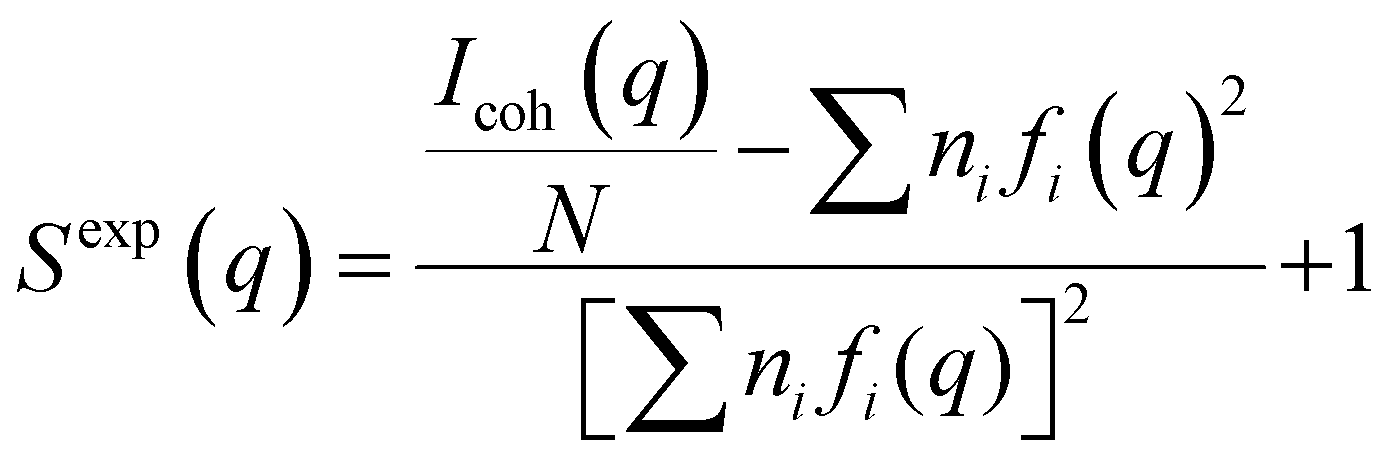A structural study on a specific Li-ion ordered complex in dimethyl carbonate-based dual-cation electrolytes†
Received
14th November 2023
, Accepted 26th December 2023
First published on 5th January 2024
Abstract
Dimethyl carbonate (DMC) is a linear carbonate solvent commonly used as an electrolyte for electric double-layer capacitors (EDLCs) and Li-ion batteries. However, there are serious problems with the use of DMC as an electrolyte solvent: (1) low ionic conductivity when using Li salts (e.g. LiBF4) and (2) liquid–liquid phase separation when using spiro-type quaternary ammonium salts (e.g. SBPBF4). Dual-cation electrolytes, i.e., bi-salt (SBPBF4 and LiBF4) in DMC, are promising candidates to avoid the phase separation issue and to enhance the total and Li+ conductivities. Herein, we reported a specific Li-ion structure in DMC-based dual-cation electrolytes by combining high-energy X-ray total scattering (HEXTS) and all-atom molecular dynamics (MD) simulations. Quantitative radial distribution function analysis based on experimental and simulation results revealed that the phase-separated SBPBF4/DMC (i.e., the bottom phase of 1 M SBPBF4/DMC) forms long-range ion ordering based on the structured SBP+–BF4− ion pairs. When adding LiBF4 salt into SBPBF4/DMC (i.e., dual-cation electrolyte), the ordered SBP+–BF4− structure disappeared owing to the formation of Li-ion solvation complexes. We found that in the dual-cation electrolyte Li ions form multiple Li+–Li+ ordered complexes in spite of relatively low Li-salt concentration (1 M), being a promising Li+-conducting medium with reduced Li salt usage and low viscosity.
Introduction
Electrical energy storage systems (EESS) with good safety and high power are critical for the efficient use of energy and management of energy supply. In addition, EESS are key technologies towards a sustainable society. Electric double-layer capacitors (EDLCs), lithium-ion batteries, and hybrid capacitors are among the energy storage devices under development for commonly used EESS.1–9 These EESS use several aprotic solvents as electrolytes to achieve the desired electrochemical performance. Some of the most employed solvents include cyclic carbonates (propylene carbonate – PC and ethylene carbonate – EC), linear carbonates (dimethyl carbonate – DMC, ethyl methyl carbonate – EMC, and diethyl carbonate – DEC), and others (acetonitrile – AN and sulfolane – SL).10–15 Among these solvents, linear carbonates such as DMC have a low viscosity (0.59 cP at 20 °C)10 and a low polarizability, resulting in a lower charge-transfer resistance of the Li+ insertion/extraction reaction (ca. 40 kJ mol−1) compared to another aprotic solvent based system (ca. 50–60 kJ mol−1).16 Thus, using a linear carbonate as a main solvent is beneficial for high power applications. However, the combination of DMC solvent and chemically stable BF4-based salts presents some critical issues: (i) using LiBF4 salts results in significantly low ionic conductivity (0.5 mS cm−1) compared to LiBF4/PC systems (3 mS cm−1).17,18 In addition, (ii) using quaternary ammonium salts (i.e., 1,1′-spiropyrrolidinium (or 5-azoniaspiro[4.4]nonane tetrafluoroborate – SBPBF4)) or ionic liquids (i.e. 1-ethyl-3-methylimidazolium tetrafluoroborate – EMIBF4) enhances the ionic conductivity (up to 15–20 mS cm−1); however, undesired liquid–liquid phase separation is a common problem in this case. For instance, 1 M SBPBF4/DMC yields phase separation into nearly pure DMC as the upper phase and 2 M SBPBF4/DMC as the bottom phase.17
Regarding the phase separation behavior, Wang et al. reported that mixing strong Lewis acid cations (e.g. Li+) might minimize this issue in a phase-separated co-solvent of water and DMC.19 Recently, we proposed that introducing a dual-cation system comprising Li salt (LiBF4) and additional electrolytic solvents (SBPBF4) can generate a stable DMC-based electrolyte solution; that is, mixing 1 M LiBF4 into 1 M SBPBF4/DMC (that is phase-separated at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol%) produces a single-phase solution with moderate–high ionic conductivity (5.9 mS cm−1) relative to a single Li salt system (1 M LiBF4/DMC).17 Moreover, the DMC-based dual-cation system showed a high Li+ conductivity of 1.4 mS cm−1 (defined as the product of total ionic conductivity and the Li+ transport number) compared to PC-based dual-cation electrolytes (0.9 mS cm−1), although both electrolytes have similar overall ionic conductivities. Owing to these improvements, the DMC-based dual-cation system achieved high-rate performance (88% capacity retention at 50 mA cm−2) in Li+-based energy storage systems.17 However, to the best of our knowledge, the origin of the phase separation behavior in SBPBF4/DMC and the high Li+ conduction in DMC-based dual-cation electrolytes is still unknown. Several studies indicated that the ionic hydrophobicity may cause such specific liquid–liquid phase separation,20 even though the detailed mechanism and solution structure are yet to be unraveled. Considering that DMC is a promising solvent for energy storage systems in high-power applications, the understanding of the origin of the phase separation behavior and ways to increase the ionic conduction is pivotal.
:1 vol%) produces a single-phase solution with moderate–high ionic conductivity (5.9 mS cm−1) relative to a single Li salt system (1 M LiBF4/DMC).17 Moreover, the DMC-based dual-cation system showed a high Li+ conductivity of 1.4 mS cm−1 (defined as the product of total ionic conductivity and the Li+ transport number) compared to PC-based dual-cation electrolytes (0.9 mS cm−1), although both electrolytes have similar overall ionic conductivities. Owing to these improvements, the DMC-based dual-cation system achieved high-rate performance (88% capacity retention at 50 mA cm−2) in Li+-based energy storage systems.17 However, to the best of our knowledge, the origin of the phase separation behavior in SBPBF4/DMC and the high Li+ conduction in DMC-based dual-cation electrolytes is still unknown. Several studies indicated that the ionic hydrophobicity may cause such specific liquid–liquid phase separation,20 even though the detailed mechanism and solution structure are yet to be unraveled. Considering that DMC is a promising solvent for energy storage systems in high-power applications, the understanding of the origin of the phase separation behavior and ways to increase the ionic conduction is pivotal.
Herein, we thus investigated the solution structure of DMC-based dual-cation electrolytes using quaternary ammonium or imidazolium salts and LiBF4via the combined high-energy X-ray total scattering (HEXTS) with all-atom molecular dynamics (MD) simulations. Based on the radial distribution function analysis, we propose a specific Li-ion ordered structure, which triggers high Li+ conduction, in the DMC-based dual-cation system. Our findings may provide a new perspective on the use of the DMC solvent and specific salts, such as Li and quaternary ammonium salts, as well as ionic liquids and other Mg2+- and Ca2+-based salts.
Experimental
Materials
LiBF4 (Kishida Chemicals), SBPBF4 (Carlit Holdings), and EMIBF4 (Kishida Chemicals) were used as electrolytic salts. DMC and PC (Kishida Chemicals) were used as solvents. All the chemicals were used as received without any post treatment or purification process. The salts and solvents were mixed in a volumetric flask in an Ar-filled glovebox (Unico, dew point: −70 °C) to obtain the single-cation (LiBF4/DMC, SBPBF4/DMC, and EMIBF4/DMC) and dual-cation (SBPBF4/DMC and EMIBF4/DMC with LiBF4) electrolytes.
Measurements
The ionic conductivity was measured using an ionic conductivity meter (Mettler Toledo, S230 and InLab 710) at 298 K. High-energy X-ray total scattering (HEXTS) measurements were carried out using high-energy X-ray diffraction apparatus (BL04B2 beamline at SPring-8, JASRI, Japan) at room temperature with monochromatized 61.6 keV X-rays by a Si (200) monochromator.21 The methodology for performing HEXTS measurements has been detailed in our previous reports.22–24 The X-ray scattering intensities were corrected for factors (absorption, polarization, and incoherent scatterings) to obtain coherent scattering intensities, [Icoh(q)]. The experimental X-ray structure factor [Sexp(q)] per stoichiometric volume and radial distribution function [Gexp(r)] were obtained according to eqn (1) and (2):| |  | (1) |
| |  | (2) |
where ni is the number of atom i, fi(q) is the atomic scattering factor of atom i, ρ0 is the density of atom i, N is the number of atoms in stoichiometric volume, and qmax is the maximum value of q (herein, 25 Å−1). All-atom molecular dynamics (MD) simulations were conducted under the isothermal–isobaric (NPT) ensemble at 298 K and 1 atm in a cubic cell. The simulation time was 10 ns for all the systems examined to be in the equilibrium state. The methodology for the MD simulations has been outlined in our previous studies.25–27 The composition of ions and solvents in the cubic box, and the resulting density (g cm−3) at the equilibrium state are listed in Table S1 (ESI†). The force field parameters and partial charges used herein are described in detail in the ESI† (Fig. S1). Using the trajectories from the MD simulations, we determined the X-ray-weighted S(q) and G(r) functions [SMD(q) and GMD(r), respectively] to compare with the experimental functions; the details are described in the ESI.† Density functional theory (DFT) calculations were performed using Gaussian 09 software.28 The optimized geometries of the Li-ion complexes, i.e., [Li(BF4)n(DMC)m]1−n, were obtained via the calculation at the B3LYP/6-311** level, followed by normal frequency analysis. The binding energy (DEbind) for the Li(BF4)n(DMC)m complex was calculated as the self-consistent field (SCF) energy difference between the complex and its individual components (Li+, BF4−, and DMC) according to the following relation: DEbind = ESCF(complex) − mESCF(BF4) − nESCF(DMC), which was corrected by the basis set superposition error using the counterpoise method.29
Results and discussion
Phase behavior of DMC-based electrolytes
Fig. 1 shows the phase behavior of the single-cation (LiBF4/DMC, SBPBF4/DMC, EMIBF4/DMC) and the dual-cation (LiBF4/DMC with SBPBF4 or EMIBF4) electrolytes. In general, the DMC-based electrolytes with LiX salts (X: Cl, BF4, PF6, etc.) yield a stable single-phase solution as shown in Fig. 1a (left);17,30 however, when using the quaternary ammonium salt (SBPBF4) or imidazolium-based ionic liquid (EMIBF4) as an alternative to Li salt, phase separation into upper and bottom phases occurred (Fig. 1a, center and right). In 1 M SBPBF4/DMC and 1 M EMIBF4/DMC, the upper phase was a nearly pure DMC solvent and thus exhibits an extremely low ionic conductivity (0.002–0.008 mS cm−1), whereas the bottom phase was a salt-rich solution (ca. 2 M) that exhibits high ionic conductivity (σ = 16–17 mS cm−1). This phase separation was particularly stable, as it settles to a partitioned state even after shaking the mixture. The resulting solutions have different densities in each phase (upper: 1.066 g cm−3, bottom: 1.155 g cm−3, in 1 M SBPBF4/DMC) according to our previous work.17 We found that 1 M SBPBF4/DMC turns to a single-phase solution when adding LiBF4 salt to the phase-separated system as shown in Fig. 1b. For example, the addition of 0.4 M LiBF4 salt yielded a 9:16 vol% phase-separated solution (Fig. 1b, left): nearly pure DMC (upper phase, σ = 0.008 mS cm−1) and 1 M SBPBF4/DMC with 0.6 M of LiBF4 (bottom phase, σ = 16 mS cm−1). Further increasing LiBF4 concentration up to 1 M made it a complete single-phase solution (Fig. 1b, center), i.e., 1 M SBPBF4/DMC containing 1 M of LiBF4: σ = 5.9 mS cm−1. A similar behavior was observed in the 1 M EMIBF4/DMC with 1 M LiBF4 system (Fig. 1b, right). The mixture of LiBF4 and SBPBF4 or EMIBF4 in DMC exhibited high ionic conductivity (σ = 5.9–6.3 mS cm−1), which was 10 times higher than that in the single Li salt solution (1 M LiBF4/DMC: 0.5 mS cm−1). In addition, compared to the conventional PC-based dual-cation system (1 M SBPBF4/PC with 1 M LiBF4), the DMC-based system offers a higher Li+ transference number (tLi+, PC: 0.17, DMC: 0.23) and Li+ conductivity (PC: 0.9 mS cm−1, DMC: 1.4 mS cm−1), even though both electrolytes without SBPBF4 have similar ionic conductivity (ca. 6 mS cm−1).17 To obtain the detailed structures of ions in the DMC-based electrolytes (particularly the Li-ion solvation structure), we conducted a combined HEXTS with MD simulations.
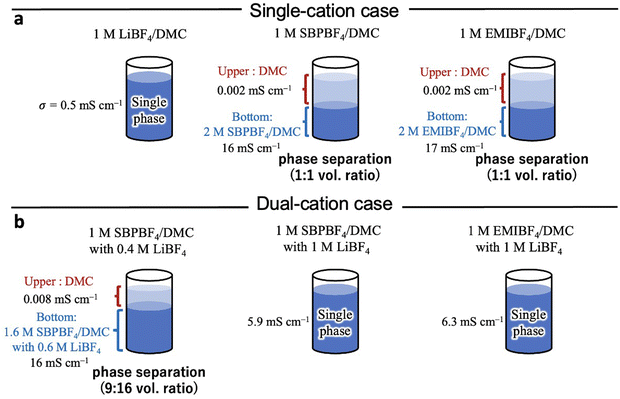 |
| | Fig. 1 Liquid phase behaviors and ionic conductivities (σ) of (a) the single-cation (1 M LiBF4/DMC, 1 M SBPBF4/DMC, 1 M EMIBF4/DMC) and (b) the dual-cation (1 M SBPBF4/DMC with 0.4 or 1 M LiBF4, 1 M EMIBF4/DMC with 1 M LiBF4) electrolytes. Note that 1 M SBPBF4 and 1 M SBPBF4/DMC with 0.4 M LiBF4 showed phase separation at 1:1 and 9:16 volume ratios, respectively. Detailed physicochemical parameters (ionic conductivity, viscosity, density, and concentration) were described in our previous report.17 | |
Ion solvation structure in DMC-based electrolytes
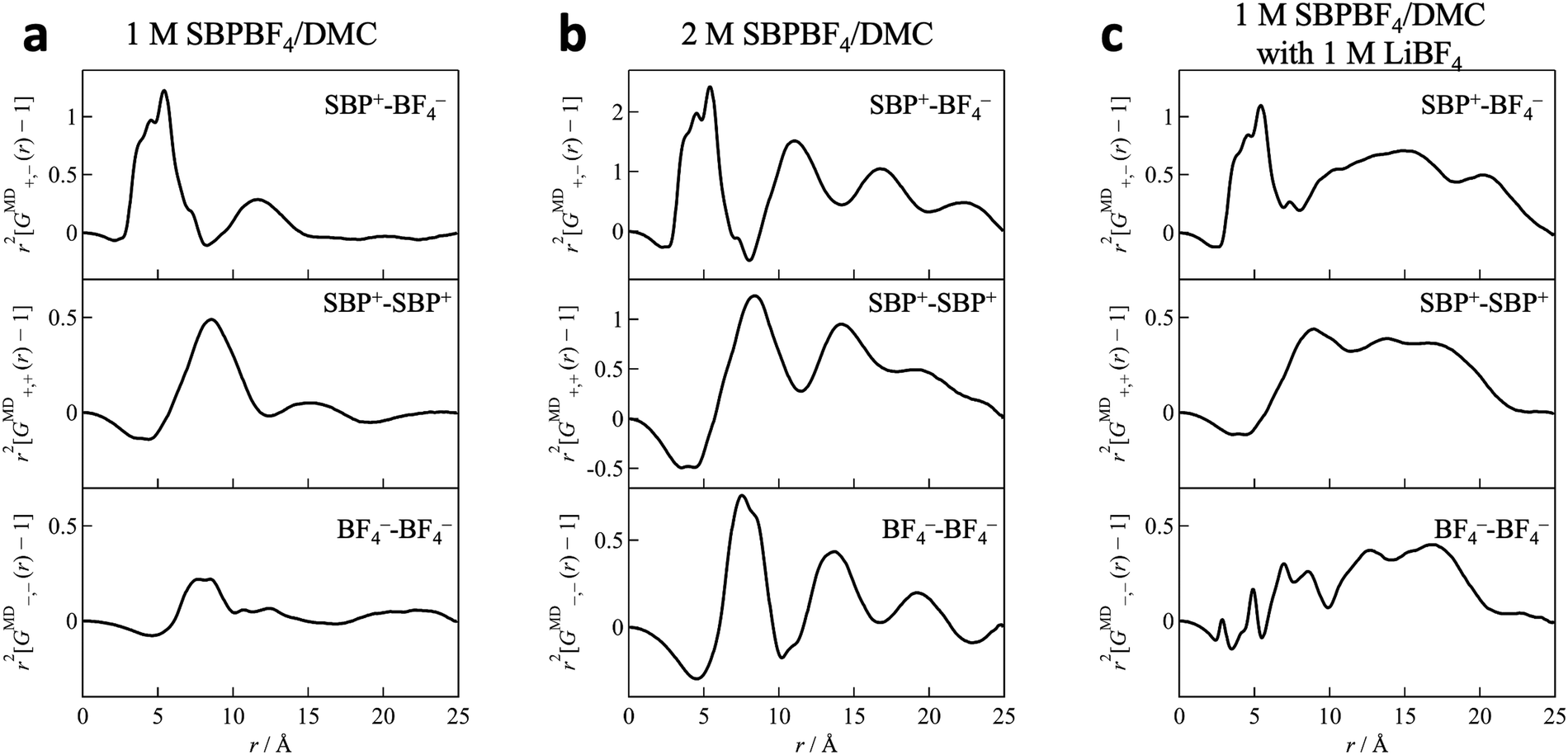
Fig. 2 shows the X-ray structure factor S(q) and its Fourier transform G(r) as an r2-weighted difference form (r2[G(r) − 1]) obtained from HEXTS measurements and MD simulations for 1 M SBPBF4/DMC solution with LiBF4 salt (cLi = 1 M). It is clear that in both S(q) and G(r) the simulated results (shown with solid red lines in Fig. 2) represent the HEXTS experimental values (open black circles). This strongly suggested that the force field parameters used in the current MD simulations are reasonable for describing the solution structure at the molecular level. Additional HEXTS and MD simulation results (1 M LiBF4/DMC, 2 M SBPBF4/DMC, and the bottom phase of 1 M SBPBF4/DMC with 0.4 M LiBF4) are provided in the ESI† (Fig. S2–S4). To obtain more insights into the solution structure, we divided total GMD(r) values into the respective contributions of cation–cation (SBP+–SBP+; r2[GMD+,+(r) − 1]), anion–anion (BF4−–BF4−; r2[GMD−,−(r) − 1]), and cation–anion (SBP+–BF4−; r2[GMD+,−(r) − 1]) interactions, which are shown in Fig. 3. Here, note that we performed the MD simulations for the 1 M SBPBF4/DMC system as a model system, which is a phase-separated solution (nearly pure DMC and 2 M SBPBF4/DMC phases) in actual solution, to gain fundamental knowledge of the solvated SBP cations. In all systems, we observed a sharp peak around 5 Å in the r2[GMD+,−(r) − 1] function, corresponding to nearest-neighbor cation–anion interactions. This indicates that the SBP cations are electrostatically interacted with BF4 anions to form ion pairs in DMC-based solutions, irrespective of SBPBF4 salt concentration. In the long r-range (>5 Å), the 2 M SBPBF4/DMC system showed several subsequent peaks for cation–anion correlations at ∼11 Å, ∼17 Å, and ∼22 Å (Fig. 3b, top); however, only the second neighbor peak (∼11 Å) was found in the 1 M SBPBF4/DMC system (Fig. 3a, top). This result suggests that structuredness in solutions based on the SBP+–BF4− ion pairs depends on the salt concentration, i.e., concentrated SBPBF4 salt in DMC-based solution produces a highly ionic ordered structure, like ionic liquids,31,32 which is discussed in detail in the ESI† (long-range ordering in the EMIBF4 ionic liquid; Fig. S5). We note that in the dual-cation case (1 M SBPBF4/DMC with 1 M LiBF4; Fig. 3c) the long-range ion–ion ordering disappeared by adding LiBF4 salt. This result implies that the structured SBP+–BF4− ion-pairs are ruptured owing to the formation of Li-ion solvation complexes, which is discussed in a later section. We expected that this ordered structure in SBPBF4/DMC solution is related to the poor dissolution capability of SBPBF4 salts, in other words the poor solvation power of the DMC solvent molecule, originating from the bulkiness and low polarization of the SBP cation and the low dielectric constant of DMC (3.1).10 Indeed, the aforementioned dissolution process including phase separation does not occur in other linear carbonates (EMC and DEC) and cyclic carbonates (PC and EC) with high dielectric constants (60–90).10,20,33,34
 |
| | Fig. 2 (a) X-ray structure factor S(q) and (b) radial distribution function in the difference form, r2[G(r) − 1], obtained from the HEXTS measurements (open black circles) and the MD simulations (solid red lines) for 1 M SBPBF4/DMC with 1 M LiBF4. | |
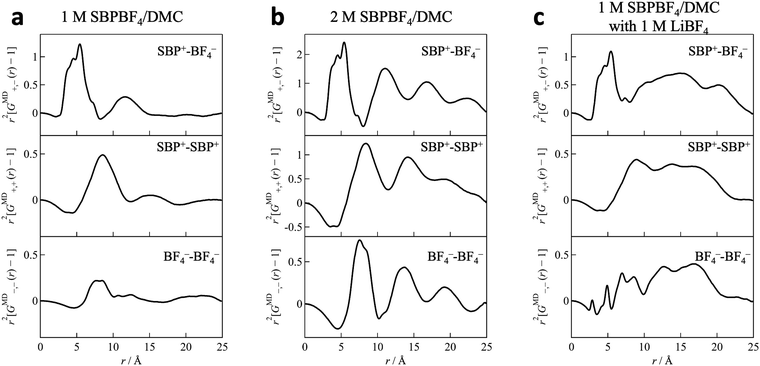 |
| | Fig. 3 Partial distribution functions for cation–cation (SBP+–SBP+; r2[GMD+,+(r) − 1]), anion–anion (BF4−–BF4−; r2[GMD−,−(r) − 1]), and cation–anion (SBP+–BF4−; r2[GMD+,−(r) − 1]) correlations in (a) 1 M SBPBF4/DMC, (b) 2 M SBPBF4/DMC, and (c) 1 M SBPBF4/DMC with 1 M LiBF4 solutions. Note that the single-phase solution of (a) 1 M SBPBF4/DMC can be obtained only in the current model simulation because its actual solution shows a phase separation into neat DMC and 2 M SBPBF4/DMC at 1:1 vol%. | |
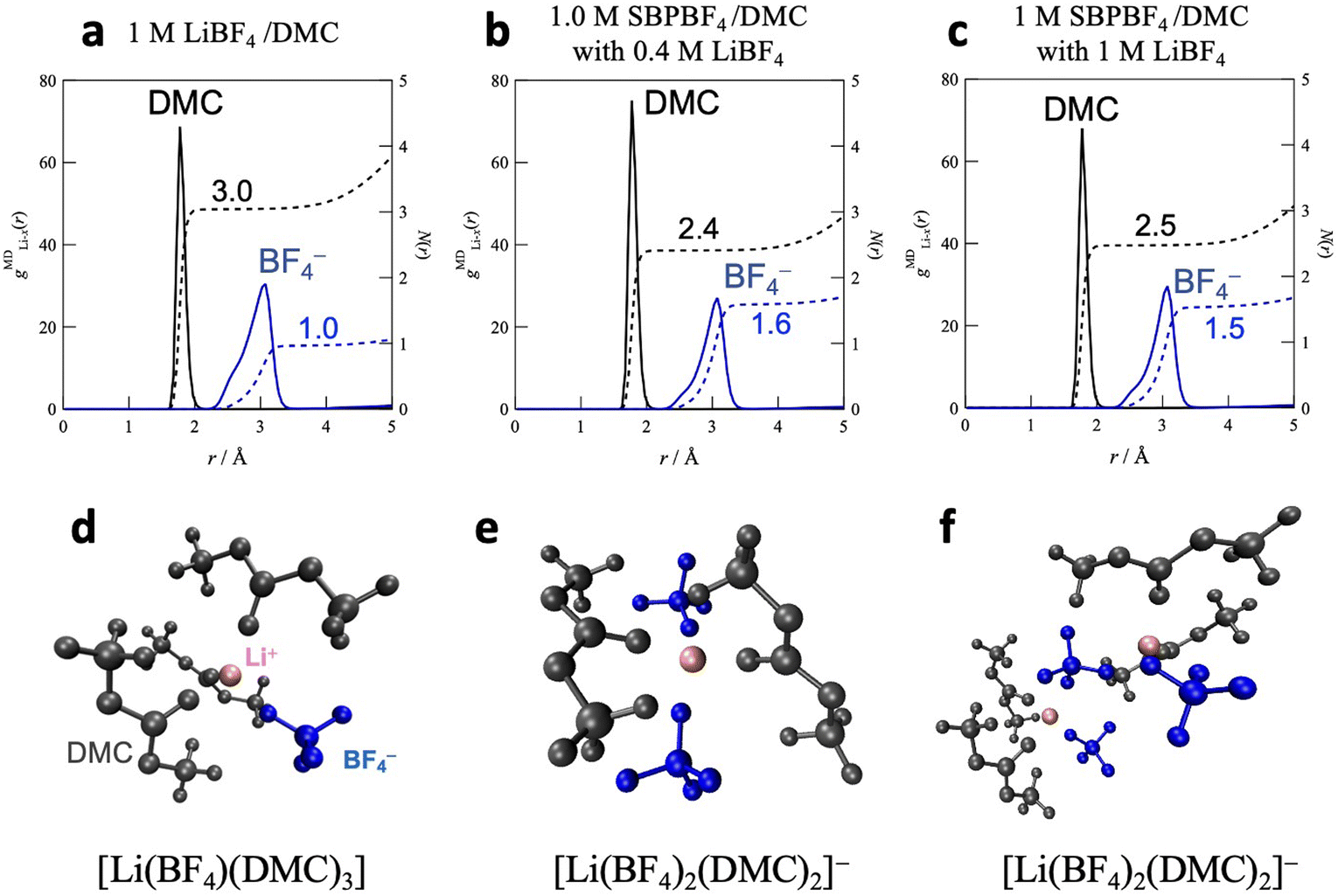
Fig. 4 shows the atom–atom pair correlation functions, gMDX–Y(r), for the O atoms (DMC) and F (BF4−) atoms around the Li ions in the DMC-based dual-cation systems, 1 M SBPBF4/DMC with LiBF4 (cLi = 0.4 and 1 M), together with the single-cation solution, 1 M LiBF4/DMC. In all electrolytes, nearest neighbor interactions for Li–DMC (Li–O) and Li–BF4 (Li–B) appeared at 1.9 and 3.0 Å, respectively, which originated from the first solvation sphere of Li ions. The average coordination numbers N(r) were calculated by integrating the corresponding gMDX–Y(r) values up to a given r: ∼2.1 Å and ∼3.6 Å for Li–DMC and Li–BF4 systems, respectively. In 1 M LiBF4/DMC (Fig. 4a), the N(r) values are 3.0 and 1.0 for Li–DMC and Li–BF4 interactions, respectively, suggesting that the Li ions are coordinated with three DMC molecules and one BF4 anion to form the charge-neutral Li-ion complex [Li(BF4)1(DMC)3] (Fig. 4d). When LiBF4 coexisted with SBPBF4 in the dual-cation electrolytes (Fig. 4b and c), the coordination number of DMC decreased to 2.4–2.5, while that of the BF4 anion increased to 1.5–1.6, irrespective of the Li salt concentration, indicating the formation of the charged [Li(BF4)2(DMC)2]− complex as an average structure (Fig. 4e and f). This result is consistent with the Li-ion coordination structure determined based on quantitative analysis of Raman spectra as reported in our previous work,17 that is, [Li(BF4)1(DMC)3] in 1 M LiBF4/DMC and [Li(BF4)2(DMC)2]− in 1 M SBPBF4/DMC with 0.4–1 M LiBF4. We thus concluded that the SBPBF4 salt triggers a change in the Li ion complexes from the neutral to the negatively charged state in DMC-based solutions. This may lead to a higher ionic conductivity in the dual-cation electrolyte (σ = 5.9 mS cm−1) compared with the single-cation LiBF4/DMC (σ = 0.5 mS cm−1), as mentioned above (Fig. 1).
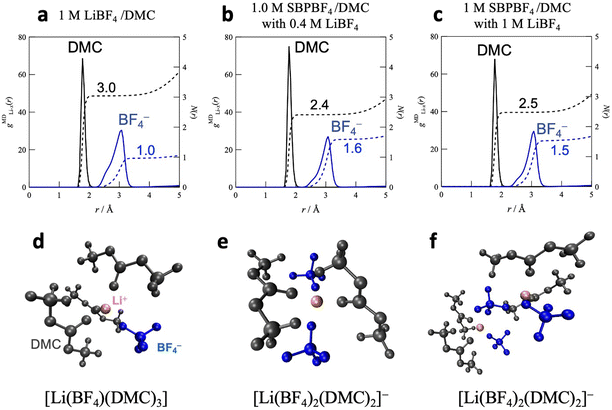 |
| | Fig. 4 Atom–atom pair correlation function, gMDX–Y(r), for the O atoms (DMC) and the F atoms (BF4−) around Li ions in (a) 1 M LiBF4/DMC, (b) the bottom phase of 1.0 M SBPBF4/DMC with 0.4 M LiBF4, and (c) 1 M SBPBF4/DMC with 1 M LiBF4. (d)–(f) Typical snapshots of Li-ion complexes confirmed in the simulation box for their solutions. | |
Here, we focus on the coordination manner of BF4 anions around the central Li ion in Li-ion complexes. The BF4 anion can act as both monodentate and bidentate ligands. If the BF4 anion coordinates with a Li ion via two F atoms (bidentate ligand), the Li ions need to form a [Li(BF4)1(solvent)2] complex; this is because the total coordination number of Li ions is approximately 4 as is well known. In the current HEXTS and MD studies, however, this is not the case, i.e., the BF4 anions bind as a monodentate ligand to form [Li(mono-BF4)1(DMC)3] (single-cation) and [Li(mono-BF4)2(DMC)2]− (dual-cation) complexes. This result was reasonable from the viewpoint of stabilization energy in the Li-ion complex formation by DFT calculations (Fig. 5). We calculated the binding energy (DEbind) for the possible complex [Li(mono-BF4)1(DMC)3] and then compared it with that for the model bidentate-type complex [Li(bi-BF4)1(DMC)2]. The energy difference from the monodentate-type to the bidentate-type complex was estimated to be −16 kJ mol−1. This indicates that the monodentate-type complex is more stable than the bidentate one, which agrees with the MD results in this work.
 |
| | Fig. 5 Optimized geometries of possible [Li(BF4)(DMC)2] (bidentate) and [Li(BF4)(DMC)3] (monodentate) complexes by DFT calculations and their energy difference (ΔΔEbind) calculated from their binding energies ΔEbind. | |
Fig. 6 shows the Li+–Li+ correlations, gMDLi–Li(r), in single- and dual-cation electrolytes. In the single-cation electrolyte (1 M LiBF4/DMC; Fig. 6a), a major broad peak was found at 8.9 Å, followed by around ∼17 Å, though there was a small peak at 5.2 Å. This 8.9 Å-peak originated from the correlation between mononuclear Li-ion solvation clusters (herein the [Li(BF4)1(DMC)3] complex),35 and thus we concluded no closest Li+–Li+ correlation over the long-r range. On the other hand, the dual-cation electrolyte (Fig. 6b) exhibits strong closest Li+–Li+ peaks at 4–5 Å (overlapping 4.4 Å with 5.3 Å) and the subsequent clear peaks around 9 and 13 Å. This strongly suggests that a multiple ordered (or polymeric) Li-ion complex was formed in the dual-cation electrolyte, which is similar to the specific Li-ion ordered structure formed in highly concentrated electrolytes for Li-ion batteries, i.e., organic and/or ionic liquid-based electrolytes with an extremely high concentration of Li salt.35–38Fig. 6c displays a typical snapshot obtained from the MD simulations for the dual-cation electrolyte; a Li-ion ordering linked via BF4 anions was clearly evident. Such an ordering formation might help to show high Li+ conductivity in dual-cation electrolyte systems, e.g., Li-ion hopping conduction, as reported in highly concentrated electrolytes.36,39–41 Indeed, DMC-based dual-cation electrolytes can achieve higher Li+ conductivity (∼1.5 mS cm−1) than single-cation (0.2 mS cm−1) or ordinary cyclic carbonate systems (PC-based dual-cation electrolytes, ca. 0.9 mS cm−1).17 We point out that, compared to the highly concentrated electrolyte (>3 M Li salt), our developed DMC-based dual-cation system allows the long-range Li+–Li+ ordered structure and high Li+ conduction at a relatively low Li+ concentration (ca. 1 M Li salt). Therefore, the DMC-based dual-cation system is a promising electrolyte with high Li+ conductivity while presenting reduced Li salt concentration and low viscosity for optimum processability. Controlling the solution structure by incorporating several types of ions into the DMC solvent can be an effective strategy for achieving high power not only for Li+-based electrolytes, but also for electrolytes with relatively high solvation energy such as Mg2+- and Ca2+-based systems.
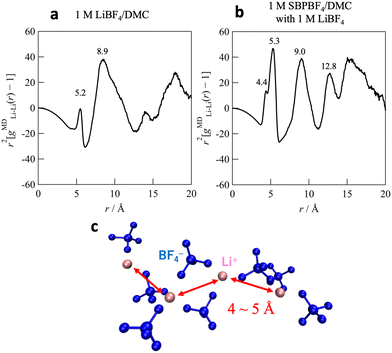 |
| | Fig. 6 Atom–atom pair correlation functions between Li ions [gMDLi–Li(r)] for (a) 1 M LiBF4/DMC and (b) 1 M SBPBF4/DMC with LiBF4 (cLi = 1 M). (c) Typical MD snapshots of Li-ion complexes found in 1 M SBPBF4/DMC with LiBF4. | |
Conclusions
We performed structural studies based on combined HEXTS experiments with MD simulations to demonstrate the unique solution structure in DMC-based dual-cation electrolytes. The radial distribution function G(r) obtained from both HEXTS and MD results suggested an ion ordered structure based on SBP+–BF4− ion pairs in the phase-separated 2 M SBPBF4/DMC; in contrast, there was no ion ordering in 1 M LiBF4/DMC solution. The ion ordered structure in SBPBF4/DMC disappeared by adding LiBF4 salt to form Li-ion solvation complexes alternatively, resulting in a single-phase solution, i.e., dual-cation electrolyte. In dual-cation electrolyte (1 M SBPBF4/DMC with 1 M LiBF4), Li ions aggregate via BF4 anions to form multiple Li-ion ordered complexes despite low Li salt concentration, and the structural feature was similar to those reported in highly concentrated Li-ion battery electrolytes using an organic solvent and Li salt (>3 M). We thus propose that DMC-based dual-cation electrolytes exhibit unique electrolytic properties arising from their specific solution structures to show great potential in the design of novel electrolytes with improved electrochemical properties, which are not limited to Li+-based electrolytes.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
This study was supported by JSPS KAKENHI (grant no. JP19H00882 (KN), JP20H02823 (KF), 21K05241 (EI), 21K20554 (YC), 22K14759 (YC), and 23H02066 (KF)) and the Hitachi Metals and Materials Science Foundation (YC). The synchrotron radiation experiments were performed at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (proposal no. 2021B1200 (KN), 2022A1174 (KN), 2023A1289 (KF), and 2023A1623 (SS)).
Notes and references
- N. Omar, M. Daowd, O. Hegazy, M. Al Sakka, T. Coosemans, P. Van den Bossche and J. Van Mierlo, Electrochim. Acta, 2012, 86, 305–315 CrossRef CAS.
- C. Schütter, S. Pohlmann and A. Balducci, Adv. Energy Mater., 2019, 9, 1900334 CrossRef.
- S. Dong, N. Lv, Y. Wu, G. Zhu and X. Dong, Adv. Funct. Mater., 2021, 31, 2100455 CrossRef CAS.
- A. Muzaffar, M. B. Ahamed, K. Deshmukh and J. Thirumalai, Renewable Sustainable Energy Rev., 2019, 101, 123–145 CrossRef CAS.
- J. Sun, B. Luo and H. Li, Adv. Energy Sustainability Res., 2022, 3, 2100191 CrossRef.
- X. Liu, Y. Sun, Y. Tong, X. Wang, J. Zheng, Y. Wu, H. Li, L. Niu and Y. Hou, Nano Energy, 2021, 86, 106070 CrossRef CAS.
- A. Tomaszewska, Z. Chu, X. Feng, S. O'kane, X. Liu, J. Chen, C. Ji, E. Endler, R. Li and L. Liu, ETransportation, 2019, 1, 100011 CrossRef.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- H. Cheng, J. G. Shapter, Y. Li and G. Gao, J. Energy Chem., 2021, 57, 451–468 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- K. Chiba, T. Ueda and H. Yamamoto, Electrochemistry, 2007, 75, 664–667 CrossRef CAS.
- Y.-B. He, B. Li, M. Liu, C. Zhang, W. Lv, C. Yang, J. Li, H. Du, B. Zhang and Q.-H. Yang, Sci. Rep., 2012, 2, 913 CrossRef PubMed.
- E. Logan and J. Dahn, Trends Chem., 2020, 2, 354–366 CrossRef CAS.
- M. Yuan and K. Liu, J. Energy Chem., 2020, 43, 58–70 CrossRef.
- J. Xu, J. Zhang, T. P. Pollard, Q. Li, S. Tan, S. Hou, H. Wan, F. Chen, H. He, E. Hu, K. Xu, X.-Q. Yang, O. Borodin and C. Wang, Nature, 2023, 614, 694–700 CrossRef CAS PubMed.
- Y. Kondo, T. Abe and Y. Yamada, ACS Appl. Mater. Interfaces, 2022, 14, 22706–22718 CrossRef CAS PubMed.
- Y. Chikaoka, R. Ochi, K. Fujii, T. Ariga, M. Sakurai, A. Matsumoto, T. Ueda, E. Iwama and K. Naoi, J. Phys. Chem. C, 2022, 126, 14389–14398 CrossRef CAS.
- S. Hwang, D.-H. Kim, J. H. Shin, J. E. Jang, K. H. Ahn, C. Lee and H. Lee, J. Phys. Chem. C, 2018, 122, 19438–19446 CrossRef CAS.
- F. Wang, O. Borodin, M. S. Ding, M. Gobet, J. Vatamanu, X. Fan, T. Gao, N. Eidson, Y. Liang and W. Sun, Joule, 2018, 2, 927–937 CrossRef CAS.
- A. B. McEwen, S. F. McDevitt and V. R. Koch, J. Electrochem.
Soc., 1997, 144, L84 CrossRef CAS.
- S. Kohara, K. Suzuya, Y. Kashihara, N. Matsumoto, N. Umesaki and I. Sakai, Nucl. Instrum. Methods Phys. Res., Sect. A, 2001, 467–468, 1030–1033 CrossRef CAS.
- S. Sawayama, A. Morinaga, H. Mimura, M. Morita, Y. Katayama and K. Fujii, ACS Appl. Mater. Interfaces, 2021, 13, 6201–6207 CrossRef CAS PubMed.
- K. Fujii, R. Kanzaki, T. Takamuku, Y. Kameda, S. Kohara, M. Kanakubo, M. Shibayama, S.-I. Ishiguro and Y. Umebayashi, J. Chem. Phys., 2011, 135, 244502 CrossRef PubMed.
- K. Fujii, Y. Soejima, Y. Kyoshoin, S. Fukuda, R. Kanzaki, Y. Umebayashi, T. Yamaguchi, S. Ishiguro and T. Takamuku, J. Phys. Chem. B, 2008, 112, 4329–4336 CrossRef CAS PubMed.
- K. Hirosawa, K. Fujii, K. Hashimoto and M. Shibayama, Macromolecules, 2017, 50, 6509–6517 CrossRef CAS.
- M. Shibata, S. Sawayama, M. Osugi and K. Fujii, J. Mol. Liq., 2022, 366, 120255 CrossRef CAS.
- M. Sogawa, S. Sawayama, J. Han, C. Satou, K. Ohara, M. Matsugami, H. Mimura, M. Morita and K. Fujii, J. Phys. Chem. C, 2019, 123, 8699–8708 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. Asada, T. Fujimori, K. Fujii, R. Kanzaki, Y. Umebayashi and S. Ishiguro, J. Raman Spectrosc., 2007, 38, 417–426 CrossRef CAS.
- H. Lee, S. Hwang, M. Kim, K. Kwak, J. Lee, Y.-K. Han and H. Lee, J. Phys. Chem. Lett., 2020, 11, 10382–10387 CrossRef CAS PubMed.
- Y. Umebayashi, H. Hamano, S. Seki, B. Minofar, K. Fujii, K. Hayamizu, S. Tsuzuki, Y. Kameda, S. Kohara and M. Watanabe, J. Phys. Chem. B, 2011, 115, 12179–12191 CrossRef CAS PubMed.
- R. Kanzaki, T. Mitsugi, S. Fukuda, K. Fujii, M. Takeuchi, Y. Soejima, T. Takamuku, T. Yamaguchi, Y. Umebayashi and S.-I. Ishiguro, J. Mol. Liq., 2009, 147, 77–82 CrossRef CAS.
- K. Chiba, T. Ueda and H. Yamamoto, Electrochemistry, 2007, 75, 668–671 CrossRef CAS.
- T. Sukizaki, S. Fukuda, T. Yamaguchi, K. Fujii, R. Kanzaki, K. Chiba, H. Yamamoto, Y. Umebayashi and S.-I. Ishiguro, Electrochemistry, 2007, 75, 628–634 CrossRef CAS.
- K. Fujii, M. Matsugami, K. Ueno, K. Ohara, M. Sogawa, T. Utsunomiya and M. Morita, J. Phys. Chem. C, 2017, 121, 22720–22726 CrossRef CAS.
- K. Dokko, D. Watanabe, Y. Ugata, M. L. Thomas, S. Tsuzuki, W. Shinoda, K. Hashimoto, K. Ueno, Y. Umebayashi and M. Watanabe, J. Phys. Chem. B, 2018, 122, 10736–10745 CrossRef CAS PubMed.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- K. Shigenobu, M. Shibata, K. Dokko, M. Watanabe, K. Fujii and K. Ueno, Phys. Chem. Chem. Phys., 2021, 23, 2622–2629 RSC.
- S. Kondou, M. L. Thomas, T. Mandai, K. Ueno, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2019, 21, 5097–5105 RSC.
- Y. Ugata, M. L. Thomas, T. Mandai, K. Ueno, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2019, 21, 9759–9768 RSC.
- Y. Watanabe, Y. Ugata, K. Ueno, M. Watanabe and K. Dokko, Phys. Chem. Chem. Phys., 2023, 25, 3092–3099 RSC.
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Tomoya
Tashiro
c,
Saki
Sawayama
c,
Ayana
Kobayashi
a,
Ayuna
Matsumoto
a,
Etsuro
Iwama
*ab,
Tomoya
Tashiro
c,
Saki
Sawayama
c,
Ayana
Kobayashi
a,
Ayuna
Matsumoto
a,
Etsuro
Iwama