last, E. Rhoades and A. D. Miranker, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9460–9465 CrossRef CAS PubMed.
- M. Birol, S. Kumar, E. Rhoades and A. D. Miranker, Nat. Commun., 2018, 9, 1312 CrossRef PubMed.
- Y. A. Domanov and P. K. J. Kinnunen, J. Mol. Biol., 2008, 376, 42–54 CrossRef CAS PubMed.
- C. C. Lee, Y. Sun and H. W. Huang, Biophys. J., 2012, 102, 1059–1068 CrossRef CAS PubMed.
- A. Martel, L. Antony, Y. Gerelli, L. Porcar, A. Fluitt, K. Q. Hoffmann, I. Kiesel, M. Vivaudou, G. Fragneto and J. J. de Pablo, J. Am. Chem. Soc., 2017, 139, 137–148 CrossRef CAS PubMed.
- M. F. M. Engel, L. Khemtemourian, C. C. Kleijer, H. J. D. Meeldijk, J. Jacobs, A. J. Verkleij, B. de Kruijff, J. A. Killian and J. W. M. Hoppener, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6033–6038 CrossRef CAS PubMed.
- B. O. W. Elenbaas, S. M. Kremsreiter, L. Khemtemourian, J. A. Killian and T. Sinnige, BBA Adv., 2023, 3, 100083 CrossRef CAS PubMed.
- J. R. Brender, E. L. Lee, K. Hartman, P. T. Wong, A. Ramamoorthy, D. G. Steel and A. Gafni, Biophys. J., 2011, 100, 685–692 CrossRef CAS PubMed.
- P. E. S. Smith, J. R. Brender and A. Ramamoorthy, J. Am. Chem. Soc., 2009, 131, 4470–4478 CrossRef CAS PubMed.
- M. F. M. Sciacca, J. R. Brender, D. K. Lee and A. Ramamoorthy, Biochemistry, 2012, 51, 7676–7684 CrossRef CAS PubMed.
- J. R. Brender, S. Salamekh and A. Ramamoorthy, Acc. Chem. Res., 2012, 45, 454–462 CrossRef CAS PubMed.
- N. Bag, A. Ali, V. S. Chauhan, T. Wohland and A. Mishra, Chem. Commun., 2013, 49, 9155–9157 RSC.
- M. F. M. Sciacca, F. Lolicato, G. Di Mauro, D. Milardi, L. D’Urso, C. Satriano, A. Ramamoorthy and C. La Rosa, Biophys. J., 2016, 111, 140–151 CrossRef CAS PubMed.
- M. F. M. Sciacca, C. Tempra, F. Scollo, D. Milardi and C. La Rosa, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1625–1638 CrossRef CAS PubMed.
- D. Milardi, E. Gazit, S. E. Radford, Y. Xu, R. U. Gallardo, A. Caflisch, G. T. Westermark, P. Westermark, C. La Rosa and A. Ramamoorthy, Chem. Rev., 2021, 121, 1845–1893 CrossRef CAS PubMed.
- A. Relini, O. Cavalleri, R. Rolandi and A. Gliozzi, Chem. Phys. Lipids, 2009, 158, 1–9 CrossRef CAS PubMed.
- J. D. Knight and A. D. Miranker, J. Mol. Biol., 2004, 341, 1175–1187 CrossRef CAS PubMed.
- J. D. Knight, J. A. Hebda and A. D. Miranker, Biochemistry, 2006, 45, 9496–9508 CrossRef CAS PubMed.
- K. Sasahara, D. Hall and D. Hamada, Biochemistry, 2010, 49, 3040–3048 CrossRef CAS PubMed.
- L. Caillon, O. Lequin and L. Khemtémourian, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 2091–2098 CrossRef CAS PubMed.
- X. Zhang, J. R. S. Clair, E. London and D. P. Raleigh, Biochemistry, 2017, 56, 376–390 CrossRef CAS PubMed.
- J. A. Williamson, J. P. Loria and A. D. Miranker, J. Mol. Biol., 2009, 393, 383–396 CrossRef CAS PubMed.
- N. B. Last and A. D. Miranker, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6382–6387 CrossRef CAS PubMed.
- A. Rawat, B. K. Maity, B. Chandra and S. Maiti, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1734–1740 CrossRef CAS PubMed.
- L. Fu, G. Ma and E. C. Y. Yan, J. Am. Chem. Soc., 2010, 132, 5405–5412 CrossRef CAS PubMed.
- J. Tan, J. Zhang, Y. Luo and S. Ye, J. Am. Chem. Soc., 2019, 141, 1941–1948 CrossRef CAS PubMed.
- C. A. De Carufel, N. Quittot, P. T. Nguyen and S. Bourgault, Angew. Chem., Int. Ed., 2015, 54, 14383–14387 CrossRef CAS PubMed.
- Y. Xing, E. H. Pilkington, M. Wang, C. J. Nowell, A. Kakinen, Y. Sun, B. Wang, T. P. Davis, F. Ding and P. C. Ke, Phys. Chem. Chem. Phys., 2017, 19, 30627–30635 RSC.
- D. C. Rodriguez Camargo, K. J. Korshavn, A. Jussupow, K. Raltchev, D. Goricanec, M. Fleisch, R. Sarkar, K. Xue, M. Aichler, G. Mettenleiter, A. K. Walch, C. Camilloni, F. Hagn, B. Reif and A. Ramamoorthy, eLife, 2017, 6, e31226 CrossRef PubMed.
- S. S. Dicke, M. Maj, C. R. Fields and M. T. Zanni, RSC Chem. Biol., 2022, 3, 931–940 RSC.
- X. Dong, Q. Qiao, Z. Qian and G. Wei, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1826–1839 CrossRef CAS PubMed.
- P. H. Nguyen, A. Ramamoorthy, B. R. Sahoo, J. Zheng, P. Faller, J. E. Straub, L. Dominguez, J. E. Shea, N. V. Dokholyan, A. de Simone, B. Ma, R. Nussinov, S. Najafi, S. T. Ngo, A. Loquet, M. Chiricotto, P. Ganguly, J. McCarty, M. S. Li, C. Hall, Y. Wang, Y. Miller, S. Melchionna, B. Habenstein, S. Timr, J. Chen, B. Hnath, B. Strodel, R. Kayed, S. Lesné, G. Wei, F. Sterpone, A. J. Doig and P. Derreumaux, Chem. Rev., 2021, 121, 2545–2647 CrossRef CAS PubMed.
- M. Apostolidou, S. A. Jayasinghe and R. Langen, J. Biol. Chem., 2008, 283, 17205–17210 CrossRef CAS PubMed.
- G. L. Dignon, G. H. Zerze and J. Mittal, J. Phys. Chem. B, 2017, 121, 8661–8668 CrossRef CAS PubMed.
- Q. Qiao, G. Wei, D. Yao and Z. Song, Phys. Chem. Chem. Phys., 2019, 21, 20239–20251 RSC.
- L. Khemtemourian, H. Fatafta, B. Davion, S. Lecomte, S. Castano and B. Strodel, Front. Mol. Biosci., 2022, 9, 849979 CrossRef CAS PubMed.
- M. Christensen, K. K. Skeby and B. Schiøtt, Biochemistry, 2017, 56, 4884–4894 CrossRef CAS PubMed.
- S. M. Patil, S. Xu, S. R. Sheftic and A. T. Alexandrescu, J. Biol. Chem., 2009, 284, 11982 CrossRef CAS PubMed.
- Y. Zhang, Y. Luo, Y. Deng, Y. Mu and G. Wei, PLoS One, 2012, 7, e38191 CrossRef CAS PubMed.
- M. Pannuzzo, A. Raudino, D. Milardi, C. La Rosa and M. Karttunen, Sci. Rep., 2013, 3, 2781 CrossRef PubMed.
- X. Li, M. Wan, L. Gao and W. Fang, Sci. Rep., 2016, 6, 21614 CrossRef CAS PubMed.
- C. Poojari, D. Xiao, V. S. Batista and B. Strodel, Biophys. J., 2013, 105, 2323–2332 CrossRef CAS PubMed.
- N. Liu, M. Duan and M. Yang, Sci. Rep., 2017, 7, 7915 CrossRef PubMed.
- Z. Qian, Y. Zou, Q. Zhang, P. Chen, B. Ma, G. Wei and R. Nussinov, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1818–1825 CrossRef CAS PubMed.
- Q. Tan, H. Liu, M. Duan and S. Huo, Biochim. Biophys. Acta, Biomembr., 2021, 1863, 183691 CrossRef CAS PubMed.
- J. Zhao, Y. Luo, H. Jang, X. Yu, G. Wei, R. Nussinov and J. Zheng, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 3121–3130 CrossRef CAS PubMed.
- A. Sepehri, B. Nepal and T. Lazaridis, J. Chem. Inf. Model., 2021, 61, 4645–4655 CrossRef CAS PubMed.
- N. F. Dupuis, C. Wu, J.-E. Shea and M. T. Bowers, J. Am. Chem. Soc., 2011, 133, 7240–7243 CrossRef CAS PubMed.
- Y. Bram, A. Lampel, R. Shaltiel-Karyo, A. Ezer, R. Scherzer-Attali, D. Segal and E. Gazit, Angew. Chem., Int. Ed., 2015, 54, 2062–2067 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins: Struct., Funct., Bioinf., 2010, 78, 1950–1958 CrossRef CAS PubMed.
- J. P. M. Jämbeck and A. P. Lyubartsev, J. Phys. Chem. B, 2012, 116, 3164–3179 CrossRef PubMed.
- J. P. M. Jämbeck and A. P. Lyubartsev, J. Chem. Theory Comput., 2013, 9, 774–784 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- A. Bellet, A. Habrard and M. Sebban, Metric Learning, Springer, Cham, 2015 Search PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- S. Nose, J. Chem. Phys., 1984, 81, 511–519 CrossRef CAS.
- M. Parrinello, A. Rahman and R. A. Parrinello, Phys. Rev. Lett., 1980, 45, 1196–1199 CrossRef CAS.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- R. T. McGibbon, K. A. Beauchamp, M. P. Harrigan, C. Klein, J. M. Swails, C. X. Hernández, C. R. Schwantes, L.-P. Wang, T. J. Lane and V. S. Pande, Biophys. J., 2015, 109, 1528–1532 CrossRef CAS PubMed.
- E. N. Baker and R. E. Hubbard, Prog. Biophys. Mol. Biol., 1984, 44, 97–179 CrossRef CAS PubMed.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. Van Gunsteren and A. E. Mark, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
- R. González-Alemán, D. Hernández-Castillo, A. Rodríguez-Serradet, J. Caballero, E. W. Hernández-Rodríguez and L. Montero-Cabrera, J. Chem. Inf. Model., 2020, 60, 444–448 CrossRef PubMed.
- V. Gapsys, B. L. De Groot and R. Briones, J. Comput. Aided. Mol. Des., 2013, 27, 845–858 CrossRef CAS PubMed.
- J. P. Douliez, A. Ferrarini and E. J. Dufourc, J. Chem. Phys., 1998, 109, 2513–2518 CrossRef CAS.
- R. Gautier, A. Bacle, M. L. Tiberti, P. F. Fuchs, S. Vanni and B. Antonny, Biophys. J., 2018, 1, 436–444 CrossRef PubMed.
- M. F. M. Engel, H. A. Yigittop, R. C. Elgersma, D. T. S. Rijkers, R. M. J. Liskamp, B. De Kruijff, J. W. M. Höppener and J. Antoinette Killian, J. Mol. Biol., 2006, 356, 783–789 CrossRef CAS PubMed.
- J. R. Brender, E. L. Lee, M. A. Cavitt, A. Gafni, D. G. Steel and A. Ramamoorthy, J. Am. Chem. Soc., 2008, 130, 6424–6429 CrossRef CAS PubMed.
- R. Soong, J. R. Brender, P. M. Macdonald and A. Ramamoorthy, J. Am. Chem. Soc., 2009, 131, 7079–7085 CrossRef CAS PubMed.
- H. M. Sanders, F. Chalyavi, C. R. Fields, M. M. Kostelic, M. H. Li, D. P. Raleigh, M. T. Zanni and M. T. Marty, J. Am. Soc. Mass Spectrom., 2023, 34, 986–990 CrossRef CAS PubMed.
- R. Akter, A. Abedini, Z. Ridgway, X. Zhang, J. Kleinberg, A. M. Schmidt and D. P. Raleigh, Isr. J. Chem., 2017, 57, 750–761 CrossRef CAS PubMed.
- R. Akter, R. L. Bower, A. Abedini, A. M. Schmidt, D. L. Hay and D. P. Raleigh, ACS Chem. Biol., 2018, 13, 2747–2757 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05887e |
| This journal is © the Owner Societies 2024 |