
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-photon absorption properties of simple neutral Ir(III) complexes
Eleonora
Garoni
a,
Alessia
Colombo
 a,
Dominique
Roberto
a,
Claudia
Dragonetti
*a,
Veronique
Guerchais
b and
Kenji
Kamada
*c
a,
Dominique
Roberto
a,
Claudia
Dragonetti
*a,
Veronique
Guerchais
b and
Kenji
Kamada
*c
aDepartment of Chemistry, Università degli Studi di Milano, UdR dell’INSTM, Via Golgi 19, 20133 Milano, Italy. E-mail: claudia.dragonetti@unimi.it
bUniv. Rennes, CNRS UMR6226, F-35000 Rennes, France
cNanomaterials Research Institute (NMRI), National Institute of Advanced Industrial Science and Technology (AIST), Ikeda, Osaka 563-8577, Japan. E-mail: k.kamada@aist.go.jp
First published on 9th February 2024
Abstract
A series of neutral Ir(2-phenylpyridine)3 derivatives substituted on the para-position of the pyridyl ligands with a π-conjugated substituent possessing different donor abilities has been prepared. Their two-photon absorption properties have been determined using the Z-scan technique. Such simple iridium(III) neutral complexes, which are easy to synthesize, show good two-photon absorption activity, with relevant TPA cross sections (the best is 750 GM), giving rise to multifunctional chromophores, since they present also high second-order NLO properties.
1. Introduction
Iridium complexes are well known for their outstanding tuneable luminescence properties, that allow their application in electroluminescence devices1 and as bioimaging sensors,2,3 and for their intriguing second-order nonlinear optical (NLO) properties.4,5Iridium complexes have attracted attention also for their two-photon absorption (TPA) activity. Two-photon absorption has a lot of applications in various fields, such as 3D fluorescence imaging and control of biological systems, 3D-microfabrication and optical limiting.6
Concerning TPA activity, the Beeby group reported a series of iridium(III) tris(2-ppy) or bis(2-ppy)(acac) (acac is acetylacetonate whereas 2-ppy is 2-phenylpyridine) complexes.7 They found moderate TPA cross-section values (maximum σ(2) was 60 GM for Ir(4-pe-2-ppy)2(acac), where 4-pe-2-ppyH is 4-phenylethynyl-2-phenylpyridine) in the range between 760 and 880 nm measured by the two-photon-induced fluorescence (TPIF) method using a femtosecond Ti:sapphire oscillator (150 fs, 2.5 MHz repetition). A better TPA response was observed for the complexes bearing an acetylacetonate ligand, while substitution of 2-phenylpyridine with 2-phenylpyrimidine had a strong negative effect on the cross-section values.
Wong's group studied some Ir(ppy)3 complexes with more extended substituents on the phenylpyridine ligands.8 Complexes were characterized by very good two-photon induced emission in solution (maximum value 340 GM at 750 nm obtained by TPIF using a femtosecond optical parametric amplifier, 150 fs, 1 kHz repetition). They have been tested in vivo and they demonstrate high target-specificity to the Golgi apparatus with long residential lifetime, low cytotoxicity and intense two-photon induced emission.
Zhao et al. reported some symmetrically and unsymmetrically, para- or ortho-substituted Ir(ppy)3 complexes with oligofluorene-substituted phenylpyridines measured by TPIF using a femtosecond optical parametric amplifier (ca. 120 fs, 1 kHz repetition).9 The reported iridium complexes are among those with the largest TPA cross-sections reported in the literature. The authors demonstrated that the TPA response is proportional to the size of the conjugated ligand. When they extend from one to three fluorene units, σ(2) values are enhanced fourfold. Furthermore, if a donor carbazole group is added at the end of the ligand, TPA is further improved due to increased donor–acceptor electronic effects, reaching maxima values of slightly lower than 1200 GM.
Some of us studied the TPA properties of a neutral cis-[Ir(CO)2Cl(4-(para-di-nbutylaminostyryl)pyridine)] complex.10 The TPA cross-sectional maximum was found to be about 780 GM at 910 nm. This value is four times higher than that reported for 4-(para-di-nbutylaminostyryl)pyridine (190 GM), demonstrating how σ(2) can be highly enhanced upon coordination to the Ir metal centre. Some examples of neutral iridium(III) complexes with TPA properties and direct application in vivo were also reported by the Kawamata11 and Monnereau groups.12 The investigated complexes exhibit TPA in the near-infrared, at wavelengths suitable for biological applications, with TPA cross-section values quite low (around 20–40 GM) but sufficient to allow recognition of two-photon induced emission in cells.
The TPA response of several cationic iridium complexes was also explored. Magennis et al. studied Ir(terpy)2+ complexes, reporting fair TPA cross-section values with a maximum of 70 GM.13 The Huang group synthesized and studied a binuclear Ir(III) cationic complex bearing dimesitylboryl groups on the cyclometalated 2-phenylquinoline ligands and a push–pull conjugated bipyridine oligomer.14 The TPA spectra of the complex were determined between 730 and 840 nm obtaining a maximum value of 500 GM at 750 nm. This value is high and can be explained by the long π-extension of the bipyridyl ligand and the presence of two metal centers instead of one. Interestingly, the complex bearing boron-based substituents can be used as a one and two-photon-excited switchable phosphorescent probe for F− ions, thanks to the strong B–F interactions. The Das group studied a series of [Ir(ppy)2(bpy)]PF6 complexes with extended π-conjugated styrene substituents on the bipyridine ligand.15
The TPA cross-sections were reported at 800 nm with ps pulses and at 680 nm with fs pulses and values as high as 900 GM were reached. Nevertheless, the reported Z-scan traces reveal that two-photon absorption is not the only phenomenon that these molecules undergo when irradiated with laser pulses. The very sharp curves reveal the presence of simultaneous three-photon absorption and some distorted TPA traces indicate saturable absorption. The Chao group extensively studied the TPA response of some cationic iridium complexes with chelating ligands for applications as probes in cells, especially in mitochondria.16–18 The reported complexes have moderate TPA cross-sectional values, but they can enter cells, localize in mitochondria and signal the excessive presence of ClO− or track mitochondrial morphological changes during the early stages of cell apoptosis even under hypoxia, highlighting the potential of such complexes for applications in biomedical research.
More recently, some of us reported a novel multifunctional cyclometallated cationic iridium(III) complex [Ir(C^N-ppy-4-Ch![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHC6H4NEt2)2(N^N-bpyCHCHC6H4NEt2)][PF6] (ppy = 2-phenylpyridine; bpy = 2,2′-bipyridine) with interesting second-order nonlinear optical properties and Z-scan two-photon absorption activity; the two-photon absorption cross-sections are quite high, in the range 100–250 GM at 600–1000 nm.19
CHC6H4NEt2)2(N^N-bpyCHCHC6H4NEt2)][PF6] (ppy = 2-phenylpyridine; bpy = 2,2′-bipyridine) with interesting second-order nonlinear optical properties and Z-scan two-photon absorption activity; the two-photon absorption cross-sections are quite high, in the range 100–250 GM at 600–1000 nm.19
A two-photon cyclometalated iridium(III) complex has been presented by X. Tian et al. as a photodynamic therapy agent that targets the intracellular nucleus. Such a complex is capable of migrating sequentially from the nucleus to mitochondria and inducing dual damage under light exposure.20
Some years ago, some of us prepared a series of neutral Ir(2-phenylpyridine)3 derivatives for their second-order NLO properties (Fig. 1). All compounds are substituted in the para-position of the pyridyl ligands with a π-conjugated substituent possessing different donor abilities. These substitutions cause a permanent dipole moment along the C3 symmetry axis of the complexes (Fig. 1 inset).
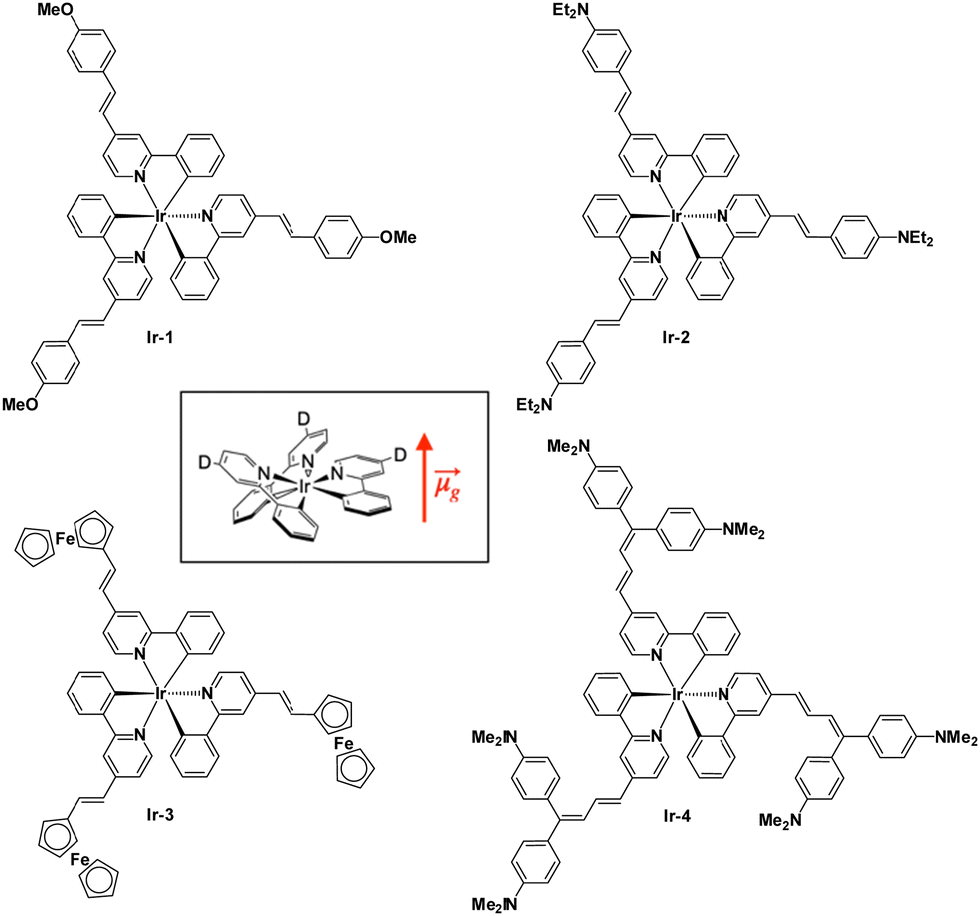 | ||
| Fig. 1 Four iridium(III) neutral complexes. | ||
They are characterized by high absolute values of μβEFISH, as determined by the electric field induced second harmonic generation technique,21,22 depending on the substituent on the position 4 of the pyridyl ring.23
It turned out that βEFISH of the complexes bearing a weak donor substituent has a negative value because of the negative value of Δμeg (difference between the excited and ground state dipole moments) due to the decreased dipole moment of the excited state.23 This is caused by the fact that the metal-to-ligand charge transfer (MLCT) transitions directed from the iridium to the pyridine are stronger than the vectorially opposed intraligand charge transfer (ILCT) transitions from the donor substituent to the pyridine ring. When the donor is stronger, the ILCT transitions prevail on the MLCT ones and the sign of βEFISH becomes positive. The value of βHLS instead takes into account also the octupolar contribution along with the dipolar ones and increases with the increasing electron-donating ability of the substituents.23 These interesting second-order nonlinear optical results prompted us to investigate the two-photon absorption properties of these fascinating complexes (Fig. 1) with the Z-scan technique. Our findings are reported in the present work.
2. Experimental
2.1 Synthetic procedure
The neutral iridium complexes presented in Fig. 1 are prepared following a reported procedure.23The biscyclometalated chloro-bridged dimers were first obtained by reaction of IrCl3·nH2O with 4-methyl-2-phenylpyridine. Upon reaction of the dimers with 4-methyl-2-phenylpyridine in glycerol at 200 °C, the tris-chelate methyl complex fac-Ir(4-methyl-2-phenylpyridine)3 was achieved. It was then treated at room temperature with the appropriate para-R-benzaldehyde in the presence of t-BuOK to obtain the desired complexes. It is not possible to synthesize the complexes directly from the styryl-substituted dimer because under the required conditions, the double bonds would be hydrogenated.
All the complexes were characterized by standard techniques.
Concerning the linear absorption,23 complexes Ir-1 and Ir-3 show similar UV-Vis spectra in dichloromethane: the more intense absorption band at 310–350 nm can be attributed to ILCT π–π* transitions of the styryl-phenylpyridine ligands, while the absorption bands at lower energy, about 370–450 nm, can be assigned to MLCT transitions and the band tailing up to 560 nm could be due to excitation to triplet charge transfer states. Complexes Ir-2 and Ir-4, bearing the amino substituents, which are strong electron-donors, show an intense band at 400–550 nm hiding the MLCT band, this is assigned to an ILCT transition in which the pyridyl ligand acts as a π*-acceptor group.23
2.2 TPA measurements
The open-aperture Z-scan method was employed for TPA spectral measurements.24,25 A femtosecond optical parametric amplifier (Spectra-Physics TOPAS Prime, repetition rate: 1 kHz) was used as a wavelength-tunable light source. Transmittance (T) of the pulsed beam through the sample media was recorded as a function of the sample position (z). The plot of the data, so-called open-aperture Z-scan trace, is usually presented with normalized transmittance (TN), where the normalization was performed to take the transmittance obtained at the sample position far from the focal point as unity. It is well known that TN of the spatially and temporally Gaussian pulses through an absorptive media of both one- and two-photon absorption can be written as24 | (1) |
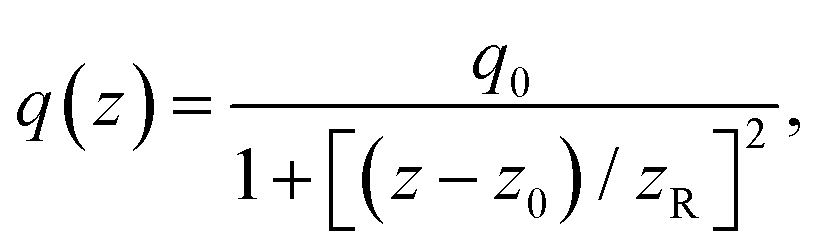 | (2) |
| q0 = β(1 − R)I0Leff. | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) TL) is the effective path length and reduced to the (physical) path length L when the one-photon absorption is negligible and TL = e−αL converges to unity or the linear (one-photon) absorption coefficient α approaches to zero. For the above equations, R is the Fresnel reflectance at the cell walls. Finally, σ(2) can be determined from the convention σ(2) = ħωβ/N where ħω is the incident photon energy and N is the number density of the molecule. On-axis peak intensity I0 for the spatially and temporally Gaussian pulse was calculated by
TL) is the effective path length and reduced to the (physical) path length L when the one-photon absorption is negligible and TL = e−αL converges to unity or the linear (one-photon) absorption coefficient α approaches to zero. For the above equations, R is the Fresnel reflectance at the cell walls. Finally, σ(2) can be determined from the convention σ(2) = ħωβ/N where ħω is the incident photon energy and N is the number density of the molecule. On-axis peak intensity I0 for the spatially and temporally Gaussian pulse was calculated by | (4) |
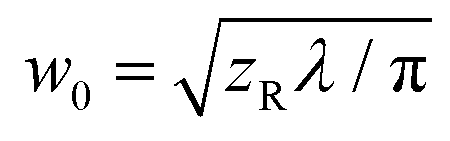 . tp is the pulse width in full width at the half maximum (FWHM) measured using an autocorrelator. Typical parameters used were P < 0.4 mW, zR = 6–10 mm, giving I0 = 70–150 GW cm−2 or less (depending on wavelength).26
. tp is the pulse width in full width at the half maximum (FWHM) measured using an autocorrelator. Typical parameters used were P < 0.4 mW, zR = 6–10 mm, giving I0 = 70–150 GW cm−2 or less (depending on wavelength).26
The neutral Ir(III) complexes were dissolved in dichloromethane and placed in 2-mm quartz cuvettes. The solubility of the complexes was good, ranging from 1.3 to 2 mM. Some expedients were necessary in order to measure properly the nonlinear absorption of these samples. Complexes Ir-2 and Ir-4 apparently undergo photochemical reactions under irradiation of the laser beam. This can be understood by looking at the shape of the Z-scan trace; it is not symmetrical and deeper than the case by stirring the sample during measurement (Fig. 2), suggesting absorption of the incident beam by a photoproduct or a long-lived intermediate. Thus, all samples were measured with stirring to remove such effect. At the short wavelength side, a tail of one-photon absorption (OPA) existed. The wavelength region is indicated by a pale blue color in (Fig. 3). The correction for the OPA was taken into account by using Leff shown above.
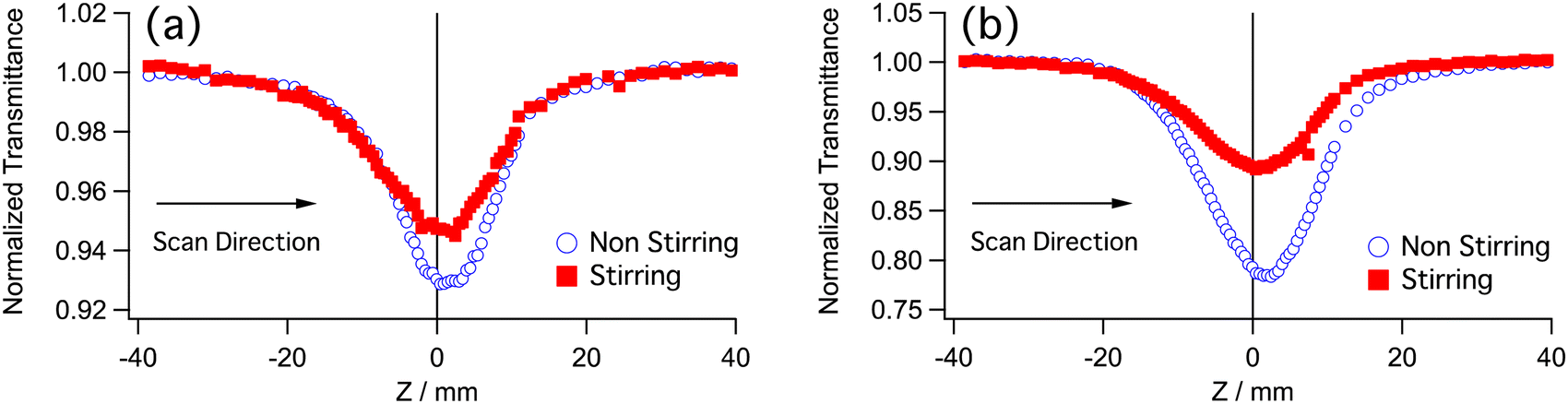 | ||
| Fig. 2 Z-scan traces of (a) Ir-2 and (b) Ir-4 without and with stirring. Wavelength was 840 nm and incident average power was 0.4 mW. The Rayleigh range was ZR = 8.3 mm. The transmittance was normalized at virtually infinite far sample position (Z > 5ZR) from the focus (Z = 0). | ||
 | ||
| Fig. 3 The two-photon absorption (circles, left and bottom axes) and one-photon absorption (plain line, right and top axes) spectra of the neutral Ir(ppy)3 complexes: the empty circles are the values obtained with the wavelength scan at a fix power, while the full markers with error bars are the ones obtained with the power scan at a fixed wavelength. The blue fill shows the λTPA region where absorbance of Z-scan sample is over 0.05. | ||
2.3 DFT calculations and spectrum simulations
OPA and TPA spectra were simulated based on the quantum chemical calculations using density functional theory (DFT). Molecular geometry was optimized by using the Coulomb-attenuating method (CAM)-B3LYP27 as a functional and LANL2DZ, involving effective core potential (EPC) considering relativistic effects, as the basis set for the Ir atom and 6-31+G(d) for other atoms. The solvent effect was considered by using the integral equation formalism (IEF) polarizable continuum model (PCM). Transition energies, transition and permanent dipole moments were calculated with the Tamm–Dancoff approximation (TDA)28-IEF-PCM-CAM-B3LYP/LANL2DZ (for Ir) and 6-31+G(d) (for others). All DFT calculations were performed by using the Gaussian 16 program package.29 Simulation of the TPA spectrum was performed by using a homemade procedure based on the formalism reported previously.303. Results and discussion
The two-photon absorption spectra of the four neutral iridium complexes Ir-1,2,3,4 are shown in Fig. 3. The σ(2) values are very high especially if compared with those of the unsubstituted Ir(2-phenylpyridine)3,7 the reported values ranging from 35 to 5 GM, between 760 and 800 nm. By adding various donating substituents, the TPA response is increased more than tenfold, depending on the strength of the donor. The complexes bearing the methoxyphenyl and ferrocenyl groups have the lowest TPA cross-section (Ir-1: σ(2)max = 170 ± 30 GM, Ir-3: σ(2)max = 440 GM both at 620 nm), demonstrating the similar donor effect of these substituents, already observed in the case of the second-order NLO properties of the compounds.31The TPA becomes more intense if the substituents are strong donor aminophenyl groups (Ir-2: σ(2)max = 500 ± 80 GM at 760 nm, Ir-4: σ(2)max = 750 ± 130 GM at 800 nm) reaching values of several hundreds of GM. The minimum values recorded, for complex Ir-1, are still much higher than for the unsubstituted complex. This behavior is of particular interest, because it demonstrates that by a careful design of the complexes and with overall small changes in the molecules’ structure, for example by simply changing the donating substituents, the TPA magnitude can be modulated over a wide range reaching high values. The largest TPA cross-sections have been obtained for Ir-4, bearing strong electron-donating substituents, with mobile electrons on the p-orbitals of the nitrogen, and with a longer π-conjugated system, due to the presence of two double bonds26 and two N,N-dimethylaniline groups. Higher TPA cross-section values (ca. 1200 GM) were reported for some neutral Ir(ppy)3 complexes by Zhao and co-workers.8 Nevertheless, they used very large and extended substituents to obtain a result that we reached with smaller complexes, using more simple ligands, and thus reducing the synthetic steps.
TPA peaks of Ir-1 and Ir-3 are located almost at twice the wavelength of OPA peaks: (TPA/OPA) 660 nm/330 nm, 800 nm/400 nm, 950 nm/750 for Ir-1, and 800 nm/400 nm, 1000 nm/500 nm for Ir-3. This shows the possibility that the TPA transitions occur in the same excited states as the OPA peaks. On the other hand, positions of the TPA peak for Ir-2 (760 nm) and Ir-4 (800 nm) are blue-shifted from twice the OPA peaks (413 and 434 nm for Ir-2 and 458 nm for Ir-4) as seen in Fig. 3. This suggests that the final state of the TPA transition in these peaks is a different excited state than that of the OPA peaks. However, the TPA spectra are very broad and have considerable magnitude for 800–1000 nm. In this region, the wavelength-doubled OPA spectra well overlap the TPA bands. This suggests that the TPA transition in this region occurs in the same excited state as the OPA transitions. The magnitude of the TPA cross section of Ir-4 between 720 and 900 nm is larger than 400 GM and higher than that of Ir-2 for this range. Both complexes possess electron-donating groups of the same nature and in Ir-4 π-conjugation is increased to a small extent with respect to Ir-2. Nevertheless, this difference is still enough to achieve a quite large increase of σ(2) and to change the energy of the excited states for the two complexes.
To understand the nature of OPA and TPA transitions of the Ir complexes, we have performed the spectral simulation based on the DFT calculation on a model molecule of Ir-2. All ethyl groups of the terminal diethylamino groups were replaced with methyl groups to save computation resources. This substitution has been known to have little effect on the calculated optical properties. Hereafter, the model molecule is called as Ir-2′. The optimized geometry of Ir-2′ has C3 symmetry (Fig. 4a) with a permanent dipole moment of ![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) g = 19.0 D along the symmetry axis (z). Unlike centrosymmetry (Ci), symmetry selection rule of C3 symmetry is not clearly exclusive to each other between OPA and TPA. C3 symmetry consists of E and A symmetry species; both are OPA-allowed, but the orientation of reaction of their transition dipole moments is in the x–y plane and along the z-axis, respectively. In contrast, a strong TPA transition can be expected for A, where the two transition dipole moments involved are parallel (x2 + y2, z2, from the index table), rather than E, where they are orthogonal ((x2 − y2, xy), (yz, xz)). Table 1 shows the calculation results of the six lowest excited states. S1 and S2 are degenerated (E) with strong OPA (oscillator strength fOS = 1.9 for both), followed by S3 and S4, both A symmetry with strong OPA (fOS = 1.36 and 0.65, respectively). The next two (S5 and S6) are degenerated (E) with negligible OPA (fOS = 0.013 and 0.015).
g = 19.0 D along the symmetry axis (z). Unlike centrosymmetry (Ci), symmetry selection rule of C3 symmetry is not clearly exclusive to each other between OPA and TPA. C3 symmetry consists of E and A symmetry species; both are OPA-allowed, but the orientation of reaction of their transition dipole moments is in the x–y plane and along the z-axis, respectively. In contrast, a strong TPA transition can be expected for A, where the two transition dipole moments involved are parallel (x2 + y2, z2, from the index table), rather than E, where they are orthogonal ((x2 − y2, xy), (yz, xz)). Table 1 shows the calculation results of the six lowest excited states. S1 and S2 are degenerated (E) with strong OPA (oscillator strength fOS = 1.9 for both), followed by S3 and S4, both A symmetry with strong OPA (fOS = 1.36 and 0.65, respectively). The next two (S5 and S6) are degenerated (E) with negligible OPA (fOS = 0.013 and 0.015).
 | ||
| Fig. 4 (a) Optimized structure of Ir-2′ in dichloromethane calculated with CAM-B3LYP and LANL2DZ for Ir and 6-31+G(d) for other atoms. (b) Simulated OPA (blue) and TPA (red) spectra. The relaxation constant was chosen to be Γ = 0.1 eV for both OPA and TPA transitions. The numbers and dotted lines show the final states of the corresponding transition and the wavelengths. (c) Decomposed components of TPA spectra into the dipolar, three-state, and cross terms. | ||
| n | Symmetry | E/eV (λ/nm) | f OS |
|---|---|---|---|
| 1 | E | 3.32 (373.9) | 1.92 |
| 2 | E | 3.32 (372.9) | 1.91 |
| 3 | A | 3.46 (358.6) | 1.36 |
| 4 | A | 3.50 (353.8) | 0.65 |
| 5 | E | 3.52 (352.4) | 0.013 |
| 6 | E | 3.52 (352.1) | 0.016 |
The simulated OPA spectrum by the DFT calculation of Ir-2′ is shown in Fig. 4b (blue curve). The largest peak (∼373 nm) is the transition to the degenerated S1 and S2 and is considered to correspond to the intense peak at 434 nm in the experimental spectrum of Ir-2, although the transition energy was overestimated. Over estimation of the CAM-B3LYP calculations is known for π-conjugated systems. Transitions to S3 and S4 are appeared as the shoulder around 355 nm in simulation likely corresponding to the peak at 413 nm in the experiment. The simulation failed to reproduce the shoulder at 500 nm; nevertheless, the simulation approximately reproduced the outline shape of the OPA spectrum except the shoulder.
Simulated TPA spectra (red curve) have a peak at 704 nm, assigned as TPA transitions to S6 and S5 and followed by to S4 and then S3. TPA transitions to S1 and S2 also appeared as the shoulder at 746 nm. The simulated spectrum has a similar spectral shape to the experimental TPA spectrum of Ir-2. The peak is located at a higher transition energy (to S4–S6) than that of the OPA peak (to S1, S2) although the shoulders have the same transition energies to the OPA peaks (to S1, S2, and S3).
Fig. 4c shows decomposed components of the simulated TPA spectrum by different terms in the theoretical formulation, where the total TPA cross section can be partitioned into dipolar (aka. two-state), three-state, and cross terms.30 Three-state term is the term that contains an intermediate state k which is neither ground state g (S0) nor the final excited state f while the terms with k = g or f are dipolar terms because only the two states (g and f) are involved for TPA. Cross-term is the mixing of these terms. The dipolar term (red dotted line) has peaks to S1, S2 (747 nm) and S3 (at 717 nm) while the three-state term (blue dashed line) is the source of the peak to S4–S6 (∼704 nm) in the total spectrum (black solid line). Dipolar term contains |fg|2|Δfg|2 factor, where fg and Δfg are the transition dipole moment and permanent dipole moment difference (Δfg ≡ f − g) between g and f, respectively. Therefore, dipolar term has a correlation with the OPA spectrum (∝|fg|2) as long as Δfg is nonzero and similar values over different f. Actually, Ir-2′ has Δfg = 4.1, 3.3, and 3.9 D, respectively, for f = S1, S2, and S3. Therefore, the three-state term dominates the short wavelength peak and the two-state term increases it contribution for the long wavelength side. The relative intensity of the three-state term and dipolar term can explain the different spectral relations experimentally observed for OPA and TPA for the Ir complexes.
Conclusions
In conclusion, our work put in evidence that simple iridium(III) neutral complexes, easy to synthetize, show a good two-photon absorption activity, along with a yet demonstrated high second-order NLO activity, to give rise to multifunctional chromophores. Clearly, the novel complexes reported here have great potential for application in photonics and biomedicine.Author contributions
Eleonora Garoni: investigation, writing – review & editing. Alessia Colombo: conceptualization, investigation, writing – review & editing. Dominique Roberto: conceptualization, investigation, writing – review & editing. Claudia Dragonetti: conceptualization, investigation, writing – original draft, writing – review & editing. Veronique Guerchais: conceptualization, writing – review & editing. Kenji Kamada: conceptualization, investigation, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Photosynergetics” (JP26107004 for Kamada) from MEXT, Japan and a Grant-in-Aid for Scientific Research 8H01943, 21H01887, and 23K04710 (K. K.) from JSPS.Notes and references
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49–82 CrossRef CAS.
- J. Shen, T. W. Rees, L. Ji and H. Chao, Coord. Chem. Rev., 2021, 443, 214016 CrossRef CAS.
- G. De Soricellis, F. Fagnani, A. Colombo, C. Dragonetti, D. Roberto, D. Marinotto, D. H. Hartnell, M. J. Hackett, M. Massi, B. Carboni and V. Guerchais, Dyes Pigm., 2023, 210, 111012 CrossRef CAS.
- A. Valore, A. Colombo, C. Dragonetti, S. Righetto, D. Roberto, R. Ugo, F. De Angelis and S. Fantacci, Chem. Commun., 2010, 46, 2414–2416 RSC.
- A. Colombo, C. Dragonetti, V. Guerchais, C. Hierlinger, E. Zysman-Colman and D. Roberto, Coord. Chem. Rev., 2020, 414, 213293 CrossRef CAS.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed.
- R. M. Edkins, S. L. Bettington, A. E. Goeta and A. Beeby, Dalton Trans., 2011, 40, 12765–12770 RSC.
- C.-L. Ho, K.-L. Wong, H.-K. Kong, Y.-M. Ho, C. T.-L. Chan, W.-M. Kwok, K. S.-Y. Leung, H.-L. Tam, M. H.-W. Lam, X.-F. Ren, A.-M. Ren, J.-K. Fengf and W.-Y. Wong, Chem. Commun., 2012, 48, 2525–2527 RSC.
- Y. Fan, D. Ding and D. Zhao, Chem. Commun., 2015, 51, 3446–3449 RSC.
- A. Colombo, C. Dragonetti, D. Roberto, A. Valore, C. Ferrante, I. Fortunati, A. L. Picone, F. Todescato and J. A. G. Williams, Dalton Trans., 2015, 44, 15712–15720 RSC.
- H. Moritomo, A. Fujii, Y. Suzuki, T. Yoshihara, S. Tobita and J. Kawamata, Jpn. J. Appl. Phys., 2016, 55, 092041 CrossRef.
- M. Lepeltier, F. Appaix, Y. Y. Liao, F. Dumur, J. Marrot, T. Le Bahers, C. Andraud and C. Monnereau, Inorg. Chem., 2016, 55, 9586–9595 CrossRef CAS PubMed.
- L. S. Natrajan, A. Toulmin, A. Chewa and S. W. Magennis, Dalton Trans., 2010, 39, 10837–10846 RSC.
- W.-J. Xu, S.-J. Liu, X. Zhao, N. Zhao, Z.-Q. Liu, H. Xu, H. Liang, Q. Zhao, X.-Q. Yu and W. Huang, Chem. – Eur. J., 2013, 19, 621–629 CrossRef CAS PubMed.
- M. Sarma, T. Chatterjee, R. Bodapati, K. N. Krishnakanth, S. Hamad, S. V. Rao and S. K. Das, Inorg. Chem., 2016, 55, 3530–3540 CrossRef CAS PubMed.
- G. Li, Q. Lin, L. Sun, C. Feng, P. Zhang, B. Yu, Y. Chen, Y. Wen, H. Wang, L. Ji and H. Chao, Biomaterials, 2015, 53, 285–295 CrossRef CAS PubMed.
- C. Jin, J. Liu, Y. Chen, L. Zeng, R. Guan, C. Ouyang, L. Ji and H. Chao, Chem. – Eur. J., 2015, 21, 12000–12010 CrossRef CAS PubMed.
- L. Sun, Y. Chen, S. Kuang, G. Li, R. Guan, J. Liu, L. Ji and H. Chao, Chem. – Eur. J., 2016, 22, 8955–8965 CrossRef CAS PubMed.
- A. Colombo, E. Garoni, C. Dragonetti, S. Righetto, D. Roberto, N. Baggi, M. Escadeillas, V. Guerchais and K. Kamada, Polyhedron, 2018, 140, 116–121 CrossRef CAS.
- X. Tian, Y. Zhu, M. Zhang, L. Luo, J. Wu, H. Zhou, L. Guan, G. Battaglia and Y. Tian, Chem. Commun., 2017, 53, 3303 RSC.
- I. Ledoux and J. Zyss, Chem. Phys., 1982, 73, 203–213 CrossRef CAS.
- S. Di Bella, C. Dragonetti, M. Pizzotti, D. Roberto, F. Tessore and R. Ugo, Top. Organomet. Chem., 2010, 28, 1–55 CrossRef CAS PubMed.
- M. Zaarour, A. Singh, C. Latouche, J. A. G. Williams, I. Ledoux-Rak, J. Zyss, A. Bouccekine, H. Le Bozec, V. Guerchais, C. Dragonetti, A. Colombo, D. Roberto and A. Valore, Inorg. Chem., 2013, 52, 7987–7994 CrossRef CAS PubMed.
- M. Sheik-Bahae, A. A. Said, T. H. Wei, D. J. Hagan and E. W. Van Stryland, IEEE J. Quantum Electron., 1990, 26, 760–769 CrossRef CAS.
- K. Kamada, K. Matsunaga, A. Yoshino and K. Ohta, J. Opt. Soc. Am. B, 2003, 20, 529–537 CrossRef CAS.
- K. Kamada, K. Ohta, Y. Iwase and K. Kondo, Chem. Phys. Lett., 2003, 372, 386–393 CrossRef CAS.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- S. Hirata, M. Head-Gordon, J. Szczepanski and M. Vala, J. Phys. Chem. A, 2003, 107, 4940–4951 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Rev. C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- K. Ohta, S. Yamada, K. Kamada, A. D. Slepkov, F. A. Hegmann, R. R. Tykwinski, L. D. Shirtcliff, M. M. Haley, P. Sałek, F. Gel’mukhanov and H. Ågren, J. Phys. Chem. A, 2011, 115, 105–117 CrossRef CAS PubMed.
- M. L. H. Green, S. R. Marder, M. E. Thompson, J. A. Bandy, D. Bloor, P. V. Kolinsky and R. J. Jones, Nature, 1987, 330, 360–362 CrossRef CAS.
| This journal is © the Owner Societies 2024 |