DOI:
10.1039/D3CP05845J
(Communication)
Phys. Chem. Chem. Phys., 2024,
26, 8043-8050
Matrix effects on the magnetic properties of a molecular spin triangle embedded in a polymeric film†
Received
30th November 2023
, Accepted 14th February 2024
First published on 14th February 2024
Abstract
Molecular triangles with competing Heisenberg interactions and significant Dzyaloshinskii–Moriya interactions (DMI) exhibit high environmental sensitivity, making them potential candidates for active elements for quantum sensing. Additionally, these triangles exhibit magnetoelectric coupling, allowing their properties to be controlled using electric fields. However, the manipulation and deposition of such complexes pose significant challenges. This work explores a solution by embedding iron-based molecular triangles in a polymer matrix, a strategy that offers various deposition methods. We investigate how the host matrix alters the magnetic properties of the molecular triangle, with specific focus on the magnetic anisotropy, aiming to advance its practical applications as quantum sensors.
Introduction
Molecular spin triangles are potential building blocks for quantum technologies.1 In the presence of antiferromagnetic interactions, their symmetry leads to preferential antiparallel spin orientations that cannot be simultaneously satisfied.2 This imbalance, combined with DMI, leads to magnetic ground states with unique and intriguing quantum properties such as spin chirality and magnetoelectric coupling.3–6 Spin-chiral states are interesting because they were shown to be protected against decoherence.3,4,7 Magnetoelectricity allows controlling the magnetic properties of quantum systems by electric fields, which can be confined more efficiently than magnetic fields. These properties motivated the investigation of several molecular spin triangles based on metals such as dysprosium,8 chromium,9 cobalt,10 copper,11 and iron.12–14 The iron-based molecular spin triangle of formula [Fe3O(O2CPh)6(py)3]ClO4·py (Fe3) was investigated by one of us using continuous wave (CW) electron paramagnetic resonance (EPR) spectroscopy under static electric fields12 and pulsed EPR spectroscopy under pulsed electric fields,15 revealing magnetoelectric effects in both solid and frozen solution states. Empirical models in this latter study, and comparisons to its CrIII analogue, highlighted the role of DMI in the development of magnetoelectricity. Previous theoretical studies3,7 also revealed the importance of DMI in the development of spin chirality.
The simplest model that successfully describes most magnetic properties of spin triangles can be expressed by the spin Hamiltonian
| | 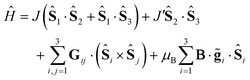 | (1) |
where
Ŝi is the spin operator,
Jij the exchange interaction and
Gij the DMI pseudovector. The first two terms in the Hamiltonian account for the asymmetry of the Heisenberg exchange interactions (expressed as Δ
J =
J − J′). The need to account for such an asymmetry became evident from very early on, from heat capacity
16 and magnetic susceptibility
17 studies on Cr
III triangles, which could only be interpreted by reducing the symmetry of the Heisenberg exchange scheme from equilateral. An isosceles scheme is normally preferred over a scalene one, since it typically accounts for the data just as successfully, without overparametrizing the problem. The third term denotes the DMI term, which is usually assumed to be
G12 =
G23 =
G31 to limit the number of parameters. Due to symmetry and by implementing the Moriya rules, we may further simplify the model by assuming (
Gx,
Gy) ∼ 0, and |
G| =
Gz, where
z is the axis perpendicular to the plane of the triangle. As was shown some time ago,
18 the magnetic anisotropy, reflected by
g⊥, is sensitive to the ratio of Δ
J and
Gz. For instance, in the simple case of three identical axial spins (principal
g-tensor elements
g0‖ and
g0⊥), which are coupled antiferromagnetically and are aligned with their
g0‖ axis parallel to the molecular
z-axis, it was shown that the ground state is a doublet, whose effective
g-value along the main axis of the molecule is
g‖eff =
g0‖.
18 In turn, the effective
g-value perpendicular to the main axis (
i.e. along the triangle plane) is given for the ground doublet by:
| | 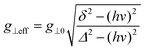 | (2) |
where
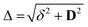
, with
δ and
D depending on the individual spin values: for
Si = 1/2,
δ = Δ
J,
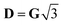
for
Si = 3/2,
δ = 2Δ
J,
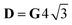
; for
Si = 5/2, as in this case,
δ = 3Δ
J,
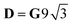
.
18
Since J, J′ and Gz depend on the molecular geometry due to their superexchange nature, small structural effects imposed by the steric constraints of the lattice can strongly influence the magnetic anisotropies of spin triangles. This property makes them an attractive framework for the construction of molecular quantum devices, to sense small structural changes in the environment.19 Magnetoelectricity further extends the versatility of spin triangles enabling magnetic (electric) sensing detected via electric (magnetic) read-out. However, before molecular spin triangles can be practically implemented in quantum sensing devices, we must first overcome challenges associated with material handling and film preparation. One possible approach involves integrating the molecules into an organic host matrix, which could facilitate handling—for instance, when preparing thin films—as well as boosting the molecule's quantum sensing properties, for example by fostering selective absorption of target analytes. A polymeric material could serve as the host matrix, wherein the quantum active element, here the molecular spin triangle, would be dispersed.20 Polymers provide a high degree of variability and can introduce additional functionalities such as electrical conductivity, photo-activation or nano-porosity. However, maintaining the magnetic properties of the molecular spin triangle is paramount. To ensure this, several critical questions must be addressed: Is the molecule stable in the polymer matrix? Does the host matrix impact the molecular structure? How are the magnetic properties affected by the host matrix? The aim of this work is to investigate the environmental effects of the molecular spin triangle Fe3 (Fig. 1a) when embedded in a polymeric matrix and subsequently deposited on the surface. We have used poly(methyl methacrylate) (PMMA, Fig. 1b) because this polymer provides a stable and inert environment, it is optically transparent and it is a suitable choice for creating uniform and reproducible films using a wide range of fabrication processes; here we limit the study to spin-coated and drop-casted samples. The prepared films are investigated by a combination of Fourier Transformed Infrared (FTIR) and multi-frequency EPR spectroscopy.
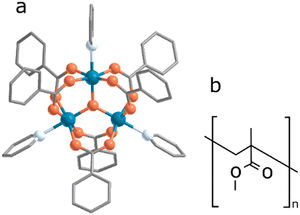 |
| | Fig. 1 (a) Crystallographic structure of Fe3; the colours correspond to: orange = oxygen, light blue = nitrogen, dark blue = iron, grey = carbon atoms. Hydrogen atoms and counter-ion molecule are omitted for clarity (the structure was first reported by Sowrey et al. in ref. 23 and a structure-based synchrotron data at 90 K was reported by Georgopoulou et al. in ref. 25). (b) Chemical structure of a single PMMA unit. | |
Experimental methods
Sample preparation
The Fe3 complex was synthesized as previously described.12 For the film preparation, two solutions were separately prepared, one for the Fe3 sample and another for the poly(methyl methacrylate) (PMMA) one (Sigma Aldrich, av. Mw 350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 by GPC, density 1.17 g mL−1 at 25 °C), in both cases using dichloromethane (Sigma Aldrich, Chromasolv Plus) as a solvent. The two solutions were then mixed together, resulting in a clear solution of 84 g L−1 of PMMA and different weight concentrations of Fe3:10% (6.3 mM), 5% (3.1 mM) and 1% (0.63 mM), expressed as a function of the PMMA content. The final solution was then drop-cast or spin-coated on previously cleaned Mylar substrates. The obtained films were dried in vacuum for several hours. The films are of a mild yellow colour with no signature of impurities, powders or particles dispersed inside. The Mylar substrates were cleaned by sonication in two consecutive baths of acetone and isopropanol, for 5 minutes in each. The spin-coater used was a Laurell spin-processor (model code WS-650Mz-23NPPB). The parameters used for the spin-coating were 3000 rpm for 1 minute and 300 rpm/second of acceleration. The film thickness was estimated by AFM, measuring the scratch made on a film that was obtained by spin-coating a 1% doped PMMA solution with a concentration of 84 g L−1 on a quartz substrate with similar lateral dimensions to the Mylar substrate used for Fe3. The quartz substrate was chosen because it is not damaged during the scratching process, unlike Mylar, which is a softer material.
000 by GPC, density 1.17 g mL−1 at 25 °C), in both cases using dichloromethane (Sigma Aldrich, Chromasolv Plus) as a solvent. The two solutions were then mixed together, resulting in a clear solution of 84 g L−1 of PMMA and different weight concentrations of Fe3:10% (6.3 mM), 5% (3.1 mM) and 1% (0.63 mM), expressed as a function of the PMMA content. The final solution was then drop-cast or spin-coated on previously cleaned Mylar substrates. The obtained films were dried in vacuum for several hours. The films are of a mild yellow colour with no signature of impurities, powders or particles dispersed inside. The Mylar substrates were cleaned by sonication in two consecutive baths of acetone and isopropanol, for 5 minutes in each. The spin-coater used was a Laurell spin-processor (model code WS-650Mz-23NPPB). The parameters used for the spin-coating were 3000 rpm for 1 minute and 300 rpm/second of acceleration. The film thickness was estimated by AFM, measuring the scratch made on a film that was obtained by spin-coating a 1% doped PMMA solution with a concentration of 84 g L−1 on a quartz substrate with similar lateral dimensions to the Mylar substrate used for Fe3. The quartz substrate was chosen because it is not damaged during the scratching process, unlike Mylar, which is a softer material.
AFM
Atomic force microscopy (AFM) experiments were performed on a Veeco Dimension 3100 apparatus mounted on an anti-vibration table. Images were taken using a PPP-NCSTR tip from Nanosensors in tapping mode. The images were processed using Gwyddion, and the root mean square roughness was calculated over the entire image.
FTIR
The experiments were carried out on a Bruker VERTEX 70v FT-IR spectrometer equipped with an Attenuated Total Reflection (ATR) unit. The measurements were performed by placing the Fe3 powder or the films on a Mylar substrate directly in contact with the diamond head. The spectra were recorded between 4500 and 400 cm−1. A background measurement was first taken and then subtracted from the subsequent spectra.
X-Band EPR
Continuous wave (CW) X-Band EPR measurements were carried out with a Bruker EMX spectrometer using an Oxford Instruments 4102ST continuous flow helium cryostat resonator (ν = 9.62 GHz). The sample was freshly prepared on Mylar substrate, which was then cut into multiple stripes of ca. 2.5 mm × 10 mm and inserted into the EPR tube. All experiments were performed at low temperatures by cooling with liquid helium.
HFEPR
CW EPR experiments at high frequencies (121.5 and 115 GHz) were conducted in a home-built spectrometer. A detailed description of the instrument is reported in ref. 21 which has been upgraded with an Oxford TeslatronPT 14T cryogen-free magnet. The signal was demodulated using a lock-in amplifier with a modulation amplitude of 20 G and a modulation frequency of 3 kHz for the Fe3 powder sample and 40 G at 1.5 kHz for the Fe3(10%) film. The Fe3 powder sample was measured as a pellet, prepared by wrapping the powder with Teflon tape and pressing it at a moderate pressure (<0.5 Tons). For the drop-cast film sample, multiple drops were deposited on a glass and air-dried. Around 20 of such drops were then collected, and pressed at a moderate pressure (<0.5 Tons) to obtain a pellet. The temperature was kept constant at 3 K for the Fe3 powder pellet and 7 K for the Fe3(10%) pellet. Unfortunately, in the latter case, it was not possible to cool down further due to technical reasons. Fits to the EPR spectra were carried out in Matlab with Easyspin v. 6.0.0–dev51 using custom routines.22
Results
Atomic force microscopy
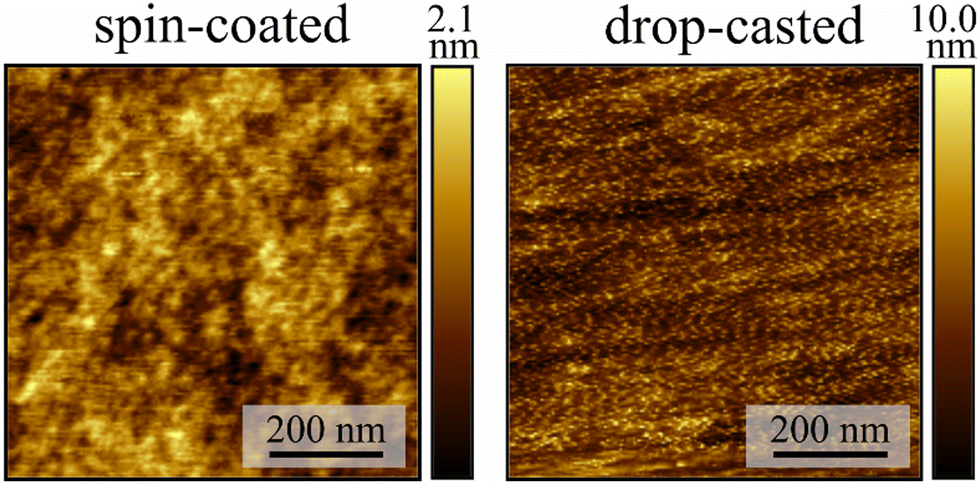
Solutions containing 84 g L−1 of PMMA and Fe3 in specific mass ratios relative to PMMA (10% w/w, Fe3(10%), 5% w/w, Fe3(5%), and 1% w/w, Fe3(1%)) were prepared using dichloromethane (dcm) as solvent. Thin films were prepared either by drop-casting (DC) or spin-coating (SC) on clean Mylar substrates (Fig. S1, ESI†). The resulting films were dried for two hours in vacuum to remove the excess solvent. These two deposition methods result in different film morphologies, as shown by atomic force microscopy (AFM) experiments on Fe3(10%) (Fig. 2). The films prepared by drop-casting show a very rough surface with a root mean square roughness value of 1.4(2) nm. On the other hand, the spin-coated films have a very smooth surface, with a root mean square roughness value of 0.31(1) nm. The thicknesses of the two types of films are quite different, with the thickness of the drop-casted film being too high (>3 μm) to be estimated by AFM, whereas the spin-coated film was estimated to be 650(50) nm thick. In both cases, the AFM images show no evidence of agglomerates or clusters, with the materials (Fe3 and PMMA) appearing to be homogeneously mixed.
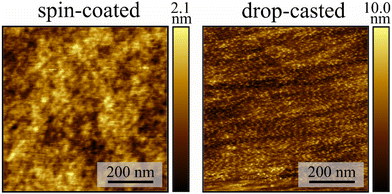 |
| | Fig. 2 AFM image of the spin-coated and drop-casted films of Fe3(10%). The colour bar represents the height of the film. | |
Infrared spectroscopy
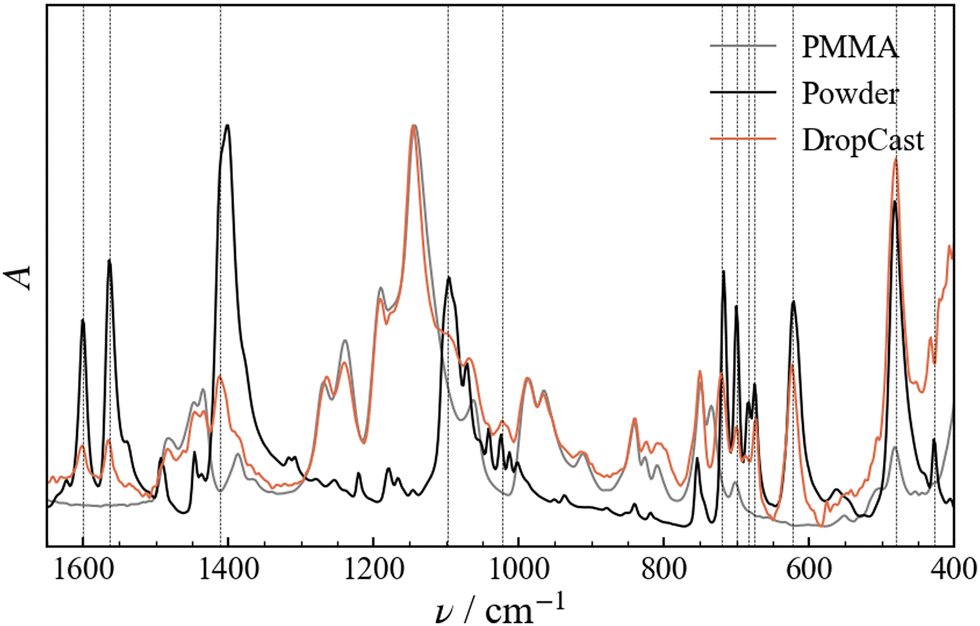
Structural changes of the molecular geometry of Fe3 can be inferred by looking at the vibrational spectrum. For this reason, FTIR experiments were carried out on Fe3 powder, SC and DC films of Fe3(10%) and a pristine PMMA film for reference. The investigated range goes from 4500 to 400 cm−1 (Fig. S2, ESI†) and the diagnostic region falls between 1650 and 400 cm−1 (Fig. 3). The Fe3 powder spectrum is fully consistent with that previously reported in literature for the same compound.23 The spectra of the SC and DC samples are for the most part superimposable (Fig. 4a). The comparison of the three samples reveals that the Fe3(10%) spectrum is the sum of the infrared PMMA and Fe3 spectra. The peaks relative to the vibrations of the Fe3 molecule are unequivocally visible in the DC spectrum wherever there are no overlaps with PMMA vibrations, for example, at 1602 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1562 (CC stretching), and 1400 cm−1 (C–H bending), which are relative to the benzoate moiety, or at 622 cm−1, which is attributed to the asymmetric stretching of the Fe3O moiety.23 From the comparison it is evident that there is no significant frequency shift between the Fe3 powder and Fe3(10%) vibrations. This observation indicates that the Fe3 molecules are successfully embedded in the polymeric matrix and no obvious modification of their structure occurs.
O stretching), 1562 (CC stretching), and 1400 cm−1 (C–H bending), which are relative to the benzoate moiety, or at 622 cm−1, which is attributed to the asymmetric stretching of the Fe3O moiety.23 From the comparison it is evident that there is no significant frequency shift between the Fe3 powder and Fe3(10%) vibrations. This observation indicates that the Fe3 molecules are successfully embedded in the polymeric matrix and no obvious modification of their structure occurs.
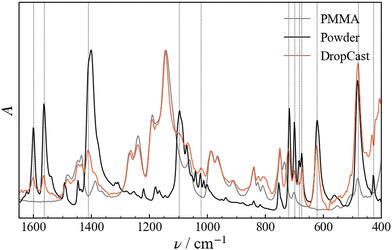 |
| | Fig. 3 Normalized FTIR spectra measured for a film of drop-cast pristine PMMA, for the Fe3 powder and for the Fe3(10%) film. Dashed lines are reported in correspondence with the peaks in the drop-cast sample that can be unequivocally attributed to Fe3. | |
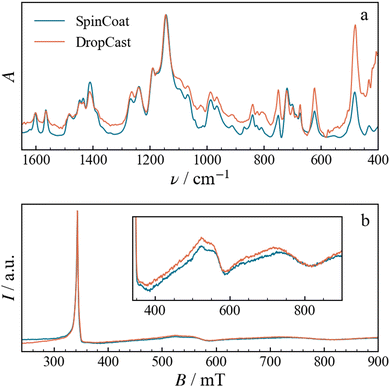 |
| | Fig. 4 (a) Normalized FTIR spectra and (b) X-Band EPR spectra, the latter recorded at 5 K, of the spin-coated and drop-cast Fe3(10%) films. | |
X-Band EPR spectroscopy
Although no structural differences are evidenced by FTIR, magnetic properties can still be different. Therefore, we recorded CW EPR spectra of Fe3 powder as well as SC and DC films at different concentrations at X-band frequency (9.6 GHz) and 5 K. The spectra of the SC and DC samples are identical, as shown for example in Fig. 4b for Fe3(10%). The magnetic properties are therefore independent of the type of deposition method, and consequently, of the film morphology and thickness. For this reason, we continue the discussion focusing only on the DC samples.
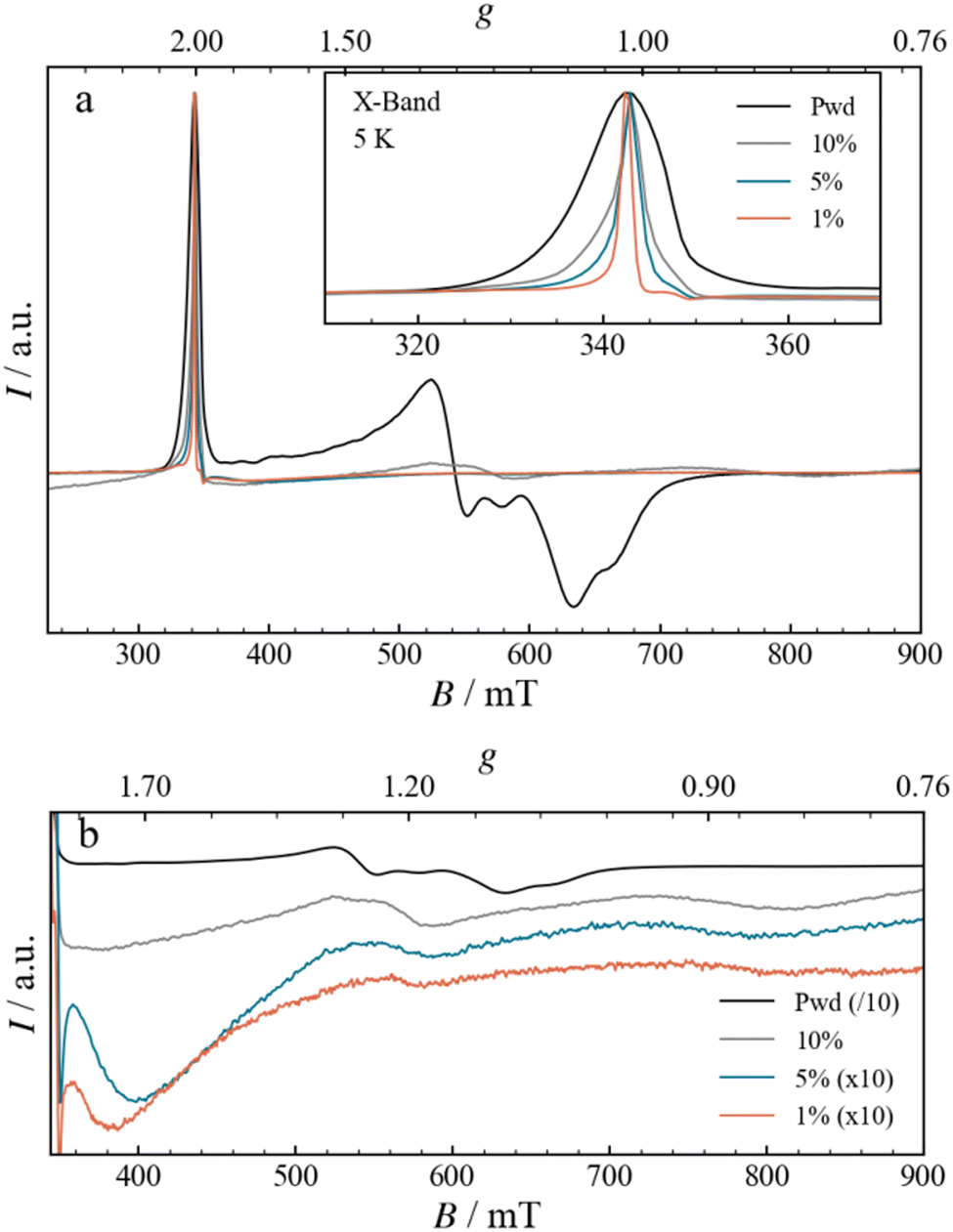
The EPR spectrum for the Fe3 powder, shown in Fig. 5, closely matches the one already reported in the literature,12 with a narrow absorption-like signal at 340 mT, attributed to g‖ (∼ 2.00), and a broad and composite derivative-like signal centred at 600 mT, spanning more than 100 mT, attributed to g⊥.24,25 Upon increasing the dilution, from Fe3(10%) to Fe3(1%), the spectra present modifications that can be analysed in terms of g‖ and g⊥ signals. The g‖ signal narrows significantly with increasing dilution, an effect already clearly visible in the Fe3(10%) DC sample (Fig. 5a, inset). The narrowing can be explained by considering a larger separation between the spins, which leads to a reduction in the intermolecular dipolar coupling. The progressive narrowing upon dilution down to Fe3(1%) allows the observation of a minor resonance, with a derivative-like shape centred at 348.3 mT (g = 1.974), a peak at 346.4 mT (g = 1.984) and a trough at 349.5 mT (g = 1.966). The high field component, g⊥, is also affected by the dilution, as shown in Fig. 5b. Indeed, the resonances become broader and shift to higher fields, with two main derivative-like features centred at 560 and 750 mT (g of 1.2 and 0.9, respectively). The FTIR spectra and the presence of the g‖ resonance at g = 2.00 strongly suggest that the structural integrity of the Fe3 triangle is maintained. Therefore, the EPR spectral differences likely point to minor structural alterations in the metallic core, which are too small to be observed by IR. When the Fe3 triangle is dissolved and dispersed into the PMMA matrix, the environment shifts from the one solely formed by perchlorate counteranions and pyridine solvate to the one formed by organic polymer chains. The altered spatial constraints imposed by the polymer matrix are expected to mainly impact the g⊥ component, which is the most sensitive one, through modifications of J, J′ and Gz, as was also observed moving from powder to solution.26 Moreover, the spatial constraints imposed by the nearby polymeric matrix could potentially prompt a variety of structural changes in the triangle, resulting in numerous conformations. Although the spectral shape of g⊥ appears to be the same upon further dilution, the signal does become less prominent. This decrease could derive from downfield shifts of the g⊥ signals beyond the range of our magnet (> 900 mT) as well as from broadening effects.
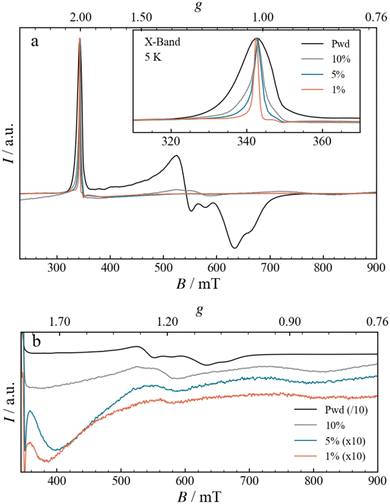 |
| | Fig. 5 (a) Normalized CW-EPR spectra measured at X-Band frequencies, at a constant temperature of 5 K, for Fe3 in the form of powder, as well as films with different concentrations of Fe3 (10, 5 and 1%) and drop-casted onto a Mylar substrate. The inset displays an enlargement of the g‖ region. (b) Enlargement of the g⊥ region, shown as a stack-plot and using different scaling factors, as indicated in the legend, to facilitate the comparison. | |
High-frequency EPR spectroscopy
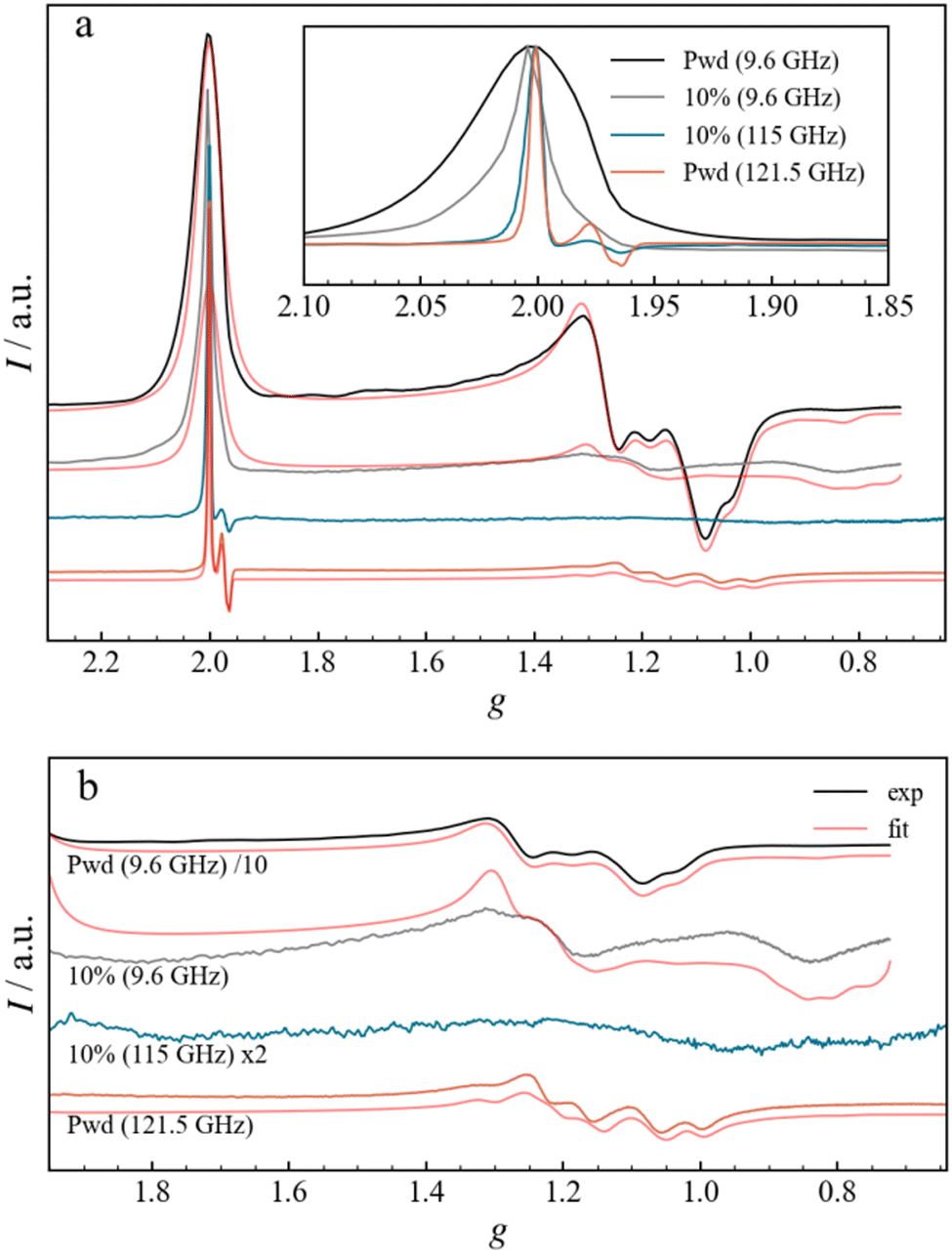
To improve the resolution of the magnetic resonance spectra, high-frequency EPR (HFEPR) experiments were carried out on Fe3 powder and Fe3(10%) DC films at the temperature of 3 and 8 K, respectively, and frequencies of 121.5 and 115.0 GHz, respectively (Fig. 6). The 121.5 GHz spectrum of the Fe3 powder shows a narrow signal at g = 2.00 (4.32 T) and a broad signal centred at g = 1.2 (7.7 T), attributed to the g‖ and g⊥ components, respectively, and in full agreement with the X-band spectrum. The small derivative signal in the g‖ region (Fig. 6a, inset) centred at around g = 1.978 is now resolved and shows a peak at g = 1.984 and a double trough at g = 1.975 and 1.969. The origin of this signal will be analysed below, but it can already be noticed that increasing the frequency has the same effect as diluting, in better resolving small g differences, albeit for different reasons. Overall, while the linewidth of the g‖ peak in the Fe3 powder spectrum changes little in absolute (B-field) terms, being ca. 9 mT, it does become sharper in g terms as the frequency is increased. The g‖ region of the HFEPR spectrum of Fe3(10%) is similar to that of Fe3 powder, also in terms of linewidth. Instead, the g⊥ line appears to be different: The diluted sample does not present any fine structure but exhibits a very broad derivative-like signal centred at g = 1.1, in agreement with the X-band measurements. This might be the result of larger g-strains, which become more relevant at higher frequencies and broaden the peaks observed in X-band at g = 1.2 and g = 0.9.
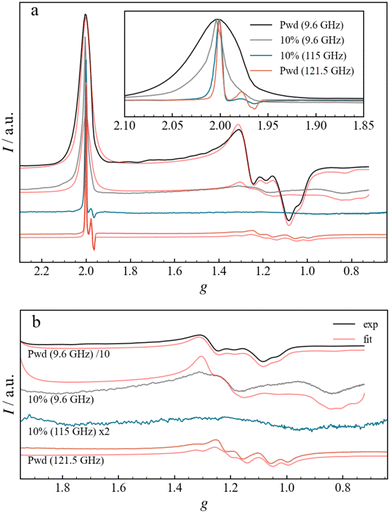 |
| | Fig. 6 (a) Normalized experimental and fitted EPR spectra for the Fe3 powder and Fe3(10%) drop-casted film measured at X-Band and at high frequency. The inset shows the enlargement of the g‖ region for the experimental spectra. (b) Enlargement of the g⊥ region, displaying both experimental and fitted curves. Different scaling factors are used, as indicated in the legend, to ensure a better comparison. For both (a) and (b), the spectra are plotted as a function of the g-factor in a stack-plot. | |
Discussion
The EPR spectrum of spin triangles depends on three parameters: (i) the values of the g-tensor elements of individual ions, (ii) ΔJ and (iii) Gz. Taking the individual spins’ gz directions parallel to the molecular z-axis (i.e. perpendicular to the triangle plane), then the effective g-value of the molecule along that axis will be g‖eff = g0‖, where g0‖ is the gz of the individual ions. In the powder EPR spectrum, this appears as an absorption-like signal centered at g = 2.00. On the other hand, the effective g-value of the molecule in the triangle's plane (g⊥eff), which is given by equation (2), parametrizes ΔJ and Gz terms. Exploration of this relation, considering a quasi-isotropic g-tensor for octahedral FeIII, reveals that g‖0‖ ≥ g⊥0 for any parameter set. Indeed, if and only if Gz ≠ 0 can there be any magnetoanisotropy. Any non-zero value of Gz shifts the g⊥ component towards higher fields; however, a magnetic asymmetry of the isotropic terms in the absence of DMI (i.e. ΔJ ≠ 0 with Gz = 0) has no such effect. Actually, and counter-intuitively, an increase of |ΔJ| has an opposing effect to Gz, shifting g⊥ back towards g‖ (i.e., at lower magnetic fields). This means that any g⊥ value can be obtained by infinite pairs of ΔJ and Gz values, which can also be understood by the fact that equation (2) does not have a unique solution since it depends on two variables.
To obtain a more quantitative description of the EPR spectra of Fe3 powder and 10% diluted films, fits were carried out based on a previously described method,27,28 which models the strain effects of g⊥ with a series of axial S = 1/2 effective spins. This method assumes that the sample consists of a series of magnetic conformers of different g⊥, which occurs due to distributions of the underlying spin Hamiltonian parameters ΔJ and Gz, according to equation (2). It should be noted that the latter parameters, ΔJ and Gz, could also be used, instead of g⊥, to model the EPR spectra, as previously demonstrated with Fe3 and with its CrIII analogue, Cr3.25,29 For instance, in the case of Cr3, simultaneous distributions of ΔJ and Gz were considered, with spectra calculated over 20 × 20 = 400 combinations of those parameters, and then summed considering the weights of the respective distributions. However, this approach requires the diagonalization of the full Hamiltonian for each individual spectrum (of dimension 216 for Fe3). Since our objective was the quantitative description of the g⊥ distribution, we opted for the direct modelling of the effective g⊥ values.
Therefore, the spectra of 25 magnetic conformers of identical g‖ and different g⊥ were calculated (this number being defined by the limitations of 30 free variables imposed by Easyspin's esfit function, setting aside five variables for other parameters, such as g‖, g-strains etc.). These fictitious spin systems were characterized by the same anisotropic g-strain, which was found to be necessary for reproducing the line shapes of the experimental spectra. These fits are shown in Fig. 7 and key parameters are shown in Table 1. The results of the fit for the Fe3 powder sample measured by HFEPR (Fig. 6b) indicate a bimodal distribution of the g⊥ component, with maxima at g ∼ 1.15 and 1.05, as shown in Fig. 7.
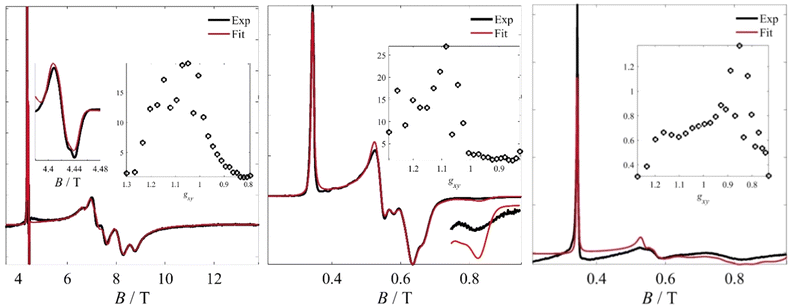 |
| | Fig. 7 Left: Fit to the 121.5 GHz spectrum of the Fe3 powder sample. The boxed inset shows the fitted bimodal probability density function for g⊥ values between 0.8 and 1.3. The smaller inset shows an expansion of the region corresponding to the “S = 3/2” component. Middle: Fit to the X-Band data of the Fe3 powder sample using the model previously described. The bottom right part shows an enlargement of the trough at 0.82 T. The boxed inset shows the calculated bimodal probability density function for g⊥ values between 0.8 and 1.3. Right: Fit to the X-band spectrum of the Fe3(10%) sample. While the g⊥ range is roughly the same and the distribution remains bimodal, the high-g⊥ component is now significantly reduced, whereas the low-g⊥ component is enhanced and shifted to even lower g-values. | |
Table 1 Parameters from simulations to distribution of axial S = 1/2 spectra
| Sample/Parameters |
g
‖
|
σ
G/mT |
σ
L/mT |
σ (g⊥) |
g
⊥ distribution maxima |
|
Fe3 pwd HFEPR |
2.000 |
3 |
4 |
0.03 |
1.15, 1.05 |
|
Fe3 pwd XBand |
2.000 |
3 |
4 |
0.03 |
1.15, 1.05 |
|
Fe3(10%) XBand |
2.000 |
4 |
3 |
0.045 |
1.15, 0.85 |
This outcome is in full agreement with the fit of the X-Band spectrum performed on the same complex, which was derived from a more elaborate dynamic spin Hamiltonian model assuming that the magnetic interactions are modulated by oxo-bridge vibrations; this had shown the presence of two systems with different vibration amplitudes.25 Best-fit parameters were g‖ = 2.000, with a g-strain σ (g⊥) = 0.030, and Gaussian and Lorentzian broadenings of σG = 3 mTpp and σL = 4 mTpp, respectively. The g ∼ 1.97 component was fitted to a minor (< 0.3%) monomeric CrIII impurity that we attribute to a starting material impurity (Fig. 7). Detailed explanations of the assignment of this impurity are given in the ESI.† To qualitatively compare the g⊥ regions between the powder and thin film samples, we carried out similar fits on the Fe3(10%) spectra measured at X-Band frequency (Fig. 6b and Fig. 7). The best-parameters obtained by the fit (g‖ = 2.000, σ(g⊥) = 0.045, σG = 4 mTpp, σL = 3 mTpp) still reveal a bimodal distribution, though the g ∼ 1.15 component is significantly reduced in relative weight and the low-g component shifts downfield to g ∼ 0.85. As this component shifts to lower g-values, its absolute intensity is decreased due to broadening effects and becomes more elusive. At the same time, we postulate that part of the spectrum crosses into magnetic fields unattainable by our magnets. The use of higher frequency allows extending the experimental magnetic field range, however, in this case, the broadening of the g-strains is large and prevents obtaining additional information from the diluted sample. This shift in g-values cannot be attributed to dipolar interactions among the Fe3 molecules, because the intermolecular distances in the matrix-diluted samples, or even in the pure powder sample, are too large (Fig. S3, ESI†). The only visible effect of such interactions is the dipolar broadening induced by decoherence. On the other hand, the g⊥-shifts can be attributed to matrix effects by considering that a larger molecular fraction becomes more equilateral, i.e. a decrease in ΔJ. This would be in line with an environment characterized by more isotropic steric constraints which allow the molecules to adopt a more equilateral conformation. We recall that in the crystal, the molecule is under the influence of disordered perchlorate anions and pyridine solvates. While on average these impose a highly symmetric crystal structure, our previous work (ref. 25) demonstrated that this is only an average static picture, with vibrational effects superimposed to influence the magnetic spectrum. It is possible that these effects are mitigated in the polymeric matrix, with a larger molecular fraction approaching the average picture.
Conclusions and perspectives
In summary, we successfully prepared thin films containing molecular spin triangles encapsulated within a polymeric matrix. The retention of the molecular structure and magnetic properties is evident from the consistent vibrational patterns and the distinctive g‖ and g⊥ signals. Notably, diluting the spin triangles within the polymer matrix led to a significant narrowing of the linewidth of the g‖ signal, suggesting a reduction in dipolar intermolecular interactions. Additionally, the magnetic anisotropy, as represented by the g⊥ signal, shifted to lower values. This shift is attributed to more isotropic strains induced by the polymer environment with respect to the crystalline one. The pronounced sensitivity of the magnetic anisotropy to its surroundings highlights the potential of molecular spin triangles as quantum sensors.
Apart from the use of the magnetoanisotropy as a magnetic probe of the matrix, the spin–lattice relaxation time T1 might also serve that purpose. In particular, T1 times depend on the intrinsic magnetic structure of the molecule, especially through Orbach processes over the doublet–doublet barrier Δ (defined in turn by ΔJ and Gz), as well as by extrinsic (lattice) effects through the Raman, direct and local-mode processes. The Orbach process should be sensitive to the same factors that affect the magnetic anisotropy through ΔJ and Gz, i.e. direct triangle-matrix interactions. This would also be the case for the other relaxation mechanisms. Such probing mechanisms may be useful for example in detecting pressure variations, which are directly transmitted to the probe via the host matrix or electric fields. The latter can be detected by CW-EPR, exploiting the magnetoelectric effect, as we have previously demonstrated for this type of molecule.12,15
On a longer-term perspective, embedding the spin triangles in a polymeric host offers several advantages relevant to quantum sensing. These advantages include enhanced stability, as the polymer matrix can act as a protective layer, potentially increasing the lifetime of the sensor. The polymer matrix can also serve as a diffusion barrier, controlling the interaction between the sensing element and the environment. Furthermore, the ease of handling during thin film fabrication enables the creation of tailored devices with specific selectivity, especially when using porous polymers designed to absorb specific molecules. We envisage in particular sensing of pressure and electric fields. The former is related to the steric constraints imposed by the matrix under pressure, which we deem could be detected magnetically. The latter is related to the magnetoelectric effect we have detected for this molecule,12 through which its magnetic properties are modified by electric fields.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the Baden-Württemberg Foundation (Competence Network Quantum Technology, project MOLTRIQUSENS) and the QUSTEC MC network for financial support. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement number 847471. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) with the project number 529038510 (L. T.). Sally Eickmeier is acknowledged by L. T. for her support with AFM experiments.
Notes and references
- S. Carretta, P. Santini, G. Amoretti, F. Troiani and M. Affronte, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 24408 CrossRef.
- A. K. Boudalis, Chem. – Eur. J., 2021, 27, 7022 CrossRef CAS PubMed.
- M. Trif, F. Troiani, D. Stepanenko and D. Loss, Phys. Rev. Lett., 2008, 101, 217201 CrossRef PubMed.
- F. Troiani, D. Stepanenko and D. Loss, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 161409 CrossRef.
- J. F. Nossa, M. F. Islam, M. R. Pederson and C. M. Canali, Phys. Rev. B: Condens. Matter Mater. Phys., 2023, 107, 245402 CrossRef CAS.
- M. Chizzini, L. Crippa, A. Chiesa, F. Tacchino, F. Petiziol, I. Tavernelli, P. Santini and S. Carretta, Phys. Rev. B: Condens. Matter Mater. Phys., 2022, 4, 43135 CAS.
- M. Trif, F. Troiani, D. Stepanenko and D. Loss, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 45429 CrossRef.
- M. Gysler, F. El Hallak, L. Ungur, R. Marx, M. Hakl, P. Neugebauer, Y. Rechkemmer, Y. Lan, I. Sheikin, M. Orlita, C. E. Anson, A. K. Powell, R. Sessoli, L. F. Chibotaru and J. van Slageren, Chem. Sci., 2016, 7, 4347 RSC.
- A. Ghirri, J. van Tol, I. Vitorica-Yrezabal, G. A. Timco and R. E. P. Winpenny, Dalton Trans., 2015, 44, 14027–14033 RSC.
- B. Kintzel, M. Fittipaldi, M. Böhme, A. Cini, L. Tesi, A. Buchholz, R. Sessoli and W. Plass, Angew. Chem., Int. Ed., 2021, 60, 8832 CrossRef CAS PubMed.
- B. Kintzel, M. Böhme, J. Liu, A. Burkhardt, J. Mrozek, A. Buchholz, A. Ardavan and W. Plass, Chem. Commun., 2018, 54, 12934 RSC.
- A. K. Boudalis, J. Robert and P. Turek, Chem. – Eur. J., 2018, 24, 14896 CrossRef CAS PubMed.
- M. Lewkowitz, J. Adams, N. S. Sullivan, P. Wang, M. Shatruk, V. Zapf and A. S. Arvij, Sci. Rep., 2023, 13, 2769 CrossRef CAS PubMed.
- B. Kintzel, M. Böhme, D. Plaul, H. Görls, N. Yeche, F. Seewald, H.-H. Klauss, A. A. Zvyagin, E. Kampert, T. Herrmannsdörfer, G. Pascua, C. Baines, H. Luetkens and W. Plass, Inorg. Chem., 2023, 62, 3420 CrossRef CAS PubMed.
- J. Robert, N. Parizel, P. Turek and A. K. Boudalis, J. Am. Chem. Soc., 2019, 141, 19765 CrossRef CAS PubMed.
- J. Wucher and J. D. Wasscher, Physica, 1954, 20, 721 CrossRef CAS.
- J. Wucher and H. M. Gijsman, Physica, 1954, 20, 361 CrossRef CAS.
- Y. V. Rakitin, Y. V. Yablokov and V. V. Zelentsov, J. Magn. Reson., 1981, 43, 288 CAS.
-
A. K. Boudalis, K. S. Kumar and M. Ruben, Comprehensive Coordination Chemistry III, Elsevier, Oxford, 2021, pp. 206 Search PubMed.
- M. Kern, L. Tesi, D. Neusser, N. Rußegger, M. Winkler, A. Allgaier, Y. M. Gross, S. Bechler, H. S. Funk, L. Te Chang, J. Schulze, S. Ludwigs and J. van Slageren, Adv. Funct. Mater., 2021, 31, 2006882 CrossRef CAS.
- P. Neugebauer, D. Bloos, R. Marx, P. Lutz, M. Kern, D. Aguilà, J. Vaverka, O. Laguta, C. Dietrich, R. Clérac and J. Van Slageren, Phys. Chem. Chem. Phys., 2018, 20, 15528–15534 RSC.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- F. E. Sowrey, C. Tilford, S. Wocadlo, C. E. Anson, A. K. Powell, S. M. Bennington, W. Montfrooij, U. A. Jayasooriya and R. D. Cannon, J. Chem. Soc., Dalton Trans., 2001, 862 RSC.
- A. N. Georgopoulou, Y. Sanakis and A. K. Boudalis, Dalton Trans., 2011, 40, 6371 RSC.
- A. N. Georgopoulou, I. Margiolaki, V. Psycharis and A. K. Boudalis, Inorg. Chem., 2017, 56, 762 CrossRef CAS PubMed.
- J. Robert, N. Parizel, P. Turek and A. K. Boudalis, Phys. Chem. Chem. Phys., 2019, 21, 19575 RSC.
- A. K. Boudalis, G. Rogez, B. Heinrich, R. G. Raptis and P. Turek, Dalton Trans., 2017, 46, 12263 RSC.
- A. K. Boudalis, Y. Sanakis, C. P. Raptopoulou, A. Terzis, J.-P. Tuchagues and S. P. Perlepes, Polyhedron, 2005, 24, 1540 CrossRef CAS.
- A. K. Boudalis, G. Rogez and P. Turek, Inorg. Chem., 2018, 57, 13259 CrossRef CAS PubMed.
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Athanassios K.
Boudalis
*a,
Athanassios K.
Boudalis
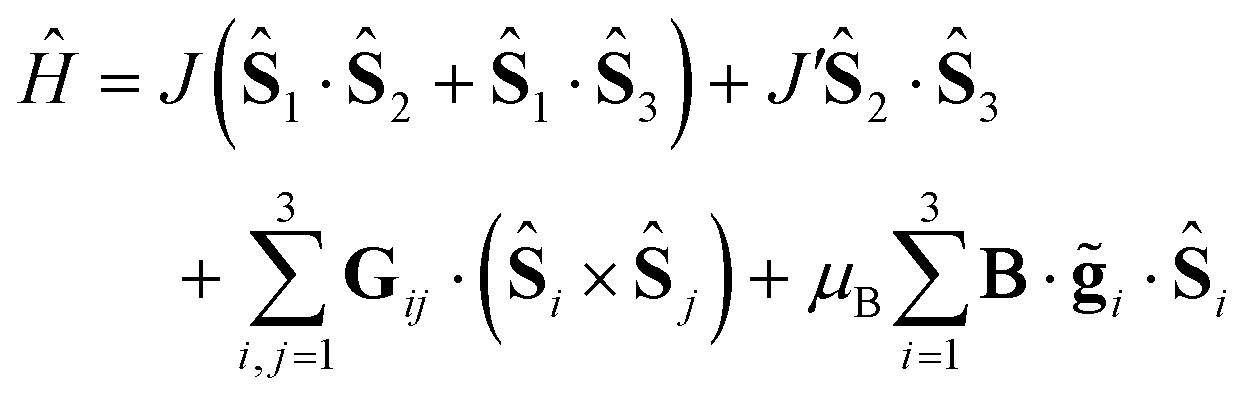

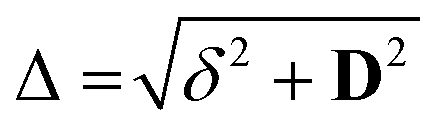 , with δ and D depending on the individual spin values: for Si = 1/2, δ = ΔJ,
, with δ and D depending on the individual spin values: for Si = 1/2, δ = ΔJ, 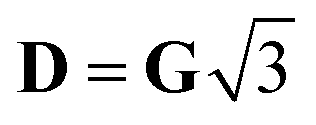 for Si = 3/2, δ = 2ΔJ,
for Si = 3/2, δ = 2ΔJ, 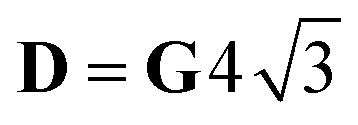 ; for Si = 5/2, as in this case, δ = 3ΔJ,
; for Si = 5/2, as in this case, δ = 3ΔJ, 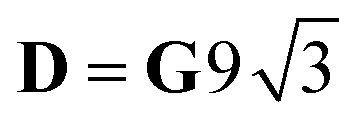 .18
.18