
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing silicon-nitride formation through ammonolysis of silanes with pseudo-halide substituents†
Anil Kumar
Tummanapelli
 ab,
Yingqian
Chen
ab and
Ming Wah
Wong
*ab
ab,
Yingqian
Chen
ab and
Ming Wah
Wong
*ab
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: chmwmw@nus.edu.sg
bApplied Materials—NUS Advanced Materials Corporate Lab, 5A Engineering Drive 1, 117411, Singapore
First published on 6th January 2024
Abstract
Considering the challenges in reactivity, potential contamination, and substrate selectivity, the ammonolysis of traditional halosilanes in silicon nitride (SiN) thin film processing motivates the exploration of alternative precursors. In this pioneering study, we employed density functional theory calculations at the M06-2X/6-311++G(3df,2p) level to comprehensively screen potential pseudo-halide substituents on silane compounds as substitutes for conventional halosilanes. Initially, we investigated the ammonolysis mechanism of halosilanes, exploring factors influencing activation barriers, with the aid of frontier molecular orbital and charge density analyses. Subsequently, a systematic screening of silane substituents from group 14 to group 16 was conducted to identify pseudo-halides with low reaction barriers. Additionally, we examined the inductive effects on pseudohalide substituents. Using cluster models to represent the silicon surface validates the realistic prediction of ammonolysis barriers with a simplified model. Our findings indicate that pseudo-halide substituents from group 16, particularly those with electron-withdrawing groups, present as practical alternatives to traditional halosilanes in SiN thin film processing, including applications such as low-temperature atomic layer deposition (ALD) techniques.
1. Introduction
Ammonolysis of silane refers to the chemical reaction between silane and ammonia (NH3). This reaction can lead to the formation of amino-functionalized silanes or silazanes, which are characterized by the presence of silicon–nitrogen (Si–N) bonds.1 These resulting products have important applications in various fields, including materials science, surface modification, and the synthesis of complex organic–inorganic hybrid materials.2,3 Due to its excellent thermal, mechanical, and electrical properties, silicon nitride (SiN) has gained significant attention for its applications in thin film coatings.2,4 In the semiconductor industry, silicon nitride thin films serve as passivation and insulating layers in devices.2–4 They offer electrical insulation, protect underlying components from environmental factors, and enhance the overall performance and reliability of semiconductor devices.In recent years, atomic layer deposition (ALD) has emerged as a crucial technique for producing thin films with precise thickness control and excellent conformality.2,4 The ALD of SiN has been extensively explored using both experimental and computational studies.1–3,5 Present SiN ALD processes rely solely on plasma activation methods instead of thermal techniques. Silane precursors like SiH4, Si2H6, or Si(SiH3)4 combined with N2/NH3 plasmas are commonly used for SiN ALD.6–8 With the ongoing scaling of semiconductor devices, there is a growing demand for thin-film deposition methods that offer improved uniformity and step coverage.5 Consequently, there is a pressing need for an ALD technique capable of depositing SiN films at low temperatures while maintaining atomic-level thickness control. In this context, halosilanes are gaining prominence as favored precursors for thermal ALD processes. Among these, the ammonolysis reaction involving tetrachlorosilane and ammonia is the most commonly employed, leading to easily synthesized and characterized aminosilanes.9,10 However, the use of traditional halide-based precursors in ALD processes has posed notable challenges, including the introduction of halogen impurities and the formation of corrosive by-products.5 Addressing these difficulties, there is a concerted effort to explore innovative approaches for the development of efficient and environmentally sustainable ALD methodologies.
In response to the existing challenges, the primary objective of this theoretical investigation is to seek potential superior alternatives to traditional halide precursors, namely, pseudo-halide precursors. This approach not only tackles the issues linked to halogen impurities, but also holds the promise of boosting the overall process efficiency. For instance, it could facilitate low-temperature ALD processes. To achieve this goal, we employed mechanistic density functional theory (DFT) calculations initially to examine the ammonolysis mechanism of conventional halosilanes and scrutinize their electronic properties and reasons for high activity (Scheme 1). Subsequently, we performed systematic screening of other silane substitutions across the periodic table, spanning from group 14 to group 16 elements (Scheme 1). Through this extensive exploration of a diverse array of silane substituents, we aimed to identify promising precursors with pseudo-halide substituents that demonstrate exceptional performance and reactivity, enabling the efficient deposition of silicon nitride thin films. Finally, for a more realistic representation of the ammonolysis reaction on the silicon surface, we delved into the investigation of two cluster models. This exploration aimed to provide insights into the surface mechanism and evaluate the applicability of the simplified ammonolysis model.
 | ||
| Scheme 1 Ammonolysis of halosilanes and potential pseudo-halide derivatives. | ||
2. Computational methods
Density functional theory (DFT) calculations were performed on the ammonolysis reactions involving SiH3X (X = halide and pseudo-halide) and NH3 (Scheme 1) based on the M06-2X density functional method.11 The choice of the M06-2X functional was made due to its better treatment of non-covalent interactions and kinetics.12–16 Optimizations and frequency calculations were performed using the M06-2X functional and the 6-311G(d,p)17 basis set. All equilibrium structures were confirmed to have zero imaginary frequencies, while transition states were verified to possess exactly one imaginary frequency. Higher-level relative energies were obtained using the larger 6-311++G(3df,2p) basis set, based on the M06-2X/6-311G(d,p) optimized geometries. Calculating activation energies using a large basis set is often necessary to achieve accurate and reliable results. Therefore, we performed additional benchmark calculations for the ammonolysis of SiH4 at the CCSD(T)18/aug-cc-pVTZ19,20//M06-2X/6-311G(d,p) level. Our finding reveals a close agreement between the M06-2X/6-311++G(3df,2p) barrier (ΔE‡ = 43.2 kcal mol−1) and the more definitive value of 45.9 kcal mol−1. This justifies that our choice of the DFT functional and large basis set is an optimal balance of accuracy and computational efficiency. Unless otherwise noted, the relative free energies (ΔG298) reported in the text correspond to the M06-2X/6-311++G(3df,2p)//M06-2X/6-311G(d,p) level of theory at 298.15 K.To gain insights into the surface reaction mechanism of chlorosilane ammonolysis, we employed two distinct cluster models, namely, CM1 (Si9H13Cl) and CM2 (Si9N5H18Cl) (Scheme 2), to represent the local environment of silicon and amine-terminated silicon surfaces, respectively. It is worth noting that these CM1 and CM2 were derived from the source paper and subsequently adapted to suit the specific requirements of our current study.21 For further elaboration and detailed information, please refer to the ESI.† The structures and energies of surface reaction pathways were examined at the M06-2X/6-311++G(3df,2p)//M06-2X/6-311G(d,p) level, allowing for a direct comparison with the simplified model of ammonolysis. Our focus was particularly on understanding the alteration of the mechanism and the predicted ammonolysis barrier.
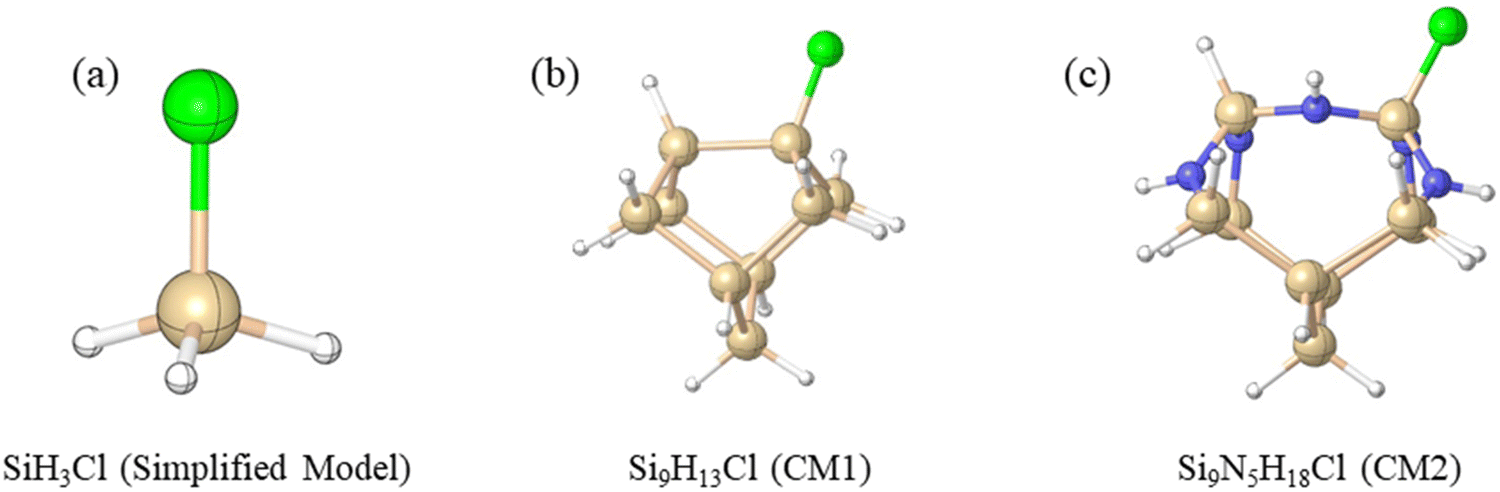 | ||
| Scheme 2 Three different theoretical models to study the ammonolysis of chlorosilane: (a) simplified model, (b) cluster model CM1, and (c) cluster model CM2. Green, blue, brown and white denote Cl, N, Si and H, respectively. | ||
All DFT calculations were performed using the Gaussian 16 suite of programs.22 Charge density analysis was performed using the natural bond orbital (NBO) approach based on the M06-2X/6-311G(d,p) wavefunction.23 Frontier molecular orbital (FMO) based HOMO–LUMO gap was also calculated to evaluate the stability and reactivity of halosilane compounds. Computed structures were illustrated using CYLView.24
3. Results and discussion
3.1 Ammonolysis mechanism of halosilanes
Our exploration commenced with an investigation into the mechanism of ammonolysis involving silanes containing various halide substituents alongside the parent silane (i.e., SiH3X, X = H, F, Cl and Br). The ammonolysis process is characterized by a concerted mechanism, facilitated by the formation of a stable zwitterionic pentavalent adduct between halosilanes and ammonia (SiH3X·NH3), as illustrated in Scheme 3. The transition state (TS) essentially involves concurrent breakage of Si–X and N–H bonds and creation of an X–H bond. The reactant adducts (axial AD1 and side AD2) are essential for the formation of the Si–N bond initially.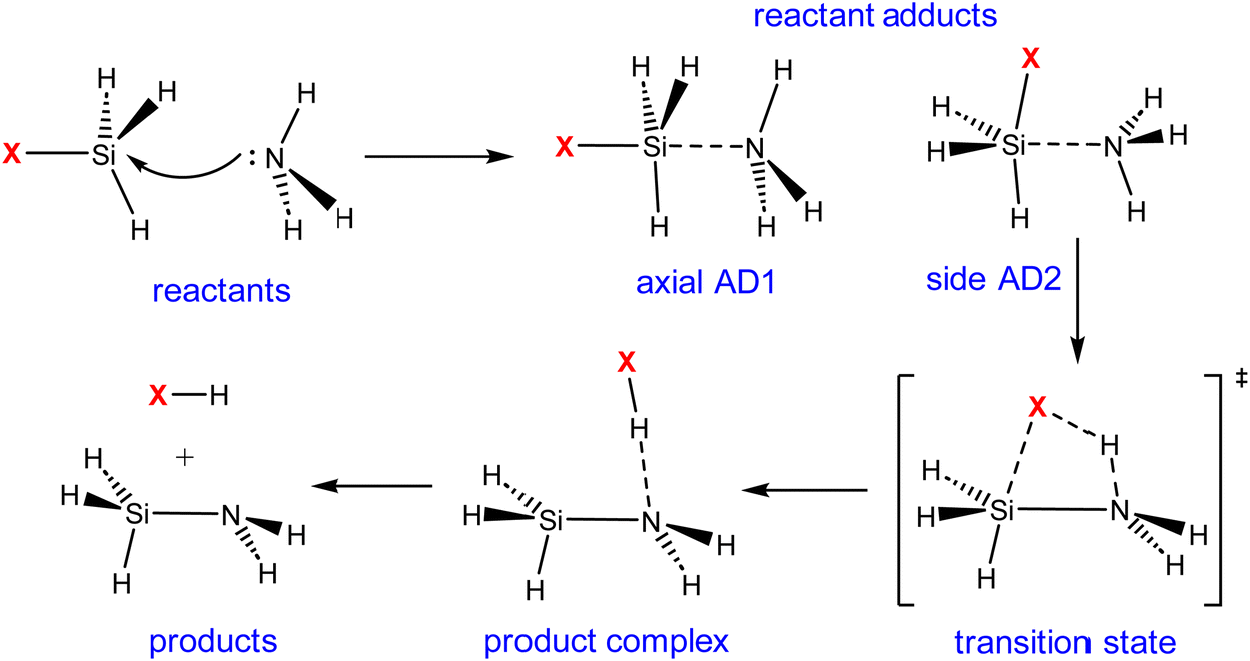 | ||
| Scheme 3 Proposed mechanism of the ammonolysis reaction between SiH3X (X = H, F, Cl, and Br) and NH3. | ||
Previous theoretical studies have focused mainly on the gas-phase reaction between silane (SiH4) and ammonia (NH3),25–42 with the most accurate predicted barrier of 49.3 kcal mol−1 at the CCSD(T)/6-311++G**//MP2/6-31G* level.41 The optimized geometries and calculated free energy reaction profiles for various ammonolysis reactions are presented in Fig. 1 and 2, respectively. While the parent reaction exhibits thermodynamic favorability (−7.5 kcal mol−1), it is characterized by a substantial barrier of 51.9 kcal mol−1. The introduction of a halogen substituent notably reduces the activation barrier. The predicted order of barriers is as follows: Br < Cl < F, with the bromine substituent demonstrating the lowest barrier at 20.6 kcal mol−1. This trend corresponds well with the electronegativity value of the halogen. We note that the 6-311++G(3df,2p) basis set is not available for the iodine atom. Hence, we opted to exclude the iodine substituent rather than using a mixed basis set to maintain consistency and avoid potential inconsistencies. Nonetheless, we calculated the barriers for the ammonolysis reaction of SiH3X (X = F, Cl, Br, and I) at the M06-2X/6-311G(d,p) level. Notably, there is a descending trend in activation barriers as we move down the halogen group, with X = F (33.9) > Cl (19.8) > Br (17.6) > I (16.9 kcal mol−1), aligning well with the expected trend (see ESI†). Intriguingly, the highest barrier occurs for the parent silane (X = H). The trend of activation energy concurs with experimental observations that thermal SiN ALD processes do not use silane precursors such as SiH4 and halosilanes (e.g., SiCl4) are the preferred precursors in such applications.43
Since the formation of a stable reactant adduct plays a crucial role in facilitating the formation of a Si–N bond, its stability is expected to be a key factor in understanding the reactivity of ammonolysis and the effect of substituents. Feng et al. have investigated the structure and stability of adducts SiH3X·NH3 (X = F, Cl, Br and I) at the G2(MP2) level of theory and demonstrated the effect of halogen atoms on the structure and stability of the pentacoordinated adduct.44 Other theoretical studies have shown that silanes can form pentacoordinated adducts with amine, arsine, phosphorus or phosphine with lone-paired electrons.45–48
Consistent with previous theoretical findings,44 there exist two distinct forms of pentacoordinated silicon adducts: axial AD1 and side AD2, as illustrated in Scheme 3. The more stable axial AD1 is characterized by NH3 approaching from the rear of the halogen, whereas the side AD2 involves a side-on approach of NH3. In both adducts, the Si–N bond formation occurs alongside the weakening of the Si–X bond (see Fig. 1). Side AD2 exhibits a notably shorter Si–N bond and a longer Si–X bond. Essentially, the geometry of side AD2 closely mirrors that of the TS. Hence, both adducts play a role in the ammonolysis reaction profile. Axial AD1 initially forms, undergoes rearrangement to become side AD2, and eventually progresses through the TS. Consistent with the trend of activation barrier, the stabilization energy of the complex aligns closely with the electronegativity of the halogen atom.
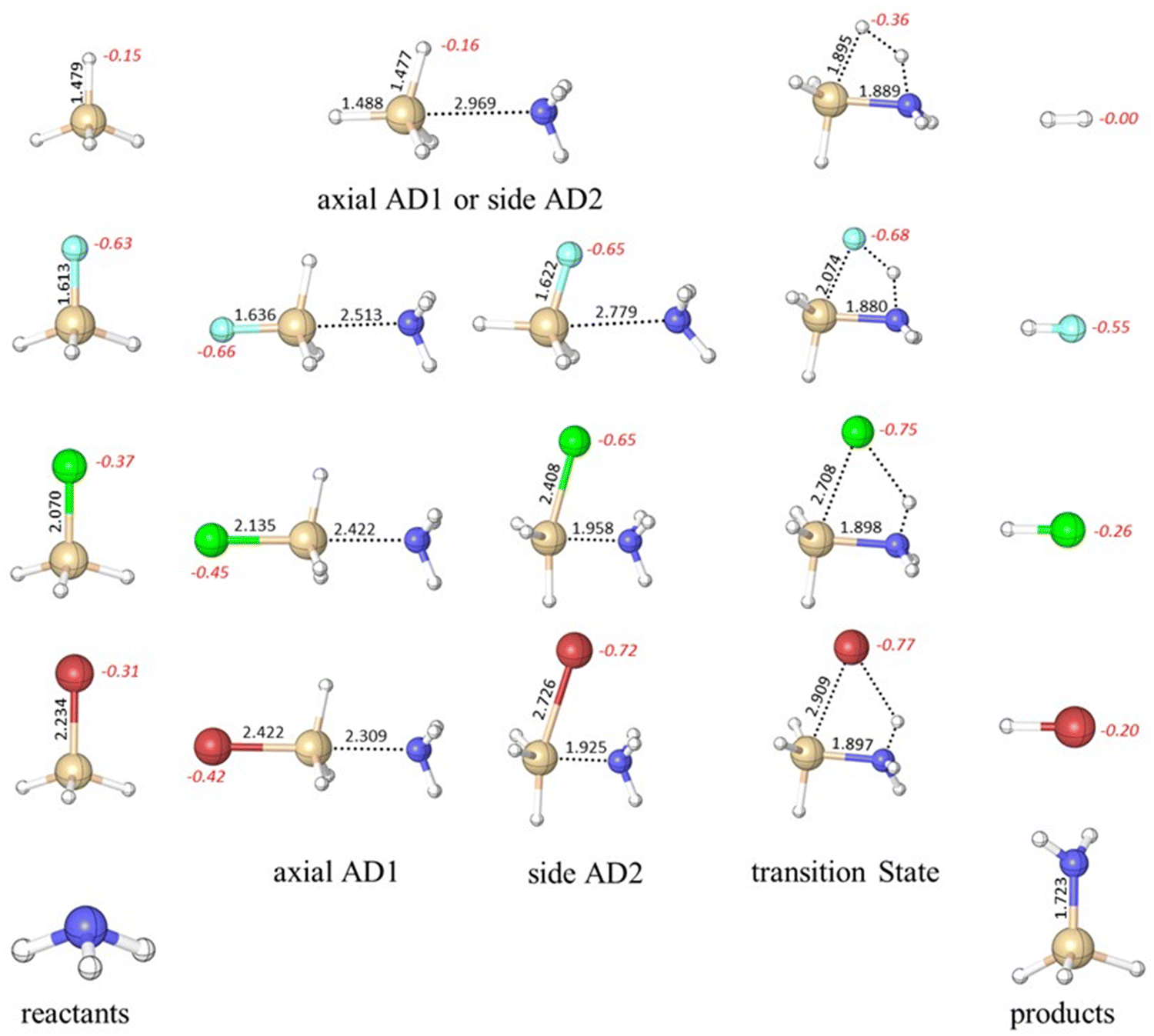 | ||
| Fig. 1 Optimized (M06-2X/6-311G(d,p)) geometries of reactants, adducts (axial AD1 and side AD2), TS, and products of the ammonolysis reaction between SiH3X (X = H, F, Cl, and Br) and NH3. Si–X and Si–N bond lengths are in Å and NBO charges on X are in italics. F, Cl, Br, N, Si, and H are represented by the colors cyan, green, red, blue, brown, and white, respectively. | ||
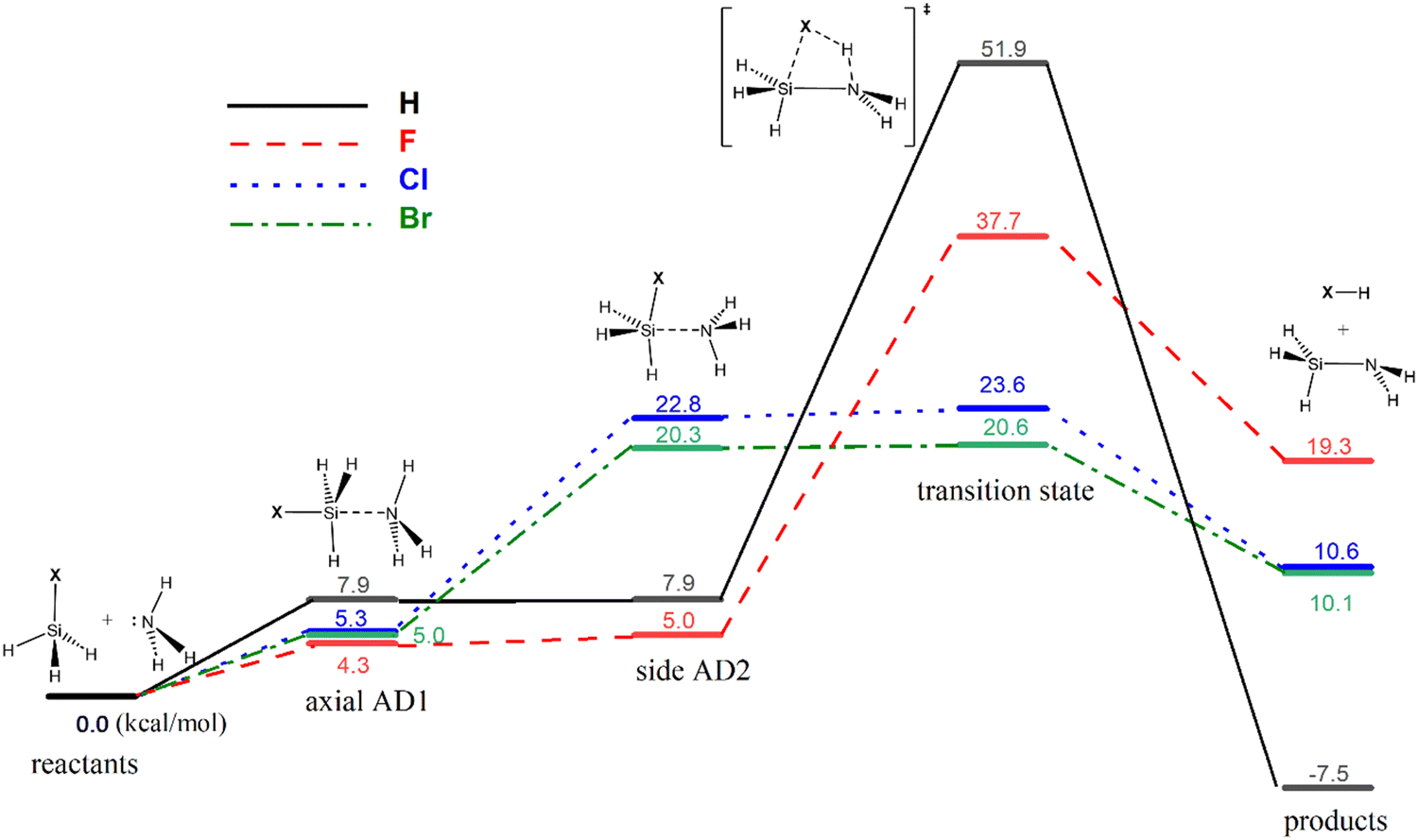 | ||
| Fig. 2 Calculated reaction free energy profiles for the ammonolysis reaction between SiH3X and NH3 for X = H, F, Cl and Br. Axial AD1 and side AD2 are identical for X = H. | ||
To assess the stability of SiH3X·NH3 complexes, a frontier molecular orbital (FMO) interaction analysis is employed. This assessment focuses on the dominant interaction of the highest occupied molecular orbital (HOMO) of ammonia (acting as the nucleophile) and the lowest unoccupied molecular orbital (LUMO) of halosilanes (acting as the electrophile). A stronger overlap between the NH3 HOMO and the SiH3X LUMO, indicated by a smaller HOMO–LUMO gap, signifies a favorable interaction and greater stability of the formed complex. Indeed, the stability of the series of halosilane adducts (axial AD1) correlates well with a smaller HOMO–LUMO gap, as shown in Table 1.
| X | ΔG298 | ΔG‡298 | HOMO–LUMO gap |
|---|---|---|---|
| H | −7.5 | 51.9 | 9.5 |
| F | 19.3 | 37.7 | 9.3 |
| Cl | 10.6 | 23.6 | 9.3 |
| Br | 10.1 | 20.6 | 9.2 |
Interestingly, the LUMO of the silane moiety exhibits Si–X antibonding character. This readily explains the weakening strength of the Si–X bond in the adduct, thereby facilitating the subsequent breaking of the Si–X bond in the TS. Essentially, the reactant complex shed light on the stability of the TS. As a result, a good correlation is also evident between the ammonolysis barrier of halosilane and the HOMO–LUMO gap (see Table 1). In summary, FMO analysis offers valuable insights for tailoring and enhancing the efficiency of ammonolysis reactions.
It is instructive to examine the charge distribution of optimized molecular structures throughout the ammonolysis reaction as it can contribute a better understanding of the reaction pathway. In this study, we employed NBO analysis to examine the atomic charge of the halogen atom throughout the entire ammonolysis process (see Fig. 1). In the case of the parent silane (X = H), the charge of the substituent remains constant from the reactant (−0.15) to the reactant complex (−0.16). However, it increases significantly in the TS to −0.36 and then decreases from TS to the product complex. This pattern suggests proton abstraction in the TS step and the subsequent formation of a stable H–H product. Remarkably, the charge on the F remained consistent throughout the entire ammonolysis process within range of −0.63 to −0.68. For SiH3Cl and SiH3Br, the charges on Cl and Br experience an increase from the reactant to the reactant complex, shifting from −0.37 to −0.45 for Cl and from −0.31 to −0.42 for Br, driven by pronounced polarization effects. This trend persists from the reactant complex to TS, where both Cl and Br atoms display heightened polarizability, facilitating proton abstraction (−0.75 for Cl and −0.77 for Br). Subsequently, a decrease in charges occurred from the TS to the product, indicating the formation of the stable H–Cl and H–Br products. Both Cl and Br substituents facilitate the abstraction of proton in the TS. The observed charge dynamics align with the mechanistic features of ammonolysis reactions, emphasizing the pivotal role of the halogen atom in the reaction path. In addition, it provides a rational basis for the design of pseudo-halide substitution.
3.2 Screening of pseudo-halides with substituents from groups 14, 15 and 16
Following a thorough exploration of ammonolysis reaction mechanisms with halide substituents (X = F, Cl, and Br), we expanded our investigation to encompass substituents ranging from group 14 to group 16, positioned adjacent to halogens. Specifically, we are examining substituents represented by X = CR3, NR2, PR2, OR, SR, and SeR, where the elements (C, N, P, O, S, and Se) display electronegativities comparable to those of halogens, with the aim of considering them as potential pseudo-halides. The objective of this screening is to identify compounds that exhibit similar reactivity to conventional halides but possess unique attributes, making them promising candidates for silicon nitride ALD.Initially, we examined the influence of R groups, particularly the inductive effects of electron-withdrawing (EWGs) and electron-donating (EDGs) groups on the ammonolysis barriers. We employed thiosilane SiH3SR variants (R = CMe3, CH3, H, F and CF3) as a case study. The results revealed that substituents with EWG such as R = CF3 display a significantly lower barrier, while those with EDG such as R = CMe3 exhibit a higher barrier compared to the parent substituent R = H (Table 2). This observation can be attributed to the fact that the EWG draws electron density from the S atom, leading to a more polarizable SR group and conversely for the EDG. As a result, a more stable zwitterionic hypervalent complex (side AD2 adduct) and corresponding TS are formed during the ammonolysis reaction for thiosilane with a EWG substituent. This stabilization effect of EWG is evident from the Si–S and Si–N bond distances in the AD2 adducts (Table 2). Intriguingly, the predicted barrier of SiH3SCF3 (21.6 kcal mol−1) is comparable to that of SiH3Br (20.6 kcal mol−1), which readily demonstrated an excellent pseudo-halide property.
| R | ΔG298 | ΔG‡298 | d(Si–S) | d(Si–N) |
|---|---|---|---|---|
| CMe3 | −1.1 | 35.4 | 2.159 | 2.862 |
| CH3 | 0.3 | 34.5 | 2.156 | 2.824 |
| H | 0.0 | 31.7 | 2.160 | 2.859 |
| F | −0.6 | 27.1 | 2.161 | 2.288 |
| CF3 | 0.4 | 21.6 | 2.571 | 1.938 |
In silico screening is extended to group-14, group-15 and group-16 substituents, specifically focusing on those with EWGs. Therefore, the choice of R groups (CR3, NR2, PR2, OR, SR, and SeR) is generally guided by the intention to modulate the reactivity of the SiH3X system. Including the halide substituents, a total of 33 substituted silanes, namely, SiH3X, where X = H, CH3, CN, CF3, CF2CN, NH2, N3, NCO, NCS, NCSe, P(CH3)2, PH2, PF2, P(CF3)2, OH, OCH3, OCN, OF, OCF3, SH, SCH3, SCMe3, SCN, SF, SCF3, SeH, SeCH3, SeCN, SeF, SeCF3, F, Cl, and Br, were examined in the investigation. The calculated ammonolysis barriers for all substituents are summarized in Fig. 3. Significant trends in ammonolysis barrier values emerge within each group, with substituents featuring electron-withdrawing groups (EWGs) displaying notably lower barrier values. Specifically, in group 14, the activation barrier trend is as follows: CN < CF2CN < CF3 < CH3. Within group 15, the trend is NCSe < NCS < P(CF3)2 < N3 < NCO < NH2 < PH2 < P(CH3)2 < PF2. As for group 16, the trend is OCN < SeCN < SCN < SeCF3 < SeF ≤ SCF3 < SeH < SF < OCF3 < OF < SeCH3 < SH < SCH3 < SCMe3 < OCH3 < OH. Chalcogen group (group 16) substituents with EWGs demonstrate barrier values similar to those of halogen group substituents, excluding fluorine. Conversely, substituents from groups 14 and 15 exhibit considerably higher activation barriers. This clearly indicates that chalcogen group substituents are the most effective pseudo-halides for ammonolysis reactions of silanes. Interestingly, X = OCN substituent is characterized by the lowest activation barrier of 17.4 kcal mol−1 among all substituents. Following OCN, among the chalcogen substituents, those containing sulfur and selenium exhibit improved predicted barriers in the sequence of SeCN < SCN < SeCF3 < SeF ≤ SCF3. To sum up, this study showcases a diverse range of potential pseudo-halide substituents, offering valuable insights for choosing superior alternatives. This knowledge significantly advances our comprehension of ammonolysis reactions and streamlines the search for optimal precursors in SiN ALD applications. A summary of computed reaction energies and barrier values for all substituents are given in Table S1 (in ESI†).
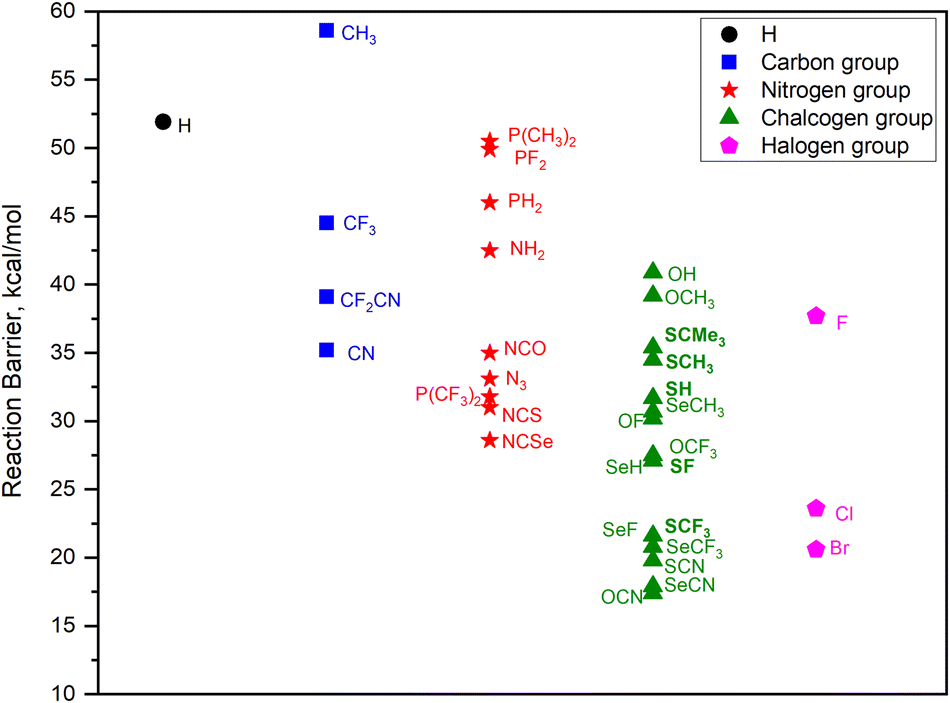 | ||
| Fig. 3 Calculated activation free energies (kcal mol−1) for ammonolysis reaction involving SiH3X with NH3, with substituents ranging from group 14 to group 17. | ||
3.3 Surface reaction mechanism using cluster models
Chlorine substituted cluster models (CM1 and CM2) were chosen to gain insights into the ammonolysis reaction on a silicon surface. The aim is to understand the underlying surface reaction mechanism and compare the reaction barrier value with that obtained from the simplified model, i.e. SiH3Cl. The schematic reaction free energy profile of ammonolysis reaction using CM1 is depicted in Fig. 4. The reactant complex of CM1 resembles the side-adduct AD2 in SiH3Cl. Notably, the CM1 model exhibits a significantly more stable reactant complex (ΔG298 = 5.5 kcal mol−1) compared to the simple model (ΔG298 = 22.8 kcal mol−1). The calculated reaction barrier for cluster model CM1 is 23.8 kcal mol−1, closely comparable to that of the simplified model (SiH3Cl, 23.6 kcal mol−1). An analogous reaction mechanism was identified for the more intricate cluster model CM2, with the reaction free energy profile given in Fig. S1 in the ESI.† The computed reaction barrier for CM2 is 27.2 kcal mol−1, aligning with both CM1 (23.8 kcal mol−1) and SiH3Cl (23.6 kcal mol−1). The reaction barrier results lend confidence in the effectiveness of the simplified ammonolysis reaction model for screening pseudo-halide alternatives. The cluster models can be employed as more realistic models to replicate real Si or SiN substrates.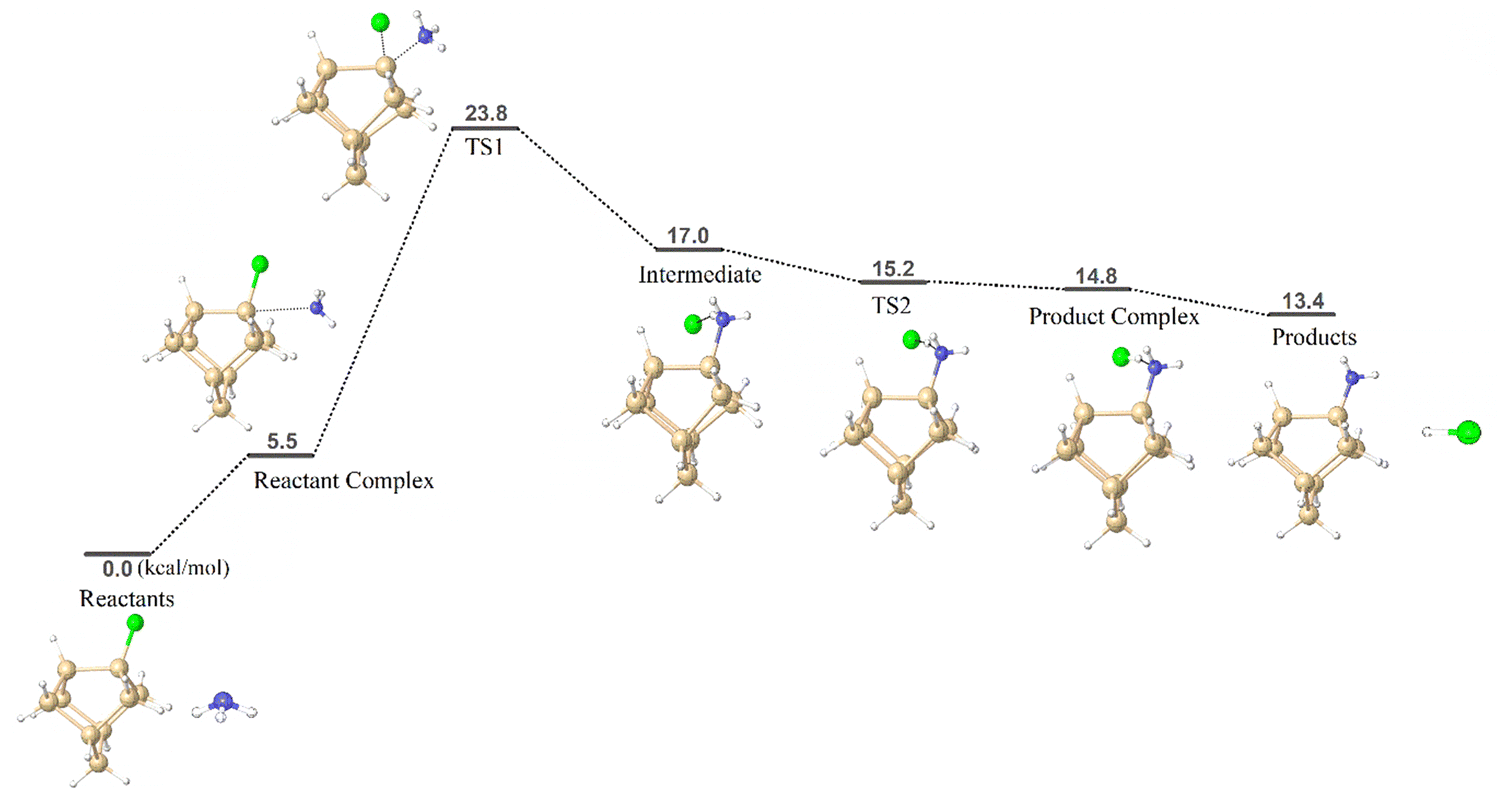 | ||
| Fig. 4 Calculated reaction free energy profile and optimized geometries of species involved in the ammonolysis reaction using cluster model CM1. | ||
4. Conclusions
In this theoretical investigation, we aimed to discover alternative precursors for traditional halosilanes in SiN film processing. Utilizing DFT calculations at the M06-2X/6-311++G(3df,2p) level, we scrutinized the ammonolysis mechanism of halosilanes (SiH3X, X = F, Cl, and Br) initially to understand the factors influencing low barriers. We then explored substituents from group 14 to group 16 to identify promising pseudo-halide candidates. Additionally, we investigated the impact of (EWGs) and (EDGs) on pseudo-halide reactivity. Employing cluster models to represent the silicon surface serves to affirm the realistic prediction of ammonolysis barriers using the simplified model. Our theoretical findings reveal that pseudo-halide substituents from group 16, particularly those with EWGs, could serve as practical alternatives to traditional halosilanes in SiN thin film processing. We believe our DFT-based study provides valuable insights into potential substitutes for halosilanes in ammonolysis reactions, paving the way for advancements in SiN film processing and semiconductor fabrication techniques, including the exploration of halide-free low-temperature ALD techniques.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by A*Star under its Applied Materials-NUS Advanced Materials Corporate Lab (ICP Ref no. I1801E0022).References
- H. Lange, G. Wötting and G. Winter, Silicon Nitride—From Powder Synthesis to Ceramic Materials, Angew. Chem., Int. Ed. Engl., 1991, 30, 1579–1597 CrossRef.
- Z. Krstic and V. D. Krstic, Silicon nitride: the engineering material of the future, J. Mater. Sci., 2012, 47, 535–552 CrossRef CAS.
- F. Ullmann and W. Gerhartz, Ullmann's Encyclopedia of Industrial Chemistry, VCH, 1986 Search PubMed.
- A. E. Kaloyeros, F. A. Jové, J. Goff and B. Arkles, Review—Silicon Nitride and Silicon Nitride-Rich Thin Film Technologies: Trends in Deposition Techniques and Related Applications, ECS J. Solid State Sci. Technol., 2017, 6, 691–714 CrossRef.
- X. Meng, Y.-C. Byun, H. S. Kim, J. S. Lee, A. T. Lucero, L. Cheng and J. Kim, Atomic Layer Deposition of Silicon Nitride Thin Films: A Review of Recent Progress, Challenges, and Outlooks, Materials, 2016, 9, 1007 CrossRef PubMed.
- S. W. King, Plasma enhanced atomic layer deposition of SiNx:H and SiO2, J. Vac. Sci. Technol., A, 2011, 29, 041501 CrossRef.
- H. Kim, H. Song, C. Shin, K. Kim, W. Jang, H. Kim, S. Shin and H. Jeon, Dielectric barrier characteristics of Si-rich silicon nitride films deposited by plasma enhanced atomic layer deposition, J. Vac. Sci. Technol., A, 2016, 35, 01A101 CrossRef.
- S. Weeks, G. Nowling, N. Fuchigami, M. Bowes and K. Littau, Plasma enhanced atomic layer deposition of silicon nitride using neopentasilane, J. Vac. Sci. Technol., A, 2015, 34, 01A140 CrossRef.
- C. Ackerhans, B. Räke, R. Krätzner, P. Müller, H. W. Roesky and I. Usón, Ammonolysis of Trichlorosilanes, Eur. J. Inorg. Chem., 2000, 827–830 CrossRef CAS.
- B. Meinel, B. Günther, A. Schwarzer and U. Böhme, Crystalline Aminoorganosilanes – Syntheses, Structures, Spectroscopic Properties, Z. Anorg. Allg. Chem., 2014, 640, 1607–1613 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals For Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, And Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, Density Functionals with Broad Applicability in Chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- L. Zhu, H. Yang and M. W. Wong, Asymmetric Nucleophilic Allylation of α-Chloro Glycinate via Squaramide Anion-Abstraction Catalysis: SN1 or SN2 Mechanism, or Both?, J. Org. Chem., 2021, 86, 8414–8424 CrossRef CAS PubMed.
- H. Yang and M. W. Wong, Application of Halogen Bonding to Organocatalysis: A Theoretical Perspective, Molecules, 2020, 25, 1045 CrossRef CAS PubMed.
- C. T. Ser, H. Yang and M. W. Wong, Iodoimidazolinium-Catalyzed Reduction of Quinoline by Hantzsch Ester: Halogen Bond or Brønsted Acid Catalysis, J. Org. Chem., 2019, 84, 10338–10348 CrossRef CAS PubMed.
- Y.-C. Ge, H. Yang, A. Heusler, Z. Chua, M. W. Wong and C.-H. Tan, Halogen-Bonding-Induced Conjugate Addition of Thiophenes to Enones and Enals, Chem. – Asian J., 2019, 14, 2656–2661 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, A Fifth-Order Perturbation Comparison of Electron Correlation Theories, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- R. A. Kendall, T. H. Dunning, Jr. and R. J. Harrison, Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- A. K. Wilson, D. E. Woon, K. A. Peterson and T. H. Dunning, Gaussian Basis Sets for Use in Correlated Molecular Calculations. IX. The Atoms Gallium through Krypton, J. Chem. Phys., 1999, 110, 7667–7676 CrossRef CAS.
- G. Fang, S. Chen, A. Li and J. Ma, Surface Pseudorotation in Lewis-Base-Catalyzed Atomic Layer Deposition of SiO2: Static Transition State Search and Born–Oppenheimer Molecular Dynamics Simulation, J. Phys. Chem. C, 2012, 116, 26436–26448 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- C. Legault, CYLview20 - Quick Guide, 2020 Search PubMed.
- H. E. O'Neal, M. A. Ring, J. G. Martin and M. T. Navio, Kinetics of Silylene Insertion into N−H Bonds and the Mechanism and Kinetics of the Pyrolysis of Dimethylsilylamine, J. Phys. Chem. A, 1998, 102, 8493–8497 CrossRef.
- C. F. Melius and P. Ho, Theoretical study of the thermochemistry of molecules in the silicon–nitrogen–hydrogen–fluorine system, J. Phys. Chem., 1991, 95, 1410–1419 CrossRef CAS.
- D. R. Meininger and B. S. Ault, Matrix Isolation Investigation of the Interaction of SiH4 with NH3 and (CH3)3N, J. Phys. Chem. A, 2000, 104, 3481–3486 CrossRef CAS.
- M. W. Schmidt, T. L. Windus and M. S. Gordon, Structural Trends in Silicon Atranes, J. Am. Chem. Soc., 1995, 117, 7480–7486 CrossRef CAS.
- A. A. Korkin, J. V. Cole, D. Sengupta and J. B. Adams, On the Mechanism of Silicon Nitride Chemical Vapor Deposition from Dichlorosilane and Ammonia, J. Electrochem. Soc., 1999, 146, 4203 CrossRef CAS.
- M. Chen, A. Zheng, H. Lu and M. Zhou, Reactions of Atomic Silicon and Germanium with Ammonia:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A Matrix-Isolation FTIR and Theoretical Study, J. Phys. Chem. A, 2002, 106, 3077–3083 CrossRef CAS.
A Matrix-Isolation FTIR and Theoretical Study, J. Phys. Chem. A, 2002, 106, 3077–3083 CrossRef CAS. - K. Yacoubi, C. Azzaro-Pantel, E. Scheid and J. P. Couderc, Analysis and Modeling of Low Pressure CVD of Silicon Nitride from a Silane-Ammonia Mixture: I. Experimental Study and Determination of a Gaseous Phase Mechanism, J. Electrochem. Soc., 1999, 146, 3009 CrossRef CAS.
- K. Yacoubi, C. Azzaro-Pantel and J. P. Couderc, Analysis and Modeling of Low Pressure CVD of Silicon Nitride from a Silane-Ammonia Mixture: II. Deposition Modeling, J. Electrochem. Soc., 1999, 146, 3018 CrossRef CAS.
- H.-J. Himmel, N. Schiefenhövel and M. Binnewies, Identification and Characterization of Monomeric, Volatile SiCl3NH2 as Product of the Reaction between SiCl4 and NH3: An Important Intermediate on the Way to Silicon Nitride?, Chem. – Eur. J., 2003, 9, 1387–1393 CrossRef CAS PubMed.
- H. Ohta, A. Nagashima, M. Hori and T. Goto, Effect of ions and radicals on formation of silicon nitride gate dielectric films using plasma chemical vapor deposition, J. Appl. Phys., 2001, 89, 5083–5087 CrossRef CAS.
- M. Yoshimoto, K. Takubo, T. Ohtsuki, M. Komoda and H. Matsunami, Deposition Mechanism of Silicon Nitride in Direct Photoassisted Chemical Vapor Deposition Using a Low-Pressure Hg Lamp, J. Electrochem. Soc., 1995, 142, 1976 CrossRef CAS.
- D. T. Murley, R. A. G. Gibson, B. Dunnett, A. Goodyear and I. D. French, Influence of gas residence time on the deposition of nitrogen-rich amorphous silicon nitride, J. Non-Cryst. Solids, 1995, 187, 324–328 CrossRef CAS.
- M. Krempp and R. Damrauer, The Gas-Phase Chemistry of Silamide Ions, Organometallics, 1995, 14, 170–176 CrossRef CAS.
- M. S. Gordon, L. P. Davis and L. W. Burggraf, The structure and stability of neutral pentacoordinated silicon compounds, Chem. Phys. Lett., 1989, 163, 371–374 CrossRef CAS.
- A. R. Rossi and J. M. Jasinski, Theoretical studies of neutral silane-ammonia adducts, Chem. Phys. Lett., 1990, 169, 399–404 CrossRef CAS.
- H. J. Emeléus and N. Miller, 175. Derivatives of monosilane. Part I. The reactions of chlorosilane with aliphatic amines, J. Chem. Soc., 1939, 819–823 RSC.
- S.-W. Hu, Y. Wang, X.-Y. Wang, T.-W. Chu and X.-Q. Liu, Gas-Phase Reactions between Silane and Ammonia: A Theoretical Study, J. Phys. Chem. A, 2003, 107, 9189–9196 CrossRef CAS.
- C. Mui, Y. Widjaja, J. K. Kang and C. B. Musgrave, Surface reaction mechanisms for atomic layer deposition of silicon nitride, Surf. Sci., 2004, 557, 159–170 CrossRef CAS.
- M. A. Hall, C. Mui and C. B. Musgrave, DFT Study of the Adsorption of Chlorosilanes on the Si(100)-2 × 1 Surface, J. Phys. Chem. B, 2001, 105, 12068–12075 CrossRef CAS.
- S. Feng, D. Feng, M. Li, Y. Zhou and P. G. Wang, Theoretical investigation on pentacoordinated silicon compounds of H3SiX (X = F, Cl, Br, I) with NH3, THEOCHEM, 2002, 618, 51–58 CrossRef CAS.
- A. B. Burg, Trimethylamine Adducts of the Chlorosilanes, J. Am. Chem. Soc., 1954, 76, 2674–2675 CrossRef CAS.
- B. J. Aylett, H. J. Emeléus and A. G. Maddock, Phosphine and arsine derivatives of monosilane, J. Inorg. Nucl. Chem., 1955, 1, 187–193 CrossRef CAS.
- H. J. Campbell-Ferguson and E. A. V. Ebsworth, Adducts formed between some halogenosilanes and the organic bases pyridine, trimethylamine, and tetramethylethylenediamine. Part I. Stoichiometry, J. Chem. Soc. A, 1966, 1508–1514 RSC.
- I. S. Ignatyev and H. F. Schaefer, Stable Hexacoordinated Neutral Complexes between Silyl Halides and Two Water or Two Ammonia Molecules: SiX4Y2 (X = H, F, Cl; Y = H2O, NH3), J. Phys. Chem. A, 2001, 105, 7665–7671 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computed reaction energies and barrier values for all substituents, individual xyz coordinate files for all calculated structures are given a compressed zip folder. See DOI: https://doi.org/10.1039/d3cp05677e |
| This journal is © the Owner Societies 2024 |