
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalysis of two overlapping fragmentation approaches in density matrix construction: GMBE-DM vs. ADMA†
Francisco
Ballesteros
 and
Ka Un
Lao
*
and
Ka Un
Lao
*
Department of Chemistry, Virginia Commonwealth University, Richmond, VA, USA. E-mail: laoku@vcu.edu
First published on 11th January 2024
Abstract
In this study, we conduct a comparative analysis of two density matrix construction methods: the generalized many-body expansion for building density matrices (GMBE-DM) based on the set-theoretical principle of inclusion/exclusion and the adjustable density matrix assembler (ADMA) based on the Mulliken–Mezey ansatz. We apply these methods to various noncovalent clusters, including water clusters, ion–water clusters, and ion-pair clusters, using both small 6-31G(d) and large def2-TZVPPD basis sets. Our findings reveal that the GMBE-DM method, particularly when combined with the purification scheme and truncation at the one-body level [GMBE(1)-DM-P], exhibits superior performance across all test systems and basis sets. In contrast, all ADMA set of methods show reasonable results only with small and compact basis sets. For example, GMBE(1)-DM-P outperforms the best ADMA method by at least 4 and 16 times with small and large basis sets, respectively, in the case of (H2O)N=6–55. This highlights the significance of the basis set choice for ADMA, which is even more critical than the fragmentation scheme, such as the size of subsystems, while GMBE-DM consistently produces accurate results irrespective of the chosen basis set. Consequently, the efficient and robust GMBE(1)-DM-P approach is recommended as a fragmentation method for generating accurate absolute and relative energies across different binding patterns and basis sets for noncovalent clusters.
1 Introduction
Quantum effects are pivotal in various scientific processes. However, determining the extent of their contribution is often challenging due to their high computational cost. Despite advancements in hardware and software, the bottleneck in electronic structure calculations, even the most efficient self-consistent field (SCF) approaches, often exhibits non-linear scaling. When incorporating electron correlation, the problem is more challenging, as the most simplest and economical among these is the second order Møller–Plesset Theory (MP2) with (N5) scaling of the system size N. To mitigate costs, the resolution of identity approximation for MP2 has proven successful, making the SCF step dominant on moderately sized systems.1 Although the MP2 component may become predominant eventually, many wave function-based methods still rely on SCF-derived information. Additionally, popular density functional theory (DFT) builds on the solution of the SCF equations.2,3 Thus, enhancing the efficiency of the SCF procedure is crucial to reducing computational expenses in quantum chemistry, particularly in mean-field theory.
(N5) scaling of the system size N. To mitigate costs, the resolution of identity approximation for MP2 has proven successful, making the SCF step dominant on moderately sized systems.1 Although the MP2 component may become predominant eventually, many wave function-based methods still rely on SCF-derived information. Additionally, popular density functional theory (DFT) builds on the solution of the SCF equations.2,3 Thus, enhancing the efficiency of the SCF procedure is crucial to reducing computational expenses in quantum chemistry, particularly in mean-field theory.
As the SCF procedure operates through iterative cycles until density, energy, or both converge, reducing the number of cycles is desirable. Initiating the SCF iteration from a high-quality starting point can achieve quicker convergence, minimizing computational costs.1 However, obtaining an accurate initial guess is challenging, particularly for systems with diverse chemical moieties, such as open-shell clusters and transition metal complexes. Many quantum chemistry codes default to the superposition of atomic density (SAD) guess,4 which relies on a block-diagonal density matrix (DM) derived from atomic densities. While the SAD scheme is efficient, its performance degrades with increasing system size,5 especially for those with extensive electron delocalization.
Considering the challenges mentioned, various alternative methods based on chemical fragmentation aim to provide high quality initial guesses for supersystem calculations by utilizing subsystem density matrices. These include the adjustable density matrix assembler (ADMA) approach,6–15 the molecular orbitals of the fragment molecular orbital (FMO-MO) method,16–19 the molecular fractionation with conjugate caps-assembled density matrix (MFCC-DM) method,5,20,21 the divide and conquer (DC) approach,22,23 and our approach based on the many-body expansion for building density matrices (MBE-DM).1 All these approaches follow a two-step fragmentation process: first, assembling the supersystem density matrix, and then computing a property related to that density matrix, typically the total energy.24 In contrast, the corresponding one-step fragmentation method involves assembling subsystem energies directly to reproduce the supersystem energy.24 Except for MBE-DM, the aforementioned two-step24 schemes have only been used with small and nondiffuse basis sets such as STO-3G and 6-31G(d). The inclusion of diffuse basis functions, for instance, has been shown to cause slow convergence or, in some cases, divergence of the DC SCF procedure.25 Additionally, the issue of the large diffuse basis set not only arises in two-step fragmentation approaches but also significantly degrades the performance of one-step fragmentation approaches with density embedding, such as FMO and effective FMO (EFMO), which have exhibited errors of several hundred kcal mol−1 in water clusters when using diffuse basis functions.26–28
Our group demonstrated a substantial reduction in the number of SCF convergence cycles when using the MBE-DM initial guess compared to other traditional techniques like SAD.1 Furthermore, this two-step MBE-DM fragmentation approach enables the prediction of high-accuracy energies directly without SCF iterations due to its high-quality density matrix and stability in large basis sets.1 The superior performance of MBE-DM is independent of system size/type, level of theory, and basis set selection.1 Thus, this makes MBE-DM advantageous over the aforementioned two-step fragmentation schemes, especially when dealing with large basis sets containing diffuse functions. Our group recently introduced the generalized many-body expansion for building density matrices (GMBE-DM), a two-step approach based on the set-theoretical derivation with overlapping fragments.29 This novel approach demonstrates remarkable accuracy in predicting both absolute and relative energies for noncovalent clusters, surpassing the corresponding one-step energy-based GMBE scheme by about an order of magnitude.29 Notably, GMBE-DM exhibits computational efficiency, achieving speeds approximately an order of magnitude faster than MBE-DM and is even faster than a supersystem calculation with the same computational resource, without the need for extensive parallelization to enhance the fragmentation method's efficiency.29
The ADMA approach of Mezey, which can be regarded as a non-self-consistent variant of the DC approach, is based on the Mulliken–Mezey ansatz6 for constructing the density matrix of large biomolecules. A large local environment for ADMA is essential to obtain accurate results but makes it much more time-consuming than running a supersystem calculation alone.8,9 For the protein crambin with 642 atoms using STO-3G as an example, ADMA is 67× more expensive than a supersystem calculation to achieve an energy error less than 1 millihartree (mH).8 Furthermore, an even larger buffer region for each fragment in ADMA is essential for basis sets with diffuse functions due to its Mulliken–Mezey scheme.7 Thus, it further decreases the efficiency of the ADMA approach. It should be noted that the Mulliken–Mezey scheme may also be related to a slow convergence or sometimes divergence of the DC SCF procedure when using diffuse basis functions.25 Lack of stability and efficiency in large basis sets render ADMA practically unusable for high-accuracy absolute or relative energy calculations. One final issue worth addressing is that the trace of the product of the density matrix obtained from ADMA and the overlap matrix, tr(PS), does not always yield the number of electrons of the investigated system.7,9,13,14 An empirical scaling factor for density matrix elements in ADMA was proposed to fulfill this condition7,9,14 but it will be shown in this work that it largely degrades the quality of density matrices.
In this work, we assess the performance of two two-step fragmentation approaches: our GMBE-DM method based on set theory and the traditional ADMA method relying on the empirical Mulliken–Mezey scheme. We use these approaches for noncovalent clusters with both small and large basis sets, exploring their performance with and without employing the purification scheme or a scaling factor on their density matrices. This study primarily focuses on two-step DM-based fragmentation approaches utilizing Gaussian basis sets. For insights into the performance of other fragmentation methodologies, please refer to the relevant literature, such as two comprehensive review articles in ref. 24 and 30 and the overlapping fragments method using the plane wave basis set in ref. 31. As pointed out below, GMBE-DM shows similar performance in different sizes of basis sets and performs much better than ADMA, especially with a large basis set. The good performance of GMBE-DM is because it strictly fulfills the set-theoretical principle of inclusion/exclusion (PIE) about the cardinality of sets. Furthermore, the choice of the basis set is very important for the performance of ADMA, in some cases perhaps more important than the fragmentation scheme.
2 Computational details
The GMBE algorithm32 begins with the partitioning of the system via a user defined distance cutoff. Through this cutoff, we can define the GMBE energy at the one-body level with intersecting fragments, GMBE(1), by invoking the PIE to avoid overcounting (or undercounting) using our robust and effective binning algorithm,29 | (1) |
 for fragment I is
for fragment I is | (2) |
We can also approximate the DM of the supersystem in an analogous form using the DMs of the subsystems via GMBE(1)-DM,29
 | (3) |
 for fragment I can be defined as
for fragment I can be defined as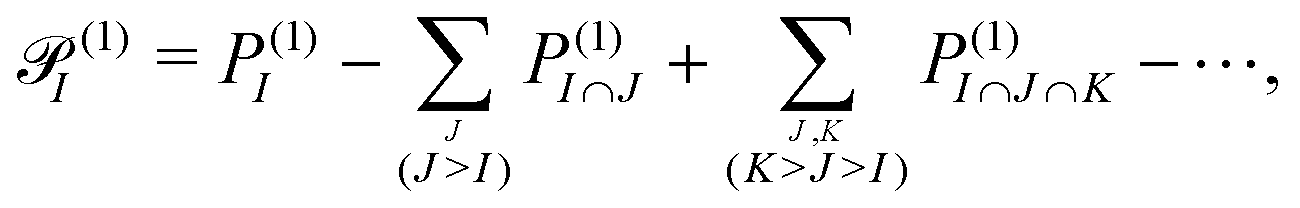 | (4) |
The ADMA method begins by constructing a series of N families where every family serves as the central family, grouping with all neighboring families within a user defined distance criterion to form a parent molecule. The DM elements of the central family within a fragment are placed in the block-diagonal position of the central family in the supersystem's DM. For the off-diagonal DM elements involving interactions between the central family with all surrounding families within a parent molecule, the components are scaled by 0.5 based on the Mulliken–Mezey scheme to avoid double counting and are placed in the corresponding off-diagonal blocks. All other non-interacting components are set to zero and therefore do not contribute to the full DM picture. This procedure is repeated for all families and eventually, the supersystem's DM with contributions from all parent molecules is obtained.6 In this way, the apparent size of the matrix is reduced, cutting the total computation time.
The further details of the GMBE-DM with the robust and efficient binning algorithm29 and the ADMA scheme6,24 can be found in their respective papers. The main procedures are summarized as follows:
1. Define the units of supersystem. For example, if the supersystem is a water hexamer (H2O)6, then each H2O molecule is considered a unit.
2. Loop over each unit to form the primitive fragment by grouping neighboring units based on a user-defined distance cutoff.
3. Apply the binning algorithm for GMBE-DM to avoid double counting of intersected fragments and generate the overall GMBE-DM recipe, ensuring adherence to the set-theoretical principle of inclusion/exclusion is satisfied. ADMA skips this step.
4. Perform SCF calculations for all ADMA subsystems from step 2 or for all GMBE-DM subsystems based on the recipe in step 3.
5. Combine the converged subsystem density matrices from step 4 according to the Mulliken–Mezey scheme for ADMA or the inclusion–exclusion principle for GMBE-DM to form the supersystem density matrix. Subsequently, either purify the density matrix if a purification scheme is employed for ADMA or GMBE-DM, or scale the density matrix by a scaling factor if a scaling is required for ADMA.
6. Build the supersystem Fock matrix from the density matrix from step 5.
7. Calculate the energy directly using the Fock matrix from step 6, the density matrix from step 5, the core Hamiltonian matrix, and the nuclear repulsion energy without the Fock diagonalization and SCF iterations.
8. Compute the energy difference between the energy obtained in step 7 and the converged supersystem SCF energy. This quantity is reported as the “error” in this work.
Since ADMA does not yield the correct trace of density matrix, which should equal the exact number of electrons for investigated systems,7,9,13,14 an empirical scaling factor has been used to offset missing or excess electrons.7,9,14 Specifically, each density matrix element is multiplied by the scalar quotient of the real number of electrons and the number obtained by tr(PS) of ADMA's density matrix. However, as shown in this work, a substantial error will be introduced for most matrix elements by using this homogeneous scaling, especially in cases where errors occur in only a small number of matrix elements. We have demonstrated that tr(PS) in (G)MBE-DM provides an exact number of electrons for investigated systems,1,29 making the empirical scaling of density matrix unnecessary.
The distance threshold of 4 Å between heavy atoms, with a maximum cardinality of 6, is used to generate subsystems for GMBE(1)-DM and ADMA calculations in this work, striking a compromise between accuracy and efficiency. Additionally, the distance threshold of 4 Å with no cardinality restriction as suggested in the original ADMA approach is also employed. To restore the fundamental properties of density matrices, such as idempotency, the one-step, non-iterative Diophantine purification method, involving matrix diagonalization, was used for ADMA.12 In this work, the iterative McWeeny purification method1,33,34 is used to purify density matrices generated by GMBE(1)-DM and ADMA with the corresponding names as GMBE(1)-DM-P and ADMA-P, respectively. ADMA-S represents the use of an empirical scaling factor to correct the trace of density matrix. The corresponding names ADMA-F, ADMA-F-P, and ADMA-F-S represent ADMA using the cardinality of all sets without restriction.
Three types of challenging noncovalent molecular clusters, including water clusters, ion–water clusters, and ion-pair clusters, were used to compare the performance and robustness of GMBE(1)-DM and ADMA. Water clusters are commonly tested for fragmentation approaches due to strong polarization and cooperative effects, even amongst smaller cluster sizes.35,36 A collection of water clusters (H2O)N=6–55, with geometries as putative global minima at the TIP4P level37 has been widely used to investigate fragmentation approaches1,29,38–40 and serves as testing systems in this work. To ensure chemical fidelity in larger systems, two large water clusters, (H2O)100 and (H2O)139 nanodrops,41,42 were used to investigate the size-dependent errors of GMBE(1)-DM and ADMA. Furthermore, ranking the relative stability of molecular cluster isomers is experimentally and computationally important, posing a challenge for many fragment-based methods.28–30,43,44 In this study, we compare the performance of GMBE(1)-DM and ADMA in ranking the relative stability of ten low-energy (H2O)20 isomers based on TIP4P structures,45 which have been studied by several fragmentation approaches.29,43,46,47 The next system type is ion–water clusters, which are even more challenging for fragmentation approaches.48–51 (H2O)30 with the ion within the cluster or at the edge of the cluster interface is used to represent ion–water clusters in this work. The charges in the system include eight anions (F−, Cl−, Br−, NO3−, H2PO4−, SCN−, CO32−, and SO42−) as well as four cations (Li+, Na+, K+, and NH4+). There are 24 ion–water clusters in total, with geometries obtained from ref. 29. The last system type is ion-pair clusters, the most challenging in this study due to the existence of both cations and anions with opposing charges throughout the cluster. Ten isomers of ethylammonium nitrate clusters, (CH3CH2NH3+)10 (NO3−)10, are considered in this work, with geometries obtained from ref. 52.
In all GMBE-DM and ADMA calculations below, each water molecule or each ion component with an integer charge is considered as a fragment. ADMA has been employed exclusively with small and nondiffuse basis sets, including STO-3G,8–10,15 3-21G,10,11 6-31G,11 6-31G(d),13 6-31G(d,p),7–9 6-311G,10,12 and 6-311G(d).10 For large and higher-quality diffuse basis sets, a very large distance cutoff is essential to generate sufficiently large parent molecules for ADMA. This accounts for the increasing influence of basis functions centered on atoms further away from the electron density of the fragment, thereby reducing the influence of the Mulliken–Mezey scheme and reproducing supersystem results. However, ADMA faces challenges with a reasonable cutoff distance for diffuse basis sets, leading to large errors and preventing the attainment of high-accuracy absolute and relative energies. In contrast, GMBE-DM employs a strict strategy based on the set-theoretical PIE, consistently performing well in both small and large basis sets. To test the basis-set dependence of ADMA and GMBE-DM, calculations will be conducted with a large def2-TZVPPD basis set and a small 6-31G(d) basis set, using the Hartree–Fock (HF) level of theory. All calculations, with SCF convergence threshold τSCF = 10−7 a.u. and integral screening threshold τints = 10−14 a.u., were performed using a locally modified version of Q-Chem.53 The calculations utilized a robust and efficient binning algorithm for generating the fragmentation recipe of GMBE, and details of the implementation can be found in ref. 29.
3 Results and discussion
3.1 Water clusters
Water clusters serve as valuable proof of concept in theoretical studies, benefiting from a plethora of benchmark works and the intricate many-body nonadditive effects that contribute to the computational complexity as the systems scale.35,54–56 In this study, we employed the energy-based GMBE(1) and the DM-based GMBE(1)-DM-(P), as well as ADMA-(F)-(S/P), to investigate (H2O)N=6–55 clusters at the HF level with 6-31G(d) and def2-TZVPPD basis sets. The results are plotted in Fig. 1 with the corresponding mean absolute errors (MAEs) shown in Table 1 and Table S1 in the (ESI†). For the small 6-31G(d) basis set, GMBE(1)-DM outperforms ADMA and ADMA-F by at least 6 and 5 times, respectively. ADMA-F exhibits slightly better performance than ADMA due to the absence of cardinality restrictions, giving subsystem sizes up to nonamer. Despite the maximum subsystem size being a hexamer in GMBE(1)-DM, it still outperforms ADMA-F, thanks to the fulfillment of the set-theoretical PIE. An interesting observation is that ADMA-(F) with a 4 Å distance threshold fails to reproduce the exact supersystem energy for (H2O)6, even though the water hexamer calculation is performed based on the ADMA-(F) recipe, which includes three hexamers, two pentamers, and one tetramer. The only way to reproduce the supersystem result with ADMA-(F) is to increase the distance threshold until all subsystems match the supersystem size. This peculiar behavior does not occur in GMBE(1)-DM, which includes only one hexamer calculation for (H2O)6, based on the set-theoretical PIE, and precisely reproduces the supersystem result. | ||
| Fig. 1 Signed error in millihartree (mH) for absolute errors of (H2O)N=6–55 clusters using various methods, including ADMA, ADMA-F, ADMA-S, ADMA-F-S, ADMA-P, ADMA-F-P, GMBE(1), GMBE(1)-DM, and GMBE(1)-DM-P with respect to the supersystem calculations at the level of HF/6-31G(d) for the upper two panels and HF/def2-TZVPPD for the lower two panels. All calculations employed a distance threshold of 4 Å between heavy atoms, with a maximum cardinality of 6, except ADMA-F, ADMA-F-S, and ADMA-F-P, where no cardinality restriction was employed. | ||
| Method | (H2O)N=6–55 | (H2O)100 | (H2O)139 | (H2O)20 | ||||
|---|---|---|---|---|---|---|---|---|
| MAE | Sign Error | Sign Error | Absolute MAE | Relative MAE | ||||
| a HF/6-31G(d). b HF/def2-TZVPPD. | ||||||||
| GMBE(1)a | 32.9 | 178.6 | 225.5 | 5.89 | 4.31 | |||
| GMBE(1)-DM-Pa | 4.1 | 12.5 | 33.5 | 0.84 | 0.15 | |||
| ADMA-Pa | 21.6 | 77.2 | 116.4 | 9.54 | 0.89 | |||
| ADMA-F-Pa | 16.6 | 77.2 | 85.4 | 6.21 | 0.95 | |||
| GMBE(1)b | 29.9 | 155.4 | 199.0 | 5.17 | 3.88 | |||
| GMBE(1)-DM-Pb | 3.7 | 14.5 | 27.2 | 0.89 | 0.15 | |||
| ADMA-Pb | 61.9 | 161.0 | 239.3 | 29.65 | 2.83 | |||
| ADMA-F-Pb | 158.5 | 161.0 | 505.8 | 31.42 | 4.82 | |||
The purification scheme significantly reduces MAEs for GMBE(1)-DM, ADMA, and ADMA-F to 4.1, 21.6, and 16.6 mH, respectively. Although both ADMA and ADMA-F outperform the energy-based GMBE(1) approach, GMBE(1)-DM-P remains vastly superior to ADMA-(F)-P. Notably, GMBE(1)-DM generates density matrices with the exact same number of electrons for the investigated systems, while ADMA and ADMA-F have up to 0.214 and 0.118 e− missing, respectively, in (H2O)N=6–55. While a homogeneous scaling factor has been proposed for ADMA-(F) to achieve correct tr(PS),7,9,14 it tends to overcorrect and leads to significant energy overestimation for water clusters in this work. In contrast, the purification scheme, which refines the idempotency property of the density matrix and ensures correct tr(PS) for water clusters, is a better scheme for refining the density matrix compared to using only a homogeneous scaling factor, which may result in a non-idempotent density by normalizing electrons missing/excess.
In the analysis employing a larger basis set, def2-TZVPPD, GMBE(1)-DM consistently performs well, with MAEs of 16.3 and 19.5 mH for small and large basis sets, respectively. It also outperforms ADMA-(F) and GMBE(1) in both basis sets. However, the performance of ADMA and ADMA-F deteriorates significantly, with a 6-fold and 7-fold decrease in accuracy when transitioning from the 6-31G(d) to the def2-TZVPPD basis set, respectively. This diminished performance is partly attributed to incorrect tr(PS), with ADMA and ADMA-F exhibiting up to 1.703 and 1.709 e− excess electrons, respectively. The discrepancies in missing or excess electrons contribute to ADMA-(F) underestimating absolute energies in 6-31G(d) while significantly overestimating them in def2-TZVPPD. A noteworthy observation is that the cardinality-unrestricted ADMA-F performs worse than ADMA with a maximum cardinality of 6. This contrasts the expected principle where a larger subset size should provide a more accurate representation of the system being partitioned. Thus, the choice of the basis set proves crucial in ADMA, surpassing the significance of the fragmentation scheme (cardinality). Even the one-step energy-based GMBE(1) approach outperforms the two-step ADMA-(F) approaches by 20 times in this large diffuse basis set. As observed in the case for the small basis set, the purification scheme enhances the performance of all DM-based approaches. However, the homogeneous scaling factor further degrades the performance of ADMA-(F). Despite only requiring subsystems of no more than six water molecules, GMBE(1)-DM-P emerges as the best fragmentation approach for water clusters as shown in Table 1 and Table S1 (ESI†), demonstrating similar MAEs for both small and large basis sets which are 4.1 and 3.7 mH, respectively. Finally, GMBE(1)-DM-P exhibits MAEs per water molecule of 0.11 and 0.10 mH in the 6-31G(d) and def2-TZVPPD basis sets, respectively, well within the threshold of “dynamical accuracy” of 0.14 mH or 0.09 kcal mol−1 or 0.37 kJ per mol per monomer.57
To further assess the robustness of the GMBE and ADMA set of methods, two large water clusters, (H2O)100 and (H2O)139, were used as test systems, and the results are presented in Table 1. Since ADMA-(F)-P performs much better than ADMA-(F) and ADMA-(F)-S, only ADMA-(F)-P is tested for both large water clusters. The absolute energies for both water clusters are underestimated by all GMBE(1), GMBE(1)-DM-P, ADMA-P, and ADMA-F-P methods. GMBE(1)-DM-P consistently performs similarly in both small and large basis sets for (H2O)100, with errors of 12.5 and 14.5 mH, respectively. These errors are at least 10 times smaller than those obtained from the energy-based GMBE(1). GMBE(1)-DM-P shows similar performance in both basis sets for (H2O)139, about 7 times better than GMBE(1). ADMA-(F)-P with 6-31G(d) performs at least two times better than ADMA-(F)-P with def2-TZVPPD. This observation further supports the notion that the ADMA set of methods only yield reasonable results by using small, nondiffuse basis sets. In both water clusters, GMBE(1)-DM-P outperforms the best ADMA method, ADMA-(F)-P. In terms of computational cost, GMBE(1)-DM-P needs 463 subsystems (68 monomers, 137 dimers, 84 trimers, 53 tetramers, 29 pentamer, and 92 hexamers) for (H2O)139. Although ADMA-P only needs 139 subsystems with size up to hexamer for (H2O)139, it performs at least 3 and 8 times worse compared to GMBE(1)-DM-P for small and large basis sets, respectively. This suggests that having additional small subsystems is crucial for satisfying PIE in GMBE(1)-DM-P to obtain accurate results. ADMA-F-P also requires 139 subsystems, with 70 subsystems larger than hexamers and sizes up to dodecamer. However, the large subsystem sizes only slightly improve its performance with the small basis set and significantly degrades its performance with the large basis set. Moreover, GMBE(1)-DM-P has demonstrated efficiency by being approximately seven times faster than the supersystem calculation in predicting energy for (H2O)139 at the level of HF/def2-TZVPPD.29 This efficiency is noteworthy, even without significant parallelization to enhance the performance of the fragmentation method. Although GMBE(1)-DM-P delivers a significantly higher-quality density matrix compared to ADMA, it performs similarly or only slightly better than ADMA in terms of SCF cycles to converge for water clusters when their respective density matrices were used as initial guesses. The sluggish SCF convergence, characterized by the oscillation behavior of the nearly convergent density matrix from GMBE(1)-DM-P, might be attributed to Pulay's direct inversion in the iterative subspace (DIIS) algorithm.58,59 This observation is consistent with the oscillatory behavior observed in DIIS using the nearly converged density matrices generated by our Grassmann interpolation scheme.60–62 Exploring a diagonalization-free procedure63 is anticipated to be an ideal approach to further expedite the SCF convergence based on this highly accurate density matrix and reduce the required SCF iterations. This synergistic approach is currently under investigation in our research group. In summary, GMBE(1)-DM-P outperforms the one-step GMBE(1) and two-step ADMA-(F)-(S/P) approaches, making it the recommended fragmentation approach for accurately and efficiently obtaining absolute energies for noncovalent clusters.
The next step is to assess the performance of GMBE and ADMA set of methods on the relative energies of ten (H2O)20 isomers, with results presented in Table 1 and Fig. 2. Notably, the energy order of the ten (H2O)20 isomers is highly dependent on the basis set, consistent with prior observations.64 Consequently, the relative energy ranking based on a small basis set may not accurately reflect the ranking for converged results. In both small and large basis sets, GMBE(1)-DM-P stands out as the only method capable of reproducing relative energy profiles for all isomers nearly indistinguishable from those obtained through supersystem calculations. The corresponding relative-energy MAE using GMBE(1)-DM-P is only 0.15 mH in both basis sets. However, GMBE(1), ADMA-P, and ADMA-F-P exhibit poor performance in predicting relative energies. Regarding the absolute energies of the ten (H2O)20 isomers, GMBE(1)-DM-P outperforms the other three approaches by at least 7 and 6 times for small and large basis sets, respectively, as shown in Table 1. Once again, ADMA-(F)-P demonstrates significantly worse performance in both absolute and relative energies when employing a large and diffuse basis set compared to a small and compact basis set. In short, GMBE(1)-DM-P consistently outperforms the ADMA set of methods in both small and large basis sets.
 | ||
| Fig. 2 Relative energies in millihartree (mH) for ten isomers of (H2O)20 at the (a) HF/6-31G(d) and (b) HF/def2-TZVPPD levels using the supersystem calculations (reference), the energy-based GMBE(1), and the DM-based GMBE(1)-DM-P, ADMA-P, and ADMA-F-P approaches. The lowest energy isomer predicted by the supersystem calculations is set to zero in both basis sets. The order of the isomer index, according to ref. 45, is based on the energy order from the supersystem calculations. | ||
3.2 Ion–water clusters and ion-pair clusters
To assess the performance of DM-based approaches on more challenging systems with increased polarization, longer-range electrostatic effects, and larger intermolecular interactions,49,65,66,67 a series of hydrated ions, (H2O)30 interacting with a single ion (eight anions and four cations) inside or outside of (H2O)30,29 were employed as testing systems. As shown in Table 2 and Table S2 in ESI† for five categories of ion–water clusters including cation only, anion only, ion inside (H2O)30, ion outside (H2O)30, and all clusters, the performance of the energy-based GMBE(1) approach, and the DM-based GMBE(1)-DM and ADMA set of methods was investigated. In both small and large basis sets, GMBE(1)-DM-P consistently outperforms GMBE(1) and ADMA-(F)-(P/S) for all five collections, except that GMBE(1) performs better than GMBE(1)-DM-P for cation-water clusters using 6-31G(d). It's noteworthy that ADMA-F-(P/S) performs significantly worse than GMBE(1)-DM-P, despite requiring subsystem sizes up to 27 bodies (out of 31 bodies in the supersystem) in (H2O)30 with H2PO4− inside, while no GMBE subsystem exceeds 6 bodies in size. In addition, GMBE(1)-DM-P exhibits consistent performance across all binding categories, with overall MAEs of 14.5 and 10.9 mH by using 6-31G(d) and def2-TZVPPD, respectively. With 6-31G(d), GMBE(1)-DM-P is at least two times better than ADMA-(F)-P. The performance widens with def2-TZVPPD, where GMBE(1)-DM-P is about one and two orders of magnitude better than ADMA-P and ADMA-F-P, respectively. The poor performance of ADMA-F-P with def2-TZVPPD is attributed to the breakdown of the purification scheme. Specifically, after purification, ADMA-(F)-P with 6-31G(d) and ADMA-P with def2-TZVPPD can give the correct tr(PS). However, the purification scheme substantially increases errors for ADMA-F with def2-TZVPPD across all categories. This discrepancy is reflected in the incorrect tr(PS) of the purified ADMA-F-P with def2-TZVPPD where F−, Cl−, Br−, SO42−, and Li+ at the edge of the (H2O)30 interface and H2PO4−, CO32−, SO42−, and K+ within (H2O)30 give extra two electrons from their ADMA-F-P density matrices. This could be explained by the fact that the McWeeny purification scheme iteratively converges to an idempotent density matrix, of which there are multiple solutions, and it is not guaranteed to obtain the correct ground state solution.68 The issue may stem from the initial guess density matrix from ADMA-F with def2-TZVPPD being significantly different from an idempotent density matrix, leading to convergence to a non-ideal solution. This problem could potentially be addressed by using the trace correcting extrapolation scheme with the correct tr(PS) as a direct constraint,69 which ensures both the idempotency condition and the correct tr(PS). However, it is not reasonable to expect that ADMA-F-P in a large basis set, even with the correct tr(PS), can outperform GMBE(1)-DM-P based on the performance of ADMA-F-P in a small basis set.| Method | Cation | Anion | Inside | Outside | Overall |
|---|---|---|---|---|---|
| a HF/6-31G(d). b HF/def2-TZVPPD. | |||||
| GMBE(1)a | 9.8 | 44.2 | 31.0 | 34.5 | 32.7 |
| GMBE(1)-DM-Pa | 16.4 | 13.3 | 16.7 | 12.0 | 14.4 |
| ADMA-Pa | 35.2 | 49.2 | 48.9 | 40.1 | 44.5 |
| ADMA-F-Pa | 22.9 | 30.9 | 29.2 | 27.3 | 28.2 |
| GMBE(1)b | 10.0 | 39.8 | 26.6 | 33.1 | 29.9 |
| GMBE(1)-DM-Pb | 8.7 | 12.0 | 12.3 | 9.4 | 10.9 |
| ADMA-Pb | 84.6 | 107.6 | 104.8 | 95.0 | 99.9 |
| ADMA-F-Pb | 1257.9 | 1331.9 | 1287.5 | 1326.9 | 1307.2 |
In the final set of systems investigated in this study, ion-pair clusters, specifically ten isomers of (CH3CH2NH3+)10 (NO3−)10, posed additional challenges for GMBE and ADMA fragmentation approaches. The MAEs at the HF/6-31G(d) level are presented in Table 3. Given the inferior performance of ADMA with def2-TZVPPD in water clusters and ion–water clusters, only the 6-31G(d) basis set was utilized in ion-pair clusters. GMBE(1)-DM-P continues to demonstrate superior performance, achieving an MAE of 32.3 mH. This result is at least 46 times better than GMBE(1) and 2 times better than ADMA-(F)-P. Notably, even with a subsystem size of up to 15 bodies, ADMA-F-P fails to provide a higher-quality description of the supersystem consisting of 20 bodies. As previously reported in our work,29 GMBE(1)-DM-P with def2-TZVPPD yields an MAE of 36.5 mH for the same ten (CH3CH2NH3+)10 (NO3−)10 isomers. The consistent performance of GMBE(1)-DM-P across small and large basis sets underscores its independence from basis set size, demonstrating its capability to deliver accurate and converged absolute and relative energies for noncovalent clusters.
| Method | Ion-pair |
|---|---|
| GMBE(1) | 1491.3 |
| GMBE(1)-DM-P | 32.3 |
| ADMA-P | 95.4 |
| ADMA-F-P | 83.9 |
4 Conclusions
In this study, we have once again demonstrated the high fidelity of the GMBE-DM protocol across various noncovalent systems, including water clusters, ion–water clusters, and ion-pair clusters, spanning a range of system sizes, complexities, and basis set sizes. Notably, GMBE(1)-DM-P consistently delivers accurate chemical representations for systems with diverse binding types at a considerably reduced computational cost compared to the supersystem calculations, a feature not achievable with the ADMA procedure. The ADMA methods, relying on the empirical Mulliken–Mezey scheme, exhibit reasonable results only with small and nondiffuse basis sets. Nevertheless, even in such cases, ADMA falls short of the accuracy achieved by GMBE(1)-DM-P, which strictly adheres to the set-theoretical PIE. As the basis sets become large and diffuse, ADMA introduces substantial errors, highlighting the critical role of basis set selection in ADMA, surpassing even the importance of the fragmentation scheme parameters such as subsystem size. Across all tested systems, GMBE(1)-DM-P consistently outperforms the ADMA set of methods, providing accurate absolute and relative energies for various basis sets and binding patterns. Hence, GMBE(1)-DM-P emerges as a highly recommended fragmentation approach for noncovalent clusters. Ongoing research in our group involves extending the GMBE-DM protocol to covalent bond systems, such as proteins, to further broaden its applicability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by start-up funds and Seed Awards from the Virginia Commonwealth University College of Humanities and Sciences. This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 using NERSC Award No. BES-ERCAP0020838. High Performance Computing resources provided by the High Performance Research Computing (HPRC) Core Facility at Virginia Commonwealth University (https://chipc.vcu.edu) were also used for conducting the research reported in this work.References
- F. Ballesteros and K. U. Lao, J. Chem. Theory Comput., 2022, 18, 179–191 CrossRef CAS PubMed.
- R. O. Jones, Rev. Mod. Phys., 2015, 87, 897–923 CrossRef.
- N. Mardirossian and M. Head-Gordon, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS.
- J. H. Van Lenthe, R. Zwaans, H. J. J. Van Dam and M. F. Guest, J. Comput. Chem., 2006, 27, 926–932 CrossRef CAS PubMed.
- X. He and K. M. Merz, J. Chem. Theory Comput., 2010, 6, 405–411 CrossRef CAS PubMed.
- P. G. Mezey, J. Math. Chem., 1995, 18, 141–168 CrossRef CAS.
- T. E. Exner and P. G. Mezey, J. Phys. Chem. A, 2002, 106, 11791–11800 CrossRef CAS.
- T. E. Exner and P. G. Mezey, J. Comput. Chem., 2003, 24, 1980–1986 CrossRef CAS PubMed.
- T. E. Exner and P. G. Mezey, J. Phys. Chem. A, 2004, 108, 4301–4309 CrossRef CAS.
- Z. Szekeres, T. Exner and P. G. Mezey, Int. J. Quantum Chem., 2005, 104, 847–860 CrossRef CAS.
- T. E. Exner and P. G. Mezey, Phys. Chem. Chem. Phys., 2005, 7, 4061–4069 RSC.
- Z. Szekeres and P. G. Mezey, Mol. Phys., 2005, 103, 1013–1015 CrossRef CAS.
- Z. Szekeres, P. G. Mezey and P. R. Surján, Chem. Phys. Lett., 2006, 424, 420–424 CrossRef CAS.
- S. Eckard and T. E. Exner, Z. Phys. Chem., 2006, 220, 927–944 CrossRef CAS.
- P. G. Mezey and Z. Antal, J. Comput. Chem., 2017, 38, 1774–1779 CrossRef CAS PubMed.
- Y. Inadomi, T. Nakano, K. Kitaura and U. Nagashima, Chem. Phys. Lett., 2002, 364, 139–143 CrossRef CAS.
- K. Tamura, T. Watanabe, T. Ishimoto, H. Umeda, Y. Inadomi and U. Nagashima, Bull. Chem. Soc. Jpn., 2008, 81, 254–256 CrossRef CAS.
- T. Watanabe, Y. Inadomi, H. Umeda, K. Fukuzawa, S. Tanaka, T. Nakano and U. Nagashima, J. Comput. Theor. Nanosci., 2009, 6, 1328–1337 CrossRef CAS.
- H. Umeda, Y. Inadomi, T. Watanabe, T. Yagi, T. Ishimoto, T. Ikegami, H. Tadano, T. Sakurai and U. Nagashima, J. Comput. Chem., 2010, 31, 2381–2388 CrossRef CAS PubMed.
- X. Chen, Y. Zhang and J. Z. H. Zhang, J. Chem. Phys., 2005, 122, 184105 CrossRef PubMed.
- X. H. Chen and J. Z. H. Zhang, J. Chem. Phys., 2006, 125, 044903 CrossRef CAS PubMed.
- W. Yang and T. Lee, J. Chem. Phys., 1995, 103, 5674–5678 CrossRef CAS.
- M. Shoji, M. Kayanuma, H. Umeda and Y. Shigeta, Chem. Phys. Lett., 2015, 634, 181–187 CrossRef CAS.
- M. S. Gordon, D. G. Fedorov, S. R. Pruitt and L. V. Slipchenko, Chem. Rev., 2012, 112, 632–672 CrossRef CAS PubMed.
- M. Kobayashi, Y. Imamura and H. Nakai, J. Chem. Phys., 2007, 127, 074103 CrossRef PubMed.
- D. G. Fedorov, L. V. Slipchenko and K. Kitaura, J. Phys. Chem. A, 2010, 114, 8742–8753 CrossRef CAS PubMed.
- D. G. Fedorov and K. Kitaura, Chem. Phys. Lett., 2014, 597, 99–105 CrossRef CAS.
- S. R. Pruitt, C. Steinmann, J. H. Jensen and M. S. Gordon, J. Chem. Theory Comput., 2013, 9, 2235–2249 CrossRef CAS PubMed.
- F. Ballesteros, J. A. Tan and K. U. Lao, J. Chem. Phys., 2023, 159, 074107 CrossRef CAS PubMed.
- J. M. Herbert, J. Chem. Phys., 2019, 151, 170901 CrossRef PubMed.
- N. Vukmirović and L.-W. Wang, J. Chem. Phys., 2011, 134, 094119 CrossRef PubMed.
- R. M. Richard and J. M. Herbert, J. Chem. Phys., 2012, 137, 064113 CrossRef PubMed.
- R. McWeeny, Rev. Mod. Phys., 1960, 32, 335–369 CrossRef.
- M. Challacombe, J. Chem. Phys., 1999, 110, 2332–2342 CrossRef CAS.
- G. A. Cisneros, K. T. Wikfeldt, L. Ojamäe, J. Lu, Y. Xu, H. Torabifard, A. P. Bartók, G. Csányi, V. Molinero and F. Paesani, Chem. Rev., 2016, 116, 7501–7528 CrossRef CAS PubMed.
- J. P. Heindel and S. S. Xantheas, J. Chem. Theory Comput., 2020, 16, 6843–6855 CrossRef CAS PubMed.
- S. Kazachenko and A. J. Thakkar, J. Chem. Phys., 2013, 138, 194302 CrossRef PubMed.
- R. M. Richard, K. U. Lao and J. M. Herbert, J. Chem. Phys., 2014, 141, 014108 CrossRef PubMed.
- R. M. Richard, K. U. Lao and J. M. Herbert, Acc. Chem. Res., 2014, 47, 2828–2836 CrossRef CAS PubMed.
- K. U. Lao, K.-Y. Liu, R. M. Richard and J. M. Herbert, J. Chem. Phys., 2016, 144, 164105 CrossRef PubMed.
- P. Miró and C. J. Cramer, Phys. Chem. Chem. Phys., 2013, 15, 1837–1843 RSC.
- ErgoSCF, 2021, https://www.ergoscf.org/xyz/h2o.php.
- K. U. Lao and J. M. Herbert, J. Chem. Theory Comput., 2018, 14, 5128–5142 CrossRef CAS PubMed.
- K.-Y. Liu and J. M. Herbert, J. Chem. Theory Comput., 2020, 16, 475–487 CrossRef PubMed.
- J. K. Kazimirski and V. Buch, J. Phys. Chem. A, 2003, 107, 9762–9775 CrossRef CAS.
- K. Wang, W. Li and S. Li, J. Chem. Theory Comput., 2014, 10, 1546–1553 CrossRef CAS PubMed.
- K. U. Lao and J. M. Herbert, J. Phys. Chem. A, 2015, 119, 235–253 CrossRef CAS PubMed.
- J. P. Heindel and S. S. Xantheas, J. Chem. Theory Comput., 2021, 17, 2200–2216 CrossRef CAS PubMed.
- K. M. Herman, J. P. Heindel and S. S. Xantheas, Phys. Chem. Chem. Phys., 2021, 23, 11196–11210 RSC.
- J. P. Heindel, K. M. Herman and S. S. Xantheas, Annu. Rev. Phys. Chem., 2023, 74, 337–360 CrossRef CAS PubMed.
- S. Schürmann, J. R. Vornweg, M. Wolter and C. R. Jacob, Phys. Chem. Chem. Phys., 2023, 25, 736–748 RSC.
- Y. Li, D. Yuan, Q. Wang, W. Li and S. Li, Phys. Chem. Chem. Phys., 2018, 20, 13547–13557 RSC.
- E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff, Z. Gan, D. Hait, P. R. Horn, L. D. Jacobson, I. Kaliman, J. Kussmann, A. W. Lange, K. U. Lao, D. S. Levine, J. Liu, S. C. McKenzie, A. F. Morrison, K. D. Nanda, F. Plasser, D. R. Rehn, M. L. Vidal, Z.-Q. You, Y. Zhu, B. Alam, B. J. Albrecht, A. Aldossary, E. Alguire, J. H. Andersen, V. Athavale, D. Barton, K. Begam, A. Behn, N. Bellonzi, Y. A. Bernard, E. J. Berquist, H. G. A. Burton, A. Carreras, K. Carter-Fenk, R. Chakraborty, A. D. Chien, K. D. Closser, V. Cofer-Shabica, S. Dasgupta, M. de Wergifosse, J. Deng, M. Diedenhofen, H. Do, S. Ehlert, P.-T. Fang, S. Fatehi, Q. Feng, T. Friedhoff, J. Gayvert, Q. Ge, G. Gidofalvi, M. Goldey, J. Gomes, C. E. González-Espinoza, S. Gulania, A. O. Gunina, M. W. D. Hanson-Heine, P. H. P. Harbach, A. Hauser, M. F. Herbst, M. Hernández Vera, M. Hodecker, Z. C. Holden, S. Houck, X. Huang, K. Hui, B. C. Huynh, M. Ivanov, Á. Jász, H. Ji, H. Jiang, B. Kaduk, S. Kähler, K. Khistyaev, J. Kim, G. Kis, P. Klunzinger, Z. Koczor-Benda, J. H. Koh, D. Kosenkov, L. Koulias, T. Kowalczyk, C. M. Krauter, K. Kue, A. Kunitsa, T. Kus, I. Ladjánszki, A. Landau, K. V. Lawler, D. Lefrancois, S. Lehtola, R. R. Li, Y.-P. Li, J. Liang, M. Liebenthal, H.-H. Lin, Y.-S. Lin, F. Liu, K.-Y. Liu, M. Loipersberger, A. Luenser, A. Manjanath, P. Manohar, E. Mansoor, S. F. Manzer, S.-P. Mao, A. V. Marenich, T. Markovich, S. Mason, S. A. Maurer, P. F. McLaughlin, M. F. S. J. Menger, J.-M. Mewes, S. A. Mewes, P. Morgante, J. W. Mullinax, K. J. Oosterbaan, G. Paran, A. C. Paul, S. K. Paul, F. Pavošević, Z. Pei, S. Prager, E. I. Proynov, Á. Rák, E. Ramos-Cordoba, B. Rana, A. E. Rask, A. Rettig, R. M. Richard, F. Rob, E. Rossomme, T. Scheele, M. Scheurer, M. Schneider, N. Sergueev, S. M. Sharada, W. Skomorowski, D. W. Small, C. J. Stein, Y.-C. Su, E. J. Sundstrom, Z. Tao, J. Thirman, G. J. Tornai, T. Tsuchimochi, N. M. Tubman, S. P. Veccham, O. Vydrov, J. Wenzel, J. Witte, A. Yamada, K. Yao, S. Yeganeh, S. R. Yost, A. Zech, I. Y. Zhang, X. Zhang, Y. Zhang, D. Zuev, A. Aspuru-Guzik, A. T. Bell, N. A. Besley, K. B. Bravaya, B. R. Brooks, D. Casanova, J.-D. Chai, S. Coriani, C. J. Cramer, G. Cserey, A. E. DePrince, R. A. DiStasio, A. Dreuw, B. D. Dunietz, T. R. Furlani, W. A. Goddard, S. Hammes-Schiffer, T. Head-Gordon, W. J. Hehre, C.-P. Hsu, T.-C. Jagau, Y. Jung, A. Klamt, J. Kong, D. S. Lambrecht, W. Liang, N. J. Mayhall, C. W. McCurdy, J. B. Neaton, C. Ochsenfeld, J. A. Parkhill, R. Peverati, V. A. Rassolov, Y. Shao, L. V. Slipchenko, T. Stauch, R. P. Steele, J. E. Subotnik, A. J. W. Thom, A. Tkatchenko, D. G. Truhlar, T. Van Voorhis, T. A. Wesolowski, K. B. Whaley, H. L. Woodcock, P. M. Zimmerman, S. Faraji, P. M. W. Gill, M. Head-Gordon, J. M. Herbert and A. I. Krylov, J. Chem. Phys., 2021, 155, 084801 CrossRef CAS PubMed.
- S. S. Xantheas, Chem. Phys., 2000, 258, 225–231 CrossRef CAS.
- Y. Chen and H. Li, J. Phys. Chem. A, 2010, 114, 11719–11724 CrossRef CAS PubMed.
- C. H. Pham, S. K. Reddy, K. Chen, C. Knight and F. Paesani, J. Chem. Theory Comput., 2017, 13, 1778–1784 CrossRef CAS PubMed.
- J. F. Ouyang and R. P. A. Bettens, J. Chem. Theory Comput., 2015, 11, 5132–5143 CrossRef CAS PubMed.
- P. Pulay, Chem. Phys. Lett., 1980, 73, 393–398 CrossRef CAS.
- P. Pulay, J. Comput. Chem., 1982, 3, 556–560 CrossRef CAS.
- J. A. Tan and K. U. Lao, J. Chem. Phys., 2023, 158, 051101 CrossRef CAS PubMed.
- J. A. Tan and K. U. Lao, J. Chem. Phys., 2023, 158, 214104 CrossRef CAS PubMed.
- J. A. Tan and K. U. Lao, Phys. Chem. Chem. Phys., 2024, 26, 1436–1442 RSC.
- A. Baldes, W. Klopper, J. Šimunek, J. Noga and F. Weigend, J. Comput. Chem., 2011, 32, 3129–3134 CrossRef CAS PubMed.
- D. Yuan, Y. Li, Z. Ni, P. Pulay, W. Li and S. Li, J. Chem. Theory Comput., 2017, 13, 2696–2704 CrossRef CAS PubMed.
- B. Hribar, N. T. Southall, V. Vlachy and K. A. Dill, J. Am. Chem. Soc., 2002, 124, 12302–12311 CrossRef CAS PubMed.
- T.-M. Chang and L. X. Dang, Chem. Rev., 2006, 106, 1305–1322 CrossRef CAS PubMed.
- Y. Marcus, Ions in Solution and their Solvation, John Wiley & Sons, 2015 Search PubMed.
- D. Bowler and M. Gillan, Comput. Phys. Commun., 1999, 120, 95–108 CrossRef CAS.
- A. M. N. Niklasson, M. Challacombe, C. J. Tymczak and K. Németh, J. Chem. Phys., 2010, 132, 124104 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional calculations and analysis. See DOI: https://doi.org/10.1039/d3cp05759c |
| This journal is © the Owner Societies 2024 |