
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Excited-state dynamics of 4-hydroxyisoindoline-1,3-dione and its derivative as fluorescent probes†
Li
Zhao
a,
Simin
Jiang
b,
Yanmei
He
bc,
Luling
Wu
 *d,
Tony D.
James
de and
Junsheng
Chen
*b
*d,
Tony D.
James
de and
Junsheng
Chen
*b
aCollege of Science, China University of Petroleum (East China), Qingdao, 266580, Shandong, China
bNano-Science Center & Department of Chemistry University of Copenhagen, Universitetsparken 5, 2100 KøbenhavnØ, Denmark. E-mail: junsheng.chen@chem.ku.dk
cDepartment of Chemical Physics and NanoLund, Lund University, P. O. Box 124, 22100 Lund, Sweden
dDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: wllcyl@126.com
eSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
First published on 3rd April 2024
Abstract
Fluorescent probes have become promising tools for monitoring the concentration of peroxynitrite, which is linked to many diseases. However, despite focusing on developing numerous peroxynitrite based fluorescent probes, limited emphasis is placed on their sensing mechanism. Here, we investigated the sensing mechanism of a peroxynitrite fluorescent probe, named BHID-Bpin, with a focus on the relevant excited state dynamics. The photoexcited BHID-Bpin relaxes to its ground state via an efficient nonradiative process (∼300 ps) due to the presence of a minimum energy conical intersection between its first excited state and ground state. However, upon reacting with peroxynitrite, the Bpin moiety is cleaved from BHID-Bpin and BHID is formed. The formed BHID exhibits strong dual band fluorescence which is caused by an ultrafast excited-state intramolecular proton transfer process (∼1 ps).
1. Introduction
Peroxynitrite (ONOO−) is generated by the combination of nitric oxide and a superoxide anion radical. Due to its strong nucleophilicity and oxidative activity, ONOO− is associated with various pathways in cells, including apoptosis and cell death.1–3 Through detecting the abnormal concentrations of ONOO−, it is easy to track the origin of diseases. For example, an increased ONOO− will be observed in acetaminophen-induced liver injury.4,5 Hydrogen peroxide (H2O2) is another unique reactive oxygen species which plays an important role in triggering a response within the cell. Abnormal levels of H2O2 are implicated in diabetes, aging and cancer.6 These reactive oxygen/nitrogen species have short lifetimes, which can be detected by fluorescent probes with the merits of high sensitivity, excellent selectivity, fast response, real-time visualization, etc.7Significant efforts have been devoted to the development of fluorescent probes based on the common fluorophores, such as coumarin,8 hemicyanine,9,10 anthracene,11 and naphthalimide,12,13 for sensing reactive oxygen/nitrogen species with reliable detection performance. The probes were designed based on different photophysical mechanisms, such as the photoinduced electron transfer (PeT) mechanism,14–18 intramolecular charge transfer (ICT),19–21 excited state intramolecular proton transfer (ESIPT),22,23 Förster resonance energy transfer (FRET),24,25 solvatochromism and aggregation-induced emission (AIE).26–28 To date, most of the studies focused on the synthesis aspect of new fluorescent probes for reactive oxygen/nitrogen species sensing, while the more fundamental studies addressing the photophysics and their sensing mechanisms are still rare. Yet a thorough understanding of their sensing mechanism is beneficial for further improvement of their performance and successful development of new fluorescent probes.
ESIPT is a crucial photophysical process, which causes double emission bands in fluorophores due to the photoisomerization of enol and keto forms.22 One of the important advantages of ESIPT fluorophores is a large Stokes shift, which can ensure the scattered excitation light to be filtered out completely. As a result, ESIPT has been widely used for the construction and development of fluorescent probes for the detection of biomarkers.29 Taking advantage of the ESIPT process, we recently developed a peroxynitrite probe (Fig. 1a, BHID-Bpin), which is non-fluorescent because the phenol is blocked by phenylboronic acid pinacol ester which inhibits the ESIPT process. Upon its oxidation and cleavage by ONOO−/H2O2, protons of the hydroxyl group in the product (BHID) can be transferred to the adjacent keto unit under photoexcitation. This unique process ensures that BHID is fluorescent with an emission centred at 515 nm in aqueous solution.30 The developed fluorescent probe exhibits high sensitivity for ONOO−/H2O2. However, a clear picture concerning its sensing mechanism remains elusive, especially its fluorescence turn on response to ONOO−. Herein, we employed time-resolved optical spectroscopy, density functional theory (DFT), time-dependent density functional theory (TDDFT) and excited-state dynamic simulations to study the underlying sensing mechanism of the reported fluorescent probe with a focus on its excited state dynamics and its fluorescence turn on response mechanism.
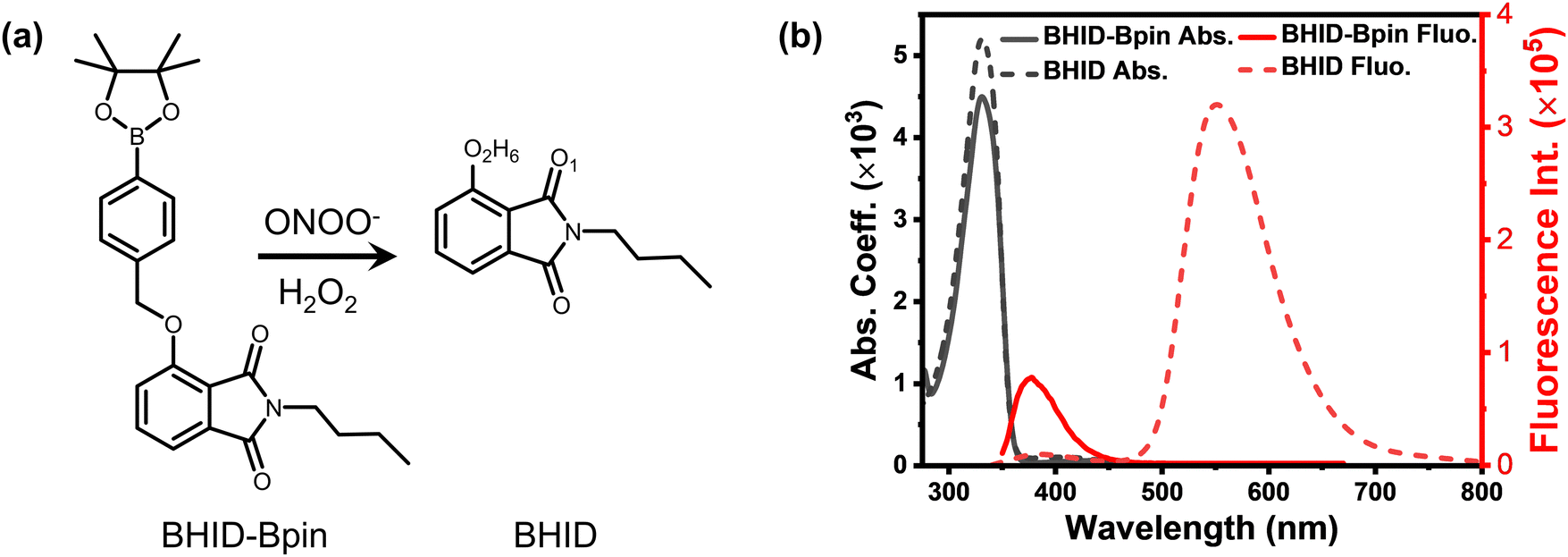 | ||
| Fig. 1 (a) Reaction between probe BHID-Bpin and ONOO−/H2O2. (b) Absorption and fluorescence spectra of BHID-Bpin and BHID in acetonitrile, λex = 320 nm, and concentration (c) = 2 × 10−5 mol L−1. | ||
2. Experimental details
2.1 Synthesis of BHID-Bpin and BHID
The probe BHID-Bpin and reference sample BHID were synthesized by following a reported procedure.30 The detailed information can be found in the ESI.†2.2 Steady-state absorption spectroscopy and fluorescence spectroscopy
Steady state absorption spectra were measured using a Cary 5000 UV/Vis/NIR spectrophotometer. Steady state fluorescence spectra and fluorescence decay curves were measured using a FluoTime 300 spectrometer. The fluorescence quantum yield (FQY) was measured based on a relative method. Quinine sulfate dissolved in 0.5 M H2SO4 was used as the reference sample with a FQY of 54.6%.31 The FQY was calculated based on the whole fluorescence spectrum range as shown in Fig. 1b.2.3 Femtosecond transient absorption (fs-TA) spectroscopy
The fs-TA experiments were performed by using a femtosecond pump–probe setup. Laser pulses (796 nm, 60 fs pulse length, 1 kHz repetition rate) were generated using a regenerative amplifier (Spitfire) seeded using a femtosecond oscillator (Tsunami, both spectra physics). For the pump, we used the TOPAS C (light conversion) to obtain pulses with a central wavelength located at 320 nm. The pump pulse energies were set to 10 nJ per pulse. The spot size was approximately 0.2 mm2 for the pump beam. For the probe, we used supercontinuum generation from a thin Sapphire plate. The mutual polarization between the pump and probe beams was set to the magic angle (54.7°) by placing a Berek compensator in the pump beam. There is no photodegradation after fs-TA experiments by checking the steady-state absorption spectra. The global analysis was performed by using the Glotaran software package (https://glotaran.org).32 A simple sequential decay model with two or three components was used to perform SVD global fitting.333. Computational methods
The most stable structures of BHID-Bpin and BHID in the ground state (S0) and the lowest singlet excited state (S1) were optimized without any constraint using DFT and TDDFT methods, respectively. The Becke three-parameter hybrid exchange functional with the Lee–Yang–Parr gradient-corrected correlation functional (B3LYP) with the long-range correction state of the coulomb-attenuating method (CAM-B3LYP)34 and the 6-31+G(d,p) basis set35 were used for all the calculations. All optimized geometries were further verified to be the global minima in the corresponding potential energy surface by carrying out vibrational frequency analysis (no imaginary frequency). The polarizable continuum model (PCM) using an integral equation formalism variant (IEF-PCM)36,37 was employed to consider the solvent effect of acetonitrile (ACN). The vertical excitation energies and corresponding oscillator strength were calculated with the TDDFT//CAM-B3LYP//6-31+G(d,p) method at the ground state stable structures with six low-lying electronic transitions. In order to study the ESIPT process of the BHID system, the S0 and S1 potential energy surfaces were scanned by keeping the O1–H6 distance (Fig. 1a) fixed at a series of values. Furthermore, the S1 dynamics simulations of BHID were performed at the TDDFT//B3LYP//6-31G(d,p) level using the NEWTON-X program38 interfaced with Gaussian 09. Here, the B3LYP functional was used for the simulation, because in general the calculation speed with the B3LYP functional is much faster than that with the CAM-B3LYP functional. The correction for the long-range interaction is not included in the B3LYP functional.34 A total of 15 trajectories were run with a time step and a maximum simulation time of 0.5 fs and 200 fs, respectively. The initial conditions were generated from a Wigner distribution, with the frequencies and normal modes calculated with the DFT//B3LYP//6-31G(d,p) method. Furthermore, the dynamics simulation results of BHID were confirmed by using the TDDFT//CAM-B3LYP//6-31+G(d,p) method with a reduced number of trajectories (7 trajectories). The excited state behaviour of the BHID-Bpin system was examined through a truncated model, which preserves the conjugated system that is essential for the electronic excitation. Such a truncated model can represent the main electronic structure of the conjugated system and reduce computational power cost. The Franck–Condon (FC) point, the minimum energy conical intersection (MECI) between S1 and S0, and the transition state (TS) between FC and MECI were optimized with the state-averaged complete active space self-consistent field (CASSCF) method with the 6-31G(d,p) basis set. Two electronic states were included with equal weights. The active space is composed of 4 electrons distributed in 4 orbitals. The active space is validated by comparing the calculated vertical excitation energy of the system with a larger active space (8 electrons distributed in 8 orbitals), employing both the 6-31G(d,p) and aug-cc-pVDZ basis sets (Table S2, ESI†), in which the obtained vertical excitation energies do not show a significant difference. It is worth noting that, to compare the effect of aqueous environments on the sensing mechanism, we also conducted calculations considering the solvent effect of water. Furthermore, to simulate the formation of intermolecular hydrogen bonds in an aqueous environment, one explicit water molecule was added in the BHID system. All the DFT and TDDFT electronic structure calculations were carried out by using the Gaussian 09 program package,39 and the CASSCF and CASPT2 methods were performed with the openMolcas software.404. Results and discussion
Considering that the interaction (e.g. hydrogen bond) between the solvent and BHID-Bpin/BHID molecules will affect the understanding of their intrinsic excited state dynamics, we carried out all the relevant optical spectroscopy measurements and theoretical calculations in the organic solvent (acetonitrile). It is free from strong solvent–solute interactions (e.g. hydrogen bond) compared to water. Furthermore, acetonitrile is a good solvent to dissolve these compounds at high concentrations (∼500 μM, for optical density to reach around 0.3 in a 1 mm optical path cuvette) for fs-TA measurements. As shown in Fig. 1b, BHID-Bpin and BHID show a similar absorption peak located at 330 nm. This indicates that the Bpin group is not conjugated with the BHID; instead, it prevents the ESIPT process to occur in BHID-Bpin. However, they show very different fluorescence (Fig. 1b) under 320 nm light excitation. BHID-Bpin exhibits an ultraviolet emission at ∼378 nm, while BHID shows dual emissions centred at ∼380 nm and ∼550 nm with a big difference of PL intensity, revealing that the ESIPT process occurs in BHID. The higher fluorescence quantum yield (FQY) of BHID (15%) compared to BHID-Bpin (0.6%) further indicates that the Bpin unit causes the fluorescence quenching in BHID-Bpin (Table 1).| Compound | τ (375 nm) | τ (560 nm) | FQY (λex = 310 nm) |
|---|---|---|---|
| BHID-Bpin | 0.3 ns | — | 0.6% |
| BHID | <0.2 ns (IRF limited) | 5.3 ns | 15% |
The fluorescence decay curves were collected by time-correlated single photon counting (TCSPC) spectroscopy. As shown in Fig. 2, the PL decay of BHID-Bpin is rather fast (∼300 ps) which is close to the instrumental response function (IRF). It could be attributed to the efficient nonradiative decay process since the FQY of BHID-Bpin is extremely low (0.6%). For BHID, the fluorescence lifetimes detected at 375 nm and 560 nm are < 200 ps and 5 ns, respectively. The fast decay of ultraviolet emission at 375 nm was not well resolved because the IRF of the TCSPC method is around 200 ps. The different fluorescence intensities and lifetimes indicate that the two fluorescence peaks (380 nm and 555 nm) originate from two different excited-state species of BHID.
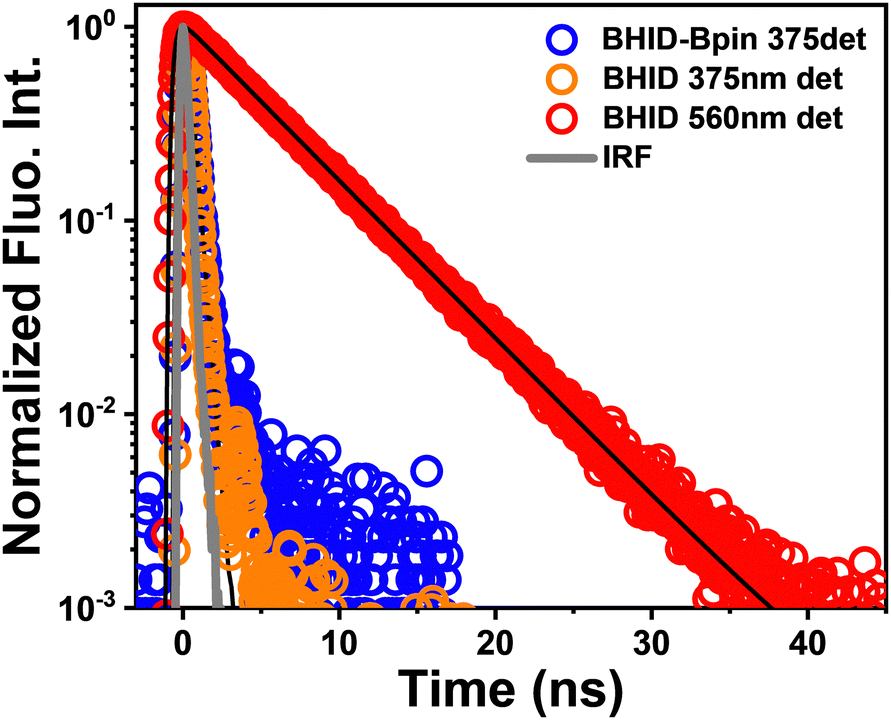 | ||
| Fig. 2 Normalized fluorescence decay curves of BHID-Bpin (λex = 300 nm and λdet = 375 nm) and BHID (λex = 300 nm and λdet = 375 nm and 560 nm). The black curves are the corresponding decay fit. The grey curve is the instrument response function (IRF). | ||
By using the femtosecond transient absorption spectroscopy (fs-TA, IRF ∼ 100 fs), the excited state dynamics of BHID-Bpin and BHID can be resolved at an ultrafast timescale. With an excitation of 320 nm, we noted that BHID-Bpin shows a broad positive signal (Fig. 3a) with two peaks located at 585 nm and 735 nm, respectively. The absence of ground state bleaching (GSB) and stimulated emission (SE) signals is because the ultraviolet absorption and fluorescence are outside of the detection window. At the very early timescale (≤500 fs, Fig. 3b), the two excited state absorption (ESA) peaks of 585 nm and 735 nm shift to 570 nm and 760 nm, respectively. After 1 ps, the ESA signals of BHID-Bpin show only intensity decay without a clear spectral shift. Based on the global analysis, we obtained two time components: 2 ps and 350 ps. The fitted curves match well with the experimental kinetics (Fig. 3c), indicating the reliability of the global analysis. The evolution decay associated spectra (EDAS, Fig. 3d) at the two time-constants show very similar shapes except for a slight spectral shift. The 350 ps time constant component matches with the PL lifetime measured by TCSPC, and we can assign it to the lifetime of the structure relaxed with minimal energy in S1. The 2 ps can be assigned to the relaxation process from the FC point to the minimum energy of S1.
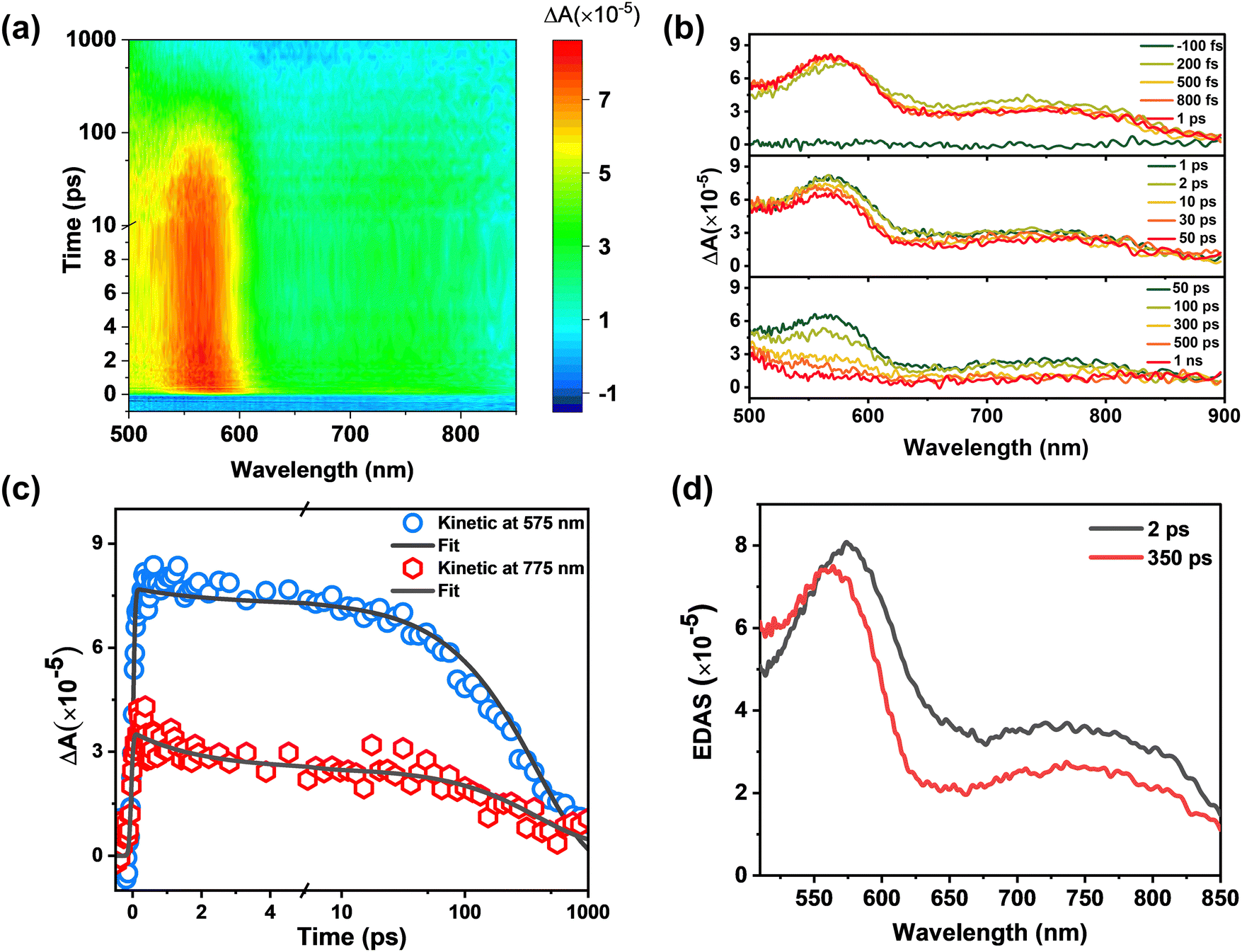 | ||
| Fig. 3 fs-TA spectra of BHID-Bpin. (a) Wavelength versus probe-delay map of the fs-TA spectra after excitation at 320 nm. The total time range is 1 ns. The change from a linear to a logarithmic scale at 10 ps is noted. (b) Transient spectra extracted from (a) over time-ranges of −100 fs to 1 ns. (c) Kinetic traces extracted at representative detection wavelengths (blue circle: 575 nm and red hexagonal: 775 nm) from the fs-TA experiment. Data overlaid by the corresponding kinetic trace extracted from the global fit (black solid lines). The change from a linear to a logarithmic scale at 5 ps is noted. (d) Evolution decay associated spectra (EDAS) extracted from the global kinetic analysis of fs-TA spectra. | ||
To gain further insight into the excited state dynamics and assign the obtained time constants, we calculated the electronic structures and electronic transitions between S0 and S1 of BHID-Bpin. As listed in Table 2 and shown in Fig. 4a, the transition between S0 and S1, with the ππ* character is mainly derived from the HOMO to LUMO transitions (72%). The transition oscillator strength (f) is 0.1252. The vertical excitation energy is calculated to be 4.09 eV (303 nm), which is in agreement with the 330 nm measured experimentally. Hence, we can assign the strong absorption λmax at 330 nm to the S0 → S1 state transition.
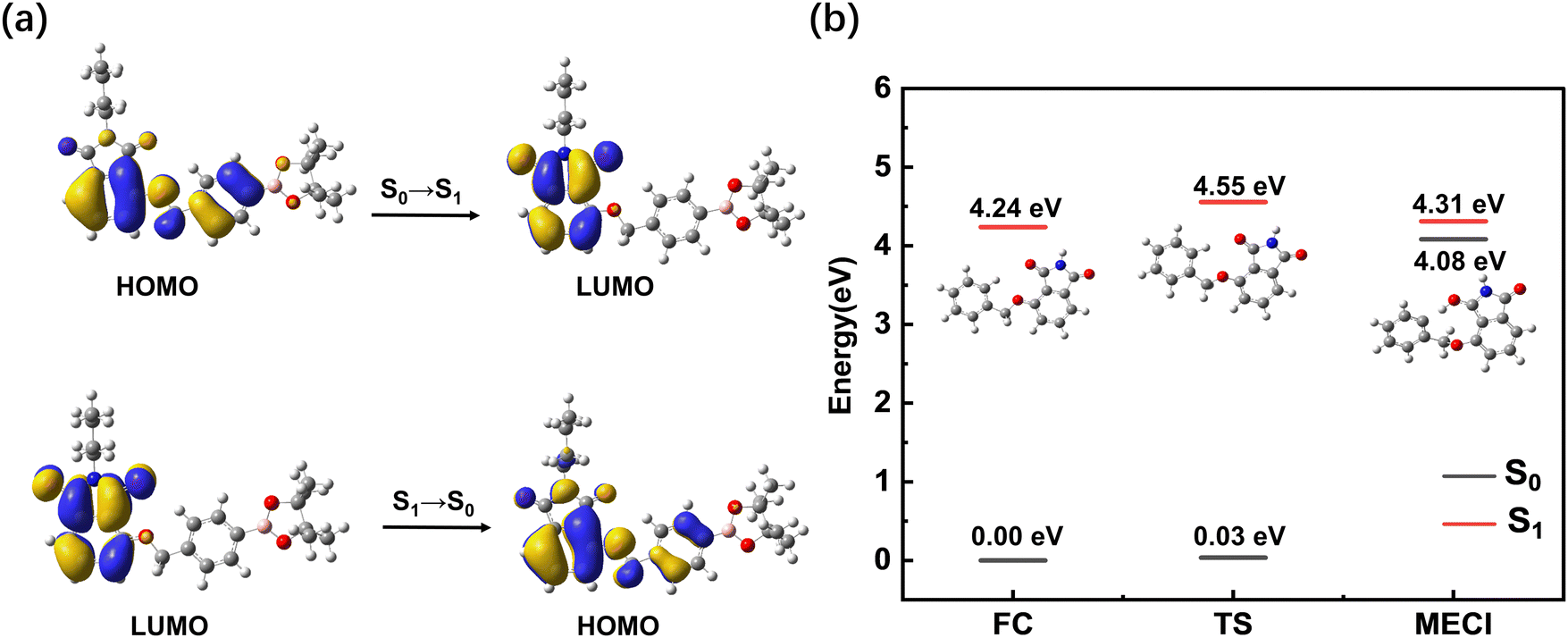 | ||
| Fig. 4 (a) Orbitals and the relevant transitions involved in the first excited state of BHID-Bpin at the TD-DFT//CAM-B3LYP//6-31+G(d,p) level. (b) Energy diagram of several critical points of the BHID-Bpin system (a truncated model) calculated with the CASPT2//SA2-CASSCF(4,4)//6-31G(d,p) method. | ||
We further optimized the stable structure of BHID-Bpin in S1 and calculated the relevant electronic transition. As listed in Table 2, the S1 → S0 transition mainly consists of the transition from the LUMO to the HOMO with a transition oscillation strength of 0.1310. The emission energy is calculated to be 3.41 eV (364 nm), which is in good agreement with the 378 nm fluorescence peak observed experimentally.
The low FQY (0.6%) observed experimentally indicates that the non-radiative process would play an important role in the deactivation process of BHID-Bpin, which is competitive with the fluorescence emission process. Thus, in S1, BHID-Bpin can relax back to S0via an efficient non-adiabatic process, in which MECI should be present between S1 and S0. To confirm the existence of such MECI, we used a truncated model (Fig. 4b, details are in the computational methods section) to carry out theoretical calculations. One MECI is located between S1 and S0, characterized by the rotation motion of the C–O bridging bond, and the H atom transfer process (Fig. S8 and Table S1, ESI†). The presence of such MECI was further confirmed using the CASPT2 method with an energy gap smaller than 0.3 eV. Fig. 4b displays the energy diagram of the FC, MECI and TS between the two. As is shown, there is an energy barrier of about 0.3 eV along the relaxation coordinate, which can be ascribed to the 350 ps time constant observed experimentally.
To understand the ESIPT process and relevant excited-state dynamics of BHID, we carried out fs-TA spectroscopy measurements of BHID with 320 nm excitation. The fs-TA spectra of BHID exhibit rich spectral dynamics (Fig. 5a). Similar to BHID-Bpin, a broad positive signal intensity appears with two peaks located at around 550 nm and 720 nm. As mentioned above, we cannot observe the GSB (330 nm) and SE (375 nm) signals since the ultraviolet absorption and emission are outside of the detection window. The SE signal of fluorescence peak 2 at around 550 nm is also absent at early time scale (Fig. 5b). After 1 ps, the SE signal of 550 nm starts to appear as shown in the upper and middle panels of Fig. 5b, which agrees well with the fluorescence peak 2 of BHID. Together with the formation of the negative SE signal, a positive ESA signal grows in at the blue side. However, we cannot resolve it completely due to the limited spectral window. After 50 ps, we observed the decay of the absolute signal intensity of both SE and ESA without a clear spectral shift. The global analysis provides three time constants: 1.3 ps, 180 ps and 5 ns. Considering that the ESIPT process is present in the system, the 1.3 ps component can be assigned to the excited enol form of BHID, which is also visualized by the latter appearance of the SE signal at 550 nm. This means that the ESIPT process has a time constant of 1.3 ps, which is in the range of reported similar systems.41,42 For 180 ps and 5 ns components, we assign them to the meta-stable state and stable state of the excited keto form, respectively.
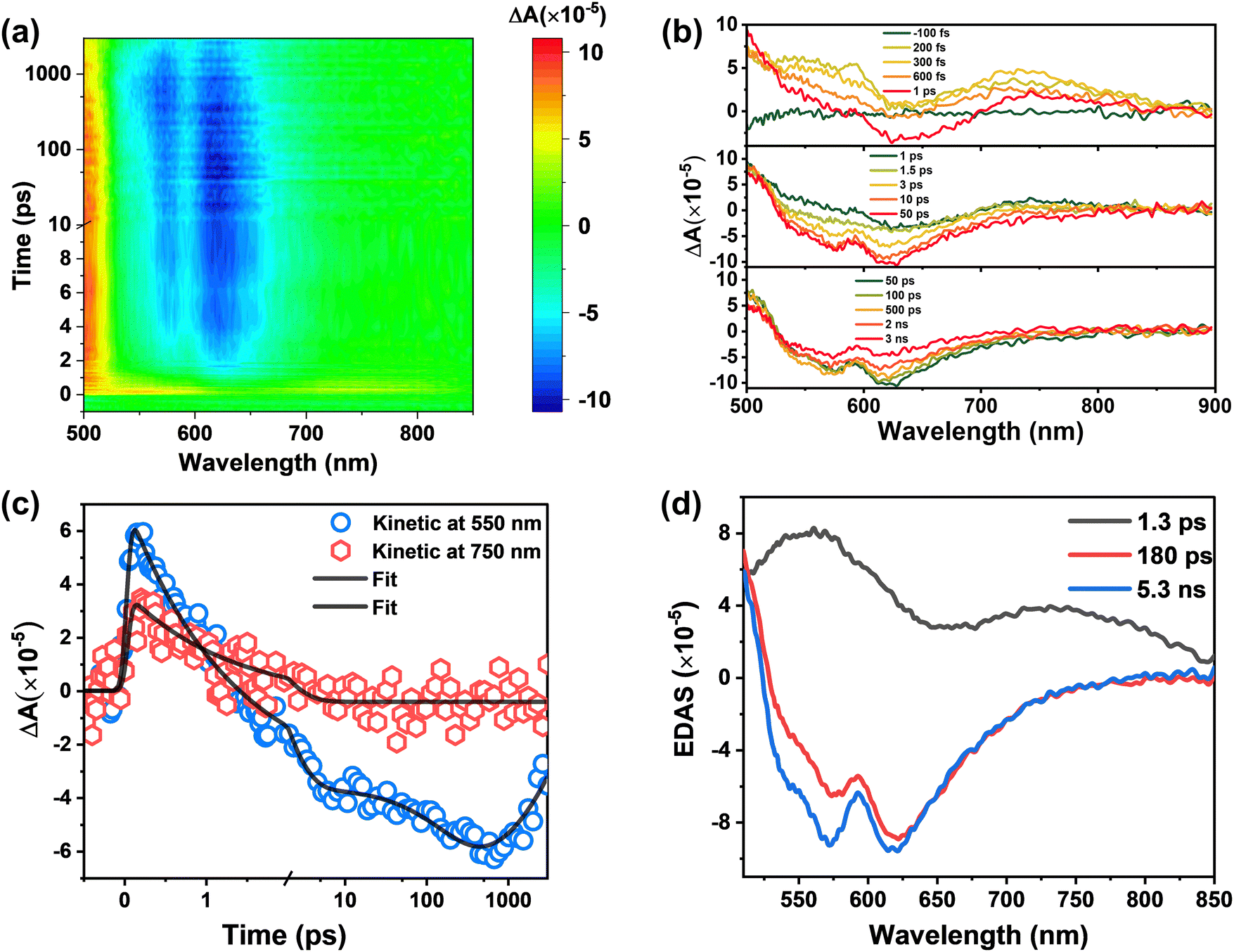 | ||
| Fig. 5 fs-TA spectra of BHID. (a) Wavelength versus probe-delay time map of the fs-TA experiment after excitation at 320 nm. The total time range is 3 ns. The change from a linear to a logarithmic scale at 10 ps is noted. (b) Transient spectra extracted from (a) over time ranges of −100 fs to 3 ns. (c) Kinetic traces extracted at representative detection wavelengths (blue circle: 550 nm; red hexagonal: 750 nm) from the fs-TA experiment. Data overlaid by the corresponding kinetic trace extracted from the global fit (black solid lines). The change from a linear to a logarithmic scale at 2 ps is noted. (d) Evolution decay associated spectra (EDAS) extracted from the global kinetic analysis of fs-TA spectra. | ||
To gain insight into the excited-state dynamics of BHID from a theoretical point of view, we optimized the structure of BHID in its S0 and S1 (Fig. S7, ESI†) with the DFT//TDDFT method. The relevant electronic transitions were calculated based on the TDDFT method and are listed in Table 3. The calculated vertical excitation energy from S0 to S1BHID is 4.95 eV (250 nm), which is overestimated compared to the experimentally observed strong absorption peak at 330 nm (∼3.76 eV, Fig. 1b). Such overestimation of vertical excitation energy is widely acknowledged for the CAM-B3LYP functional,43,44 due to the delocalization error and the inadequate treatment of long-range interactions. This electronic transition is mainly derived from the HOMO to LUMO transitions (89%) with a transition oscillator strength (f) of 0.2124. As shown in Fig. S9 (ESI†), the charge redistribution from the OH group to the oxygen atom (O6) indicated the strengthening of the hydrogen bond (O1–H6) in the excited state, which is further confirmed by the shortening of the hydrogen bond (O1–H6) from 2.181 Å (in S0) to 1.856 Å (in S1). Based on the optimized geometry of BHID in S1, we calculated its fluorescence emitting energy to be 3.37 eV (367 nm), which is consistent with the 380 nm fluorescence emission band observed experimentally (Fig. 1b). This confirms that the 380 nm fluorescence band is from BHID before the ESIPT process, namely the enol form of BHID (Fig. 6).
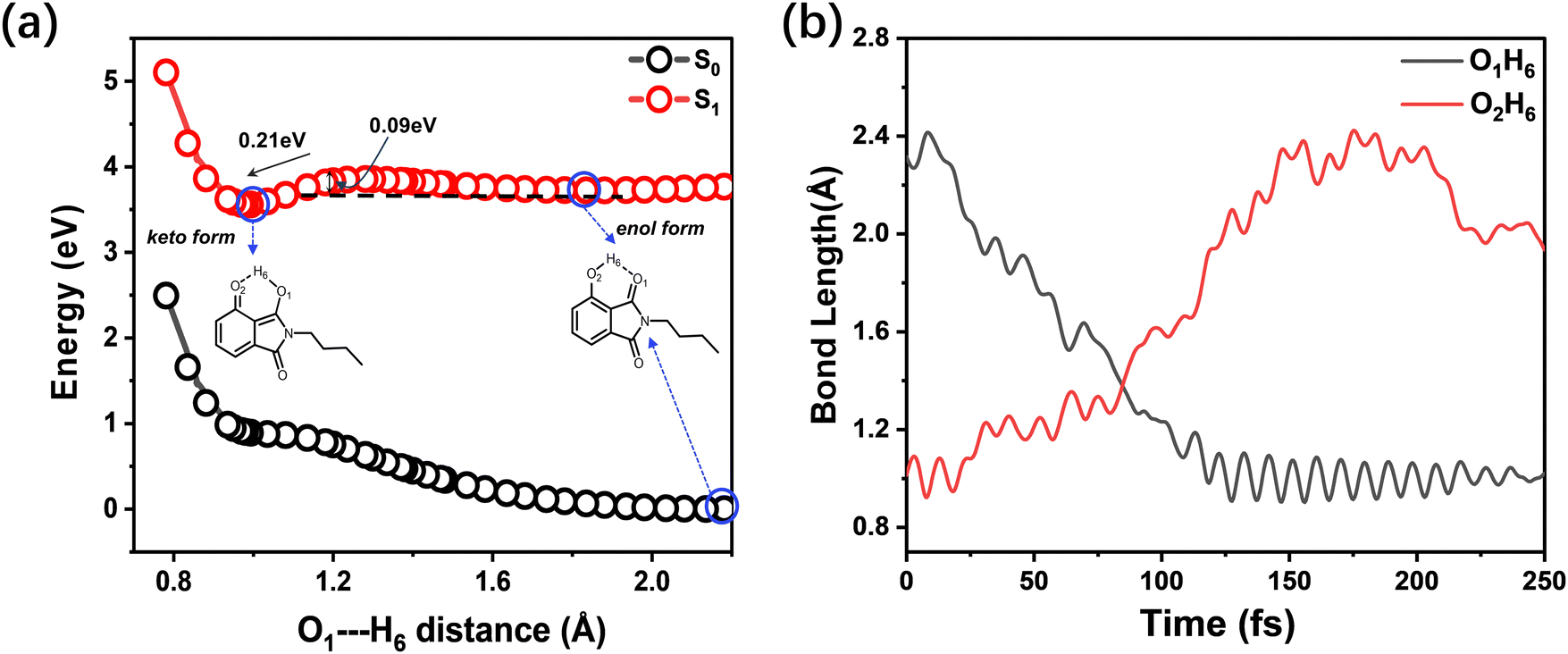 | ||
| Fig. 6 (a) Calculated potential energy curves of S1 and S0 of BHID computed at the TDDFT//CAM-B3LYP//6-31+G(d,p) level. The points marked within circles represents the most stable structures of enol- and keto-BHID in S0 and S1. (b) A typical trajectory displaying the time evolution of the O1–H6 and O2–H6 distances within 200 fs for the BHID system at the TDDFT//B3LYP//6-31G(d,p) level. | ||
The presence of dual band fluorescence and different fluorescence decay of the two bands indicate the presence of two different excited-state molecular conformers: an enol form and a keto form. Hence, in our theoretical calculation, we formed the keto form of BHID by breaking the O2H6 bond, and moving the H6 atom closer to the O1 atom to form the O1H6 bond (1.054 Å), while keeping other geometrical parameters unchanged. We optimized this structure in S1 and obtained the stable structure of the BHID keto form (Fig. S7, ESI†). In this form, the calculated S1 → S0 transition energy is 2.45 eV (504 nm), mainly consists of the transition from the LUMO to the HOMO with a transition oscillation strength of 0.2671. The results are in reasonable agreement with the experimentally measured values of 550 nm. Herein, we can assign the fluorescence peak at 550 nm to the ESIPT product: the keto form of BHID. Interestingly, we tried to optimize the structure of the BHID keto form in S0. However, all attempts ended with the BHID enol form in S0, which indicates that the relaxation process from the keto-form to the enol-form is barrierless in S0.
To estimate the energy barrier of the ESIPT process, we calculated the energy profiles of S0 and S1 for BHID with a fixed O1–H6 (Fig. 1a) distance ranging from 0.78 Å to 2.18 Å, as shown in Fig. 6a. The S0 energy profile clearly indicates that the enol form is the most stable conformer. In S0, the proton transfer from the enol to keto form is an endothermic process, requiring about 0.98 eV energy. Whereas, in S1, the keto conformer is the more stable one, which is about 0.21 eV lower than that of the enol form. A small energy barrier (∼90 meV) is present along the transformation process from the enol to keto form in S1, which is only about 3.5 times of the thermal energy (kBT = 25.7 meV). The barrier height has been further confirmed by the barrier height of the transition state (0.13 eV) along the ESIPT process, as shown in Fig. S10 (ESI†). Due to the existence of a small energy barrier, the ultrafast ESIPT reaction occurs easily,45–49 which is what have observed experimentally with about 1 ps (Fig. 5). We further performed the excited-state adiabatic dynamics simulations of BHID in S1 by using the NEWTON-X program. We tracked the bond distances of O1–H6 and O2–H6 as a function of time. This event induces an increase in the bond length of O2–H6 and a decrease in the bond length of O1–H6. The ESIPT process occurred in all of the 15 trajectories, with the H6 atom transfer time distributed in the range of 70–120 fs. In Fig. 6b, a typical trajectory displaying the time evolution of the O1–H6 and O2–H6 distances is provided. As shown, the O2–H6 bond length oscillates prior to 75 fs, and begins to increase steadily after 75 fs, indicative of the H6 atom moving away from the O2 atom. Simultaneously, the O1–H6 bond decreases, reflecting the formation of the O1–H6 bond. The theoretical calculations further confirm the presence of the ESIPT process for BHID and the formation of the stable keto-form BHID in S1, leading to the observed dual fluorescence in the experiment. These calculation results are in good agreement with those obtained based on the TDDFT//CAM-B3LYP//6-31+G(d,p) level (Fig. S11, ESI†). However, the excited-state adiabatic dynamics simulations indicate that the ESIPT process occurs at around 100 fs, which is much shorter than the experimental observation of ∼1 ps. This might be because that the real solvent environment was not considered in the simulations, in which only an IEF-PCM model approach was used to treat the solvent effect. Such an overestimated ESIPT process rate constant has been widely reported in different theoretical research studies.50,51 Nevertheless, our theoretical calculations confirm the ESIPT process which qualitatively agrees with the experimental observation.
Based on the results presented above, the excited-state dynamics of BHID-Bpin and BHID are illustrated in Fig. 7. Upon photoexcitation, the BHID-Bpin system decays to S1 minima with a time constant of about 2 ps, and relaxes to the ground state non-radiatively via MECI or radiatively via fluorescence, which gives S1 of BHID-Bpin a lifetime of 350 ps. While for the BHID system, the S1 in the enol-form can relax back to the ground state via fluorescence or changes in the keto-form conformer via the ESIPT process with a time constant of 1.3 ps. Then, the keto-form decays back to S0via fluorescence with a time-constant of about 5 ns.
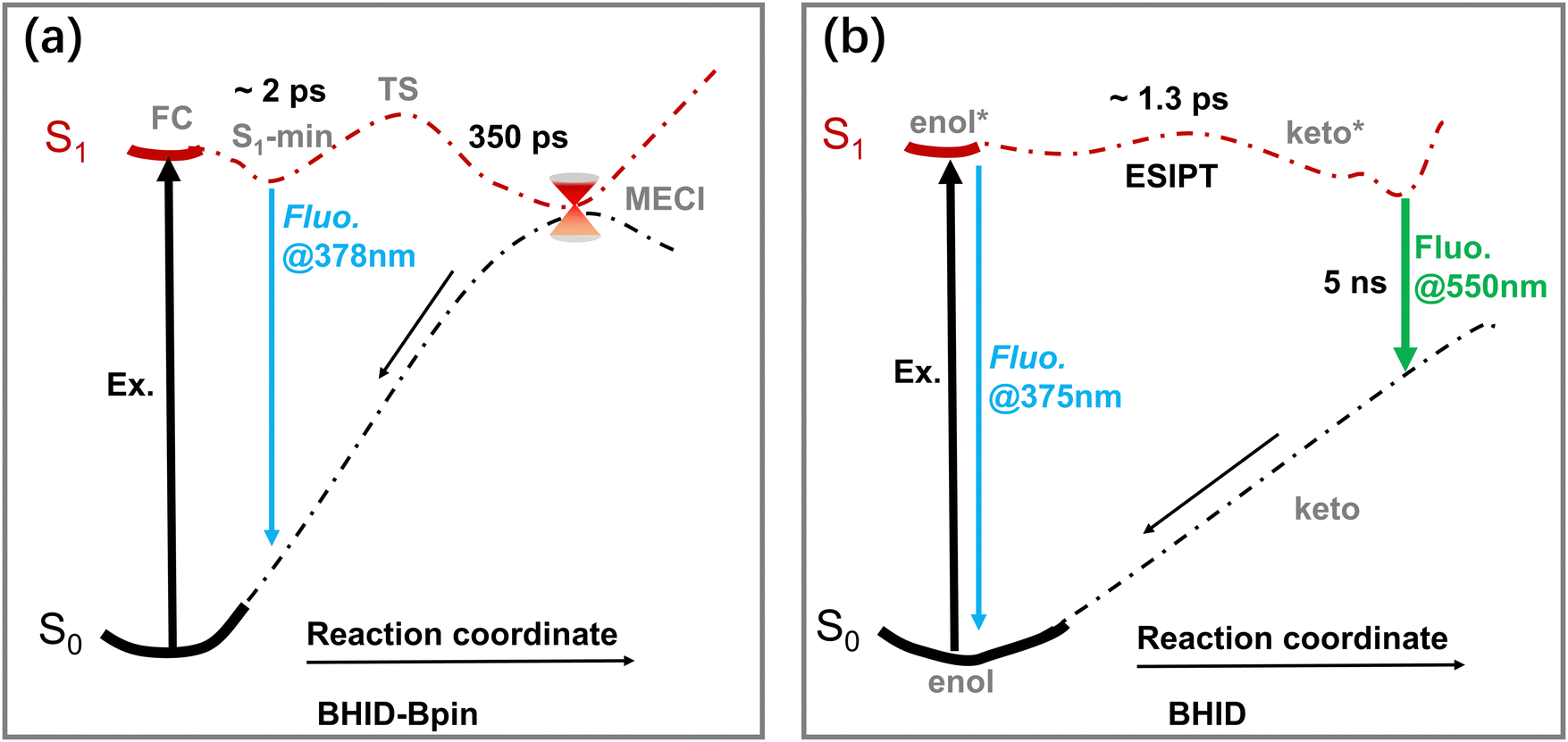 | ||
| Fig. 7 Excited-state dynamics models of BHID-Bpin (a) and BHID (b). | ||
As we mentioned above, the experimental measurements and theoretical calculations were conducted in acetonitrile. However, the peroxynitrite sensing experiment was carried out in aqueous environment. To justify, if the obtained excited-state dynamics model of BHID-Bpin and BHID in acetonitrile is applicable as they are in an aqueous environment, we conducted further computations in aqueous solutions. The absorption and fluorescence emission properties of the compounds in water (Tables S3–S5 and Fig. S12, S13, ESI†) closely match those obtained in acetonitrile, suggesting that the solvent environment does not significantly alter these properties. Our results confirm that the model obtained from the study conducted in acetonitrile is valid in aqueous solutions. Furthermore, the ESIPT process of BHID shows the same energy potential surface in acetonitrile and water (Fig. S14, ESI†). This indicates that ESIPT can occur in both environments, and this is further confirmed by the similar fluorescence peak in acetonitrile (Fig. 1b) and water (reported in ref. 30). As expected, an intermolecular complex can form between BHID and a water molecule (Fig. S15 and S16, ESI†). The presence of this intermolecular complex might influence the kinetic rate of ESIPT in different solvents.52,53 However, such a detailed study is out of the scope of the current work.
Conclusions
In conclusion, the sensing mechanism of BHID-Bpin was investigated using ultrafast optical spectroscopy and quantum chemistry calculations. The excited BHID-Bpin relaxes to its ground state via an efficient non-radiative process. After reacting with reactive oxygen/nitrogen, the Bpin moiety is cleaved and BHID is formed with a phenol group. In BHID, the ESIPT process with an energy barrier of about 90 meV can then occur at a time constant of about 1 ps, and this unique process ensures that BHID is fluorescent with dual emissions centred at 375 nm and 550 nm, respectively. We anticipate that the findings about the excited-sate dynamics for the fluorescence turn on response of BHID-Bpin to reactive oxygen/nitrogen can be valuable for the application of BHID-Bpin as a fluorescent sensor for reactive oxygen/nitrogen detection and the development of molecular fluorescent probes based on the ESIPT process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this work from the National Natural Science Foundation of China (No. 21803077) and the Novo Nordisk Foundation (NNF22OC0073582) is acknowledged. TDJ wishes to thank the University of Bath and the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University (2020ZD01) for support. YH acknowledges the support from the China Scholarship Council (No. 202006150002).References
- L. Wu, A. C. Sedgwick, X. Sun, S. D. Bull, X. P. He and T. D. James, Acc. Chem. Res., 2019, 52, 2582–2597 CrossRef CAS PubMed.
- L. C. Murfin, M. Weber, S. J. Park, W. T. Kim, C. M. Lopez-Alled, C. L. McMullin, F. Pradaux-Caggiano, C. L. Lyall, G. Kociok-Kohn, J. Wenk, S. D. Bull, J. Yoon, H. M. Kim, T. D. James and S. E. Lewis, J. Am. Chem. Soc., 2019, 141, 19389–19396 CrossRef CAS PubMed.
- J. Sun, X. Cao, W. Lu, Y. Wei, L. Kong, W. Chen, X. Shao and Y. Wang, Theranostics, 2023, 13, 1716–1744 CrossRef PubMed.
- L. Wu, J. Liu, X. Tian, R. R. Groleau, B. Feng, Y. Yang, A. C. Sedgwick, H. H. Han, Y. Wang, H. M. Wang, F. Huang, S. D. Bull, H. Zhang, C. Huang, Y. Zang, J. Li, X. P. He, P. Li, B. Tang, T. D. James and J. L. Sessler, J. Am. Chem. Soc., 2022, 144, 174–183 CrossRef CAS PubMed.
- L. Wu, J. Liu, X. Tian, R. R. Groleau, S. D. Bull, P. Li, B. Tang and T. D. James, Chem. Sci., 2021, 12, 3921–3928 RSC.
- F. Rezende, R. P. Brandes and K. Schroder, Antioxid. Redox Signaling, 2018, 29, 585–602 CrossRef CAS PubMed.
- Y. Geng, Z. Wang, J. Zhou, M. Zhu, J. Liu and T. D. James, Chem. Soc. Rev., 2023, 52, 3873–3926 RSC.
- D. Cao, Z. Liu, P. Verwilst, S. Koo, P. Jangjili, J. S. Kim and W. Lin, Chem. Rev., 2019, 119, 10403–10519 CrossRef CAS PubMed.
- H. Li, H. Kim, F. Xu, J. Han, Q. Yao, J. Wang, K. Pu, X. Peng and J. Yoon, Chem. Soc. Rev., 2022, 51, 1795–1835 RSC.
- Z. Zeng, S. S. Liew, X. Wei and K. Pu, Angew. Chem., Int. Ed., 2021, 60, 26454–26475 Search PubMed.
- G. Sivaraman, M. Iniya, T. Anand, N. G. Kotla, O. Sunnapu, S. Singaravadivel, A. Gulyani and D. Chellappa, Coord. Chem. Rev., 2018, 357, 50–104 CrossRef CAS.
- S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed.
- B. Daly, J. Ling and A. P. de Silva, Chem. Soc. Rev., 2015, 44, 4203–4211 RSC.
- R. A. Bissell, A. P. deSilva, H. Q. N. Gunaratne, P. L. M. Lynch, G. E. M. Maguire and K. R. A. S. Sandanayake, Chem. Soc. Rev., 1992, 21, 223–264 RSC.
- A. Pal, M. Karmakar, S. R. Bhatta and A. Thakur, Coord. Chem. Rev., 2021, 448, 214167 CrossRef CAS.
- S. Nagakura, J. Chem. Phys., 1995, 23, 1441–1445 CrossRef.
- H. Niu, J. Liu, H. M. O’Connor, T. Gunnlaugsson, T. D. James and H. Zhang, Chem. Soc. Rev., 2023, 52, 2322–2357 RSC.
- J. S. Chen, P. W. Zhou, S. Q. Yang, A. P. Fu and T. S. Chu, Phys. Chem. Chem. Phys., 2013, 15, 16183–16189 RSC.
- J. S. Chen, T. S. Chu, R. Z. Liu and Y. Yang, Theor. Chem. Acc., 2014, 133, 1411 Search PubMed.
- J. S. Chen, M. H. Yuan, J. P. Wang, Y. Yang and T. S. Chu, J. Phys. Chem. A, 2014, 118, 8986–8995 CrossRef CAS PubMed.
- A. C. Sedgwick, L. Wu, H. H. Han, S. D. Bull, X. P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC.
- P. Zhou and K. Han, Acc. Chem. Res., 2018, 51, 1681–1690 CrossRef CAS PubMed.
- L. Wu, C. Huang, B. P. Emery, A. C. Sedgwick, S. D. Bull, X. P. He, H. Tian, J. Yoon, J. L. Sessler and T. D. James, Chem. Soc. Rev., 2020, 49, 5110–5139 RSC.
- L. He, B. Dong, Y. Liu and W. Lin, Chem. Soc. Rev., 2016, 45, 6449–6461 RSC.
- X. Cai and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 9868–9886 CrossRef CAS PubMed.
- D. Wang and B. Z. Tang, Acc. Chem. Res., 2019, 52, 2559–2570 CrossRef CAS PubMed.
- H. Wang, Q. Li, P. Alam, H. Bai, V. Bhalla, M. R. Bryce, M. Cao, C. Chen, S. Chen, X. Chen, Y. Chen, Z. Chen, D. Dang, D. Ding, S. Ding, Y. Duo, M. Gao, W. He, X. He, X. Hong, Y. Hong, J.-J. Hu, R. Hu, X. Huang, T. D. James, X. Jiang, G.-I. Konishi, R. T. K. Kwok, J. W. Y. Lam, C. Li, H. Li, K. Li, N. Li, W.-J. Li, Y. Li, X.-J. Liang, Y. Liang, B. Liu, G. Liu, X. Liu, X. Lou, X.-Y. Lou, L. Luo, P. R. McGonigal, Z.-W. Mao, G. Niu, T. C. Owyong, A. Pucci, J. Qian, A. Qin, Z. Qiu, A. L. Rogach, B. Situ, K. Tanaka, Y. Tang, B. Wang, D. Wang, J. Wang, W. Wang, W.-X. Wang, W.-J. Wang, X. Wang, Y.-F. Wang, S. Wu, Y. Wu, Y. Xiong, R. Xu, C. Yan, S. Yan, H.-B. Yang, L.-L. Yang, M. Yang, Y.-W. Yang, J. Yoon, S.-Q. Zang, J. Zhang, P. Zhang, T. Zhang, X. Zhang, X. Zhang, N. Zhao, Z. Zhao, J. Zheng, L. Zheng, Z. Zheng, M.-Q. Zhu, W.-H. Zhu, H. Zou and B. Z. Tang, ACS Nano, 2023, 17, 14347–14405 CrossRef CAS PubMed.
- H. Gu, W. Wang, W. Wu, M. Wang, Y. Liu, Y. Jiao, F. Wang, F. Wang and X. Chen, Chem. Commun., 2023, 59, 2056–2071 RSC.
- L. Wu, X. Tian, D. J. Lee, J. Yoon, C. S. Lim, H. M. Kim and T. D. James, Chem. Commun., 2021, 57, 11084–11087 RSC.
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107–1114 CrossRef CAS.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- R. Cammi and J. Tomasi, J. Comput. Chem., 1995, 16, 1449–1458 CrossRef CAS.
- M. R. M. Barbatti and F. Plasser, et al., Newton-X: a surface-hopping, W. C. M. S. program for nonadiabatic molecular dynamics, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 26–33 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro,M. Bearpark,J. J. Heyd,E. Brothers,K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari,A. Rendell,J. C. Burant, S. S. Iyengar, J. Tomasi,M. Cossi,N. Rega,J. M. Millam,M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. A. F. Aquilante, R. K. Carlson, L. F. Chibotaru, L. D. V. M. G. Delcey, I. F. Galvan, N. Ferre, L. M. Frutos, M. G. L. Gagliardi, A. Giussani, C. E. Hoyer, G. Li, H. L. Manni, D. X. Ma, P. A. Malmqvist, T. Muller, M. O. A. Nenov, T. B. Pedersen, D. L. Peng, F. Plasser, M. R. B. Pritchard, I. Rivalta, I. Schapiro, J. Segarra, M. S. Marti, D. G. Truhlar, L. Ungur, A. Valentini, V. V. S. Vancoillie, V. P. Vysotskiy, O. Weingart, J. C. C. F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506–541 CrossRef PubMed.
- N. Alarcos, M. Gutierrez, M. Liras, F. Sanchez and A. Douhal, Phys. Chem. Chem. Phys., 2015, 17, 16257–16269 RSC.
- H. Marciniak, S. Hristova, V. Deneva, F. S. Kamounah, P. E. Hansen, S. Lochbrunner and L. Antonov, Phys. Chem. Chem. Phys., 2017, 19, 26621–26629 RSC.
- I. H. Nayyar, A. E. Masunov and S. Tretiak, J. Chem. Phys. C, 2013, 117, 18170–18189 CrossRef CAS.
- D. Jacquemin, V. Wathelet, E. A. Perpète and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420–2435 CrossRef CAS PubMed.
- M.-y Lin, Y. Li, C.-b Fu and X.-f Yu, J. Mol. Liq., 2022, 366, 120295 CrossRef CAS.
- B. K. Paul, A. Samanta and N. Guchhait, J. Mol. Struct., 2010, 977, 78–89 CrossRef CAS.
- Y. Wang and S. Yang, Chem. Phys. Lett., 2020, 761, 138024 CrossRef CAS.
- A. J. A. Aquino, F. Plasser, M. Barbatti and H. Lischhka, Croat. Chem. Acta, 2009, 82, 105–114 CAS.
- G. Yang, X. Jin, K. Chen and D. Yang, Chem. Phys. Lett., 2019, 736, 136815 CrossRef.
- S. Pijeau, D. Foster and E. G. Hohenstein, J. Phys. Chem. A, 2017, 121, 4595–4605 CrossRef CAS PubMed.
- M. C. Silfies, A. Mehmood, G. Kowzan, E. G. Hohenstein, B. G. Levine and T. K. Allison, J. Chem. Phys., 2023, 159, 104304 CrossRef CAS PubMed.
- S. Yang, Y. Zhang and K. Han, J. Lumin., 2019, 206, 46–52 CrossRef CAS.
- K. Hevalier, A. Grun, A. Stamm, Y. Schmitt, M. Gerhards and R. Diller, J. Phys. Chem. A, 2013, 117, 11233 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05777a |
| This journal is © the Owner Societies 2024 |