
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Composition dependence of X-ray stability and degradation mechanisms at lead halide perovskite single crystal surfaces†
Alberto
García-Fernández‡
 a,
Birgit
Kammlander‡
ab,
Stefania
Riva
b,
Håkan
Rensmo
b and
Ute B.
Cappel
*abc
a,
Birgit
Kammlander‡
ab,
Stefania
Riva
b,
Håkan
Rensmo
b and
Ute B.
Cappel
*abc
aDivision of Applied Physical Chemistry, Department of Chemistry, KTH – Royal Institute of Technology, 100 44 Stockholm, Sweden. E-mail: ute.cappel@physics.uu.se
bDivision of X-ray Photon Science, Department of Physics and Astronomy, Uppsala University, Box 516, 751 20 Uppsala, Sweden
cWallenberg Initiative Materials Science for Sustainability, Department of Physics and Astronomy, Uppsala University, 751 20 Uppsala, Sweden
First published on 13th December 2023
Abstract
The multiple applications of lead halide perovskite materials and the extensive use of X-ray based techniques to characterize them highlight a need to understand their stability under X-ray irradiation. Here, we present a study where the X-ray stability of five different lead halide perovskite compositions (MAPbI3, MAPbCl3, MAPbBr3, FAPbBr3, CsPbBr3) was investigated using photoelectron spectroscopy. To exclude effects of thin film formation on the observed degradation behaviors, we studied clean surfaces of single crystals. Different X-ray resistance and degradation mechanisms were observed depending on the crystal composition. Overall, perovskites based on the MA+ cation were found to be less stable than those based on FA+ or Cs+. Metallic lead formed most easily in the chloride perovskite, followed by bromide, and only very little metallic lead formation was observed for MAPbI3. MAPbI3 showed one main degradation process, which was the radiolysis of MAI. Multiple simultaneous degradation processes were identified for the bromide compositions. These processes include ion migration towards the perovskite surface and the formation of volatile and solid products in addition to metallic lead. Lastly, CsBr formed as a solid degradation product on the surface of CsPbBr3.
Introduction
Lead halide perovskites have gained great interest in the scientific community due to their useful properties in multiple applications, including photovoltaics, light emitting diodes (LEDs), photodetectors, and more recently, their use in batteries and hydrogen production.1–4 The general chemical formula for these materials is ABX3, where A represents a monovalent cation (e.g., methylammonium (MA+), CH3NH3+; formamidinium (FA+), CH(NH2)2+; Cs+), B is a divalent cation (e.g., Pb2+) and X is typically a halide (e.g., Br−, I−, Cl−).Containing heavy elements, such as lead, iodide, or bromide, enables lead halide perovskites to detect high energy radiation (e.g., X-rays or γ-rays) with high sensitivity and low detection limits.5 Therefore, perovskites have been suggested as the active material in photodetection applications, such as medical imaging, where studies indicate comparable performance to currently used materials.5 Additionally, they have been tested as extra-terrestrial solar cells for spacecraft power systems.6 Their performance under these conditions, such as absence of atmosphere (lack of moisture or oxygen) and extreme temperature fluctuations, has been tested on lead halide perovskite solar cells for a period of 10 months and shown potential.6
Despite their potential, their stability remains a challenge to address. Exposure to humidity, oxygen, illumination, and heat can all contribute to material deterioration.3 Particularly in their applications as photodetector or outer space solar cell, commercial implementation requires stability against high energy radiation.7,8 For this purpose, the X-ray stability of different perovskite compositions has to be assessed.
In addition to the previously mentioned applications, various characterization techniques which are extensively used for the study of lead halide perovskites rely on X-rays, such as powder and single crystalline X-ray diffraction (XRD), scanning X-ray fluorescence (XRF), energy dispersive X-ray analysis (EDX) and photoelectron spectroscopy (PES). To obtain reliable results, it is crucial to identify and minimize any degradation from the measurement process itself. For example, PES is extensively used to study surfaces and interfaces of lead halide perovskites9,10 and to investigate causes for their degradation when exposed to environmental stressors like humidity or heat.3,11,12 However, the interpretation of the results becomes more challenging if X-ray induced initial shifts in binding energy position,13 or the formation of metallic lead (Pb0) are present.8,14 Therefore, understanding the degradation mechanisms behind high energy radiation is necessary to prevent radiation-induced damage and to ensure a correct interpretation of measurement results.
Despite the high importance of understanding the degradation mechanisms caused by X-ray radiation, limited research has been done so far and most studies have focused on thin films. Considering the improving performance of perovskite single crystals in devices (e.g. solar cells or photodetectors),15–17 there is an interest to understand the stability behavior of single crystals both for single crystals devices and as a model system for thin films.
Some examples are a comparative X-ray stability study on CsPbBr3, CsMAPbBr3, and MAPbBr3 thin films on top of a half solar cell structure (FTO/TiO2/Perovskite), where a stability dependence on the monovalent cation was found by PES characterization.18 Specifically, the purely inorganic CsPbBr3 was found to be rather stable with no detection of metallic lead (Pb0), followed by CsMAPbBr3 which contained a steady amount of Pb0 throughout the whole experiment. In contrast, metallic lead was detected after two hours of X-ray irradiation on the MAPbBr3 thin film with the Pb0 concentration increasing over time. Density functional theory (DFT) calculations suggested that the metallic lead initiates MABr vacancies which then migrate towards the surface. Other studies in literature compared MAPbI3, FAPbI3, CsxMA1−xPbI3 and CsxMAyFA1−x−yPbI3 thin films after exposure to a controlled amount of γ radiation and observed a formation of Pb0 for all compositions except FAPbI3.19 MAPbI3 thin films were investigated via PES under ultrahigh vacuum conditions and it was reported that soft X-rays caused a rapid decomposition of the organic cation and, after long exposure, also the formation of metallic lead.20 Furthermore, a comparative stability study on MAPbI3, MAPbBr3, Cs0.15FA0.85PbI3, Cs0.1MA0.15FA0.75PbI3, CsPbI3, and CsPbBr3 thin films using PES and XRD has shown no γ-ray radiation damage on most compositions, except MAPbI3 and CsPbI3.21 MAPbI3 demonstrated degradation to PbI2 and CsPbI3 degraded to Pb0 at the surface. However, experiments on full solar cell stacks of these compositions showed that the performance of the MAPbI3 solar cell was only slightly affected by γ-rays. These results together with DFT calculations suggested that the radiation-induced degradation was reversible in a full solar cell stack for MAPbI3. Additionally, Milotti et al. investigated the X-ray stability of FAPbBr3 thin films using PES and micro-photoluminescence recently.22 They observed the formation of metallic lead and the generation of Br2 gas. However, prolonged irradiation led to re-oxidation of metallic lead and FA+ and Br− ion migration, which was reported as self-healing of the material. In our previous study, we compared the X-ray stability between CsPbBr3 and CsFAPbI3 thin films, where we reported a higher stability of CsPbBr323 and identified a distinct degradation path for each composition. In CsPbBr3, X-ray exposure resulted in defect accumulation, eventually leading to the formation of Pb0. In contrast, we found degradation of the organic cation FA+ without the formation of Pb0 in CsFAPbI3.
Polycrystalline thin films typically degrade initially at the grain boundaries under environmental stressors. In other words, variations in the film formation (and therefore grain structure) due to the preparation methods may significantly impact degradation processes.3 To avoid these external factors, single crystal surfaces instead of thin film surfaces can be studied. We have shown that clean surfaces of perovskite single crystals can be prepared in vacuum through cleaving,10,11,24 which enables us to study intrinsic behaviors of the material. Therefore, to take our previous study further and to gain insight into the behavior of lead halide perovskite surfaces with X-rays without the effects of grain boundaries, we decided to use perovskite single crystals in the work presented here. There are few studies on the X-ray stability of perovskite single crystals reported in literature, despite the implementation of single crystals in practical applications increasing considerably.25 For instance, Motoki et al. studied the X-ray stability of MAPbI3 single crystals using in-house PES.26 They compared core levels of the perovskite after exposure to X-rays for durations from 0 to 200 hours and found minor oxygen contributions. The core level spectra N 1s, I 3d and Pb 4f showed a decrease and remained constant after 40 hours, indicating X-ray induced degradation and eventually stabilization. Other studies focused on X-ray and e-beam induced degradation of MAPbBr3 single crystals using various techniques, such as surface photovoltage spectroscopy, XRD and PES. Exposing the crystals to X-rays under ambient conditions lead to quenching of free excitons into a new bound species, with the material recovering after one week under dark conditions and low humidity.27 On the other hand, both X-ray and e-beam irradiation under ultra-high vacuum (UHV) conditions induced the formation of metallic lead28,29 and the release of MABr and Br2.28
In the study presented here, the X-ray stability of clean surfaces of different lead halide perovskite single crystals was investigated via synchrotron-based PES, as PES allows following the compositional (chemical) changes on the perovskite surface and therefore to identify degradation pathways. To observe general, composition-dependent trends and compare the effects of the halide and the A-site cation, five different perovskite single crystal compositions were analyzed, and the results were divided in two main groups. MAPbBr3, MAPbI3 and MAPbCl3 were measured to study the impact of the halide on the X-ray stability (MA+-series) and MAPbBr3, FAPbBr3 and CsPbBr3 (Br-series) were measured and compared to investigate the effect of the A-site cation.
Experimental methods
Synthesis of single crystals
Perovskite single crystals, including MAPbI3, MAPbBr3, MAPbCl3, FAPbBr3, and CsPbBr3, with diameters ranging from 0.5 to 1 cm, were synthesized using the inverse temperature crystallization (ITC) method. The starting materials were used as bought without further treatment. Methylammonium chloride (CH3NH3Cl3, MACl), methylammonium iodide (CH3NH3I, MAI), methylammonium bromide (CH3NH3Br, MABr), formamidinium bromide (CH(NH2)2Br, FABr) and cesium bromide (CsBr) were purchased at Sigma Aldrich. Lead chloride (PbCl2), lead iodide (PbI2) and lead bromide (PbBr2) were bought at TCI. To prepare the precursor solutions, 1 M solutions of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 AX:PbX (A = MA/FA and X = I/Br/Cl) were made using different solvents such as γ-butyrolactone (GBL) for MAPbI3, dimethylformamide (DMF) for MAPbBr3 and FAPbBr3, dimethyl sulfoxide (DMSO) for MAPbCl3. The solutions were stirred at room temperature until all the precursors were fully dissolved. For CsPbBr3, CsBr and PbBr2 (in a molar ratio of 1:2) were dissolved on DMSO to form a saturated perovskite precursor solution. After filtration through a 0.45 μm PTFE filter, each solution was transferred to an open glass vial and heated to specific temperatures: 100 °C for MAPbI3, 80 °C for MAPbBr3 and FAPbBr3, 90 °C for MAPbCl3, and 120 °C for CsPbBr3. Once the desired temperature was reached, selected seed crystals were introduced into a new solution and allowed to grow with the vial sealed until the desired crystal size was achieved.
:1 AX:PbX (A = MA/FA and X = I/Br/Cl) were made using different solvents such as γ-butyrolactone (GBL) for MAPbI3, dimethylformamide (DMF) for MAPbBr3 and FAPbBr3, dimethyl sulfoxide (DMSO) for MAPbCl3. The solutions were stirred at room temperature until all the precursors were fully dissolved. For CsPbBr3, CsBr and PbBr2 (in a molar ratio of 1:2) were dissolved on DMSO to form a saturated perovskite precursor solution. After filtration through a 0.45 μm PTFE filter, each solution was transferred to an open glass vial and heated to specific temperatures: 100 °C for MAPbI3, 80 °C for MAPbBr3 and FAPbBr3, 90 °C for MAPbCl3, and 120 °C for CsPbBr3. Once the desired temperature was reached, selected seed crystals were introduced into a new solution and allowed to grow with the vial sealed until the desired crystal size was achieved.
The most promising crystals were chosen for characterization using photoelectron spectroscopy. The remaining crystals from the same batch were grounded using a mortar and pestle and characterized by powder X-ray diffraction analysis. Fig. S1 (ESI†) demonstrates that all the synthesized materials exhibit a single phase, free from any impurities, which is consistent with the results obtained from the single crystal XRD profiles from literature.
Photoelectron spectroscopy and beamline set-up
The PES measurements were carried out at the SMS-branch of the FlexPES30 beamline at the MAX IV synchrotron source in Lund. To measure the single crystal surfaces, the single crystals were mounted on sample plates using a two-component epoxy (EPO-TEK H20E) and cured at 100 °C for one hour under ambient conditions. The single crystals were then transferred into a preparation chamber under ultra-high vacuum (UHV) conditions and cleaved at around 1 × 10−8 mbar to obtain a clean crystal surface for the PES measurements. Subsequently, they were measured at 10−9–10−10 mbar.The X-rays for the measurements were generated using a linearly polarized undulator (period length = 54.4 mm) and monochromated via a plane grating (Zeiss SX700). To detect the ejected photoelectrons, a hemispherical spectrometer (Scienta DA30-L (W)) in normal emission mode was used. Relevant core level spectra (N 1s, C 1s, Cs 4d, Pb 4f, I 4d, Br 3d, Cl 2p, O 1s) were recorded in loops at 600 eV photon energy with a step size of 0.1 eV and a pass energy of 100 eV for each crystal composition.
All core levels were intensity normalized with respect to the Pb 4f intensity and energy calibrated against Pb 4f7/2 (Pb2+) = 138.54 eV binding energy,24 unless otherwise mentioned. The quantification was done using peak fitting with the pseudo-Voigt function with a linear, or Shirley background. All N 1s core levels were fitted using a single peak, I 4d, Cl 2p and FA-based Br 3d core levels were fitted using a single spin–orbit doublet. On the other hand, all Pb 4f core levels and Cs 4d and Br 3d core levels from CsPbBr3 were fitted with 2 peaks to account for the different contributions. As single crystal surfaces were investigated, a homogeneous distribution of their surface was assumed. The inelastic mean free path (IMFP) was estimated using the TTP-2M method. The IMFP ranged between 0.71 and 1.56 nm depending on the core level and the crystal. Details of the calculation can be found in the ESI† in Tables S1 and S2.
The sample degradation varies depending on the X-ray intensity. To avoid instant degradation through beam damage and to facilitate the study of degradation over larger timescales the beam was defocused and had a fullwidth half maximum (FWHM) of 1 × 0.4 (horizontal × vertical) mm in normal incidence, and the incident beam intensity was regulated via the exit slit. Considering a Gaussian intensity distribution, the standard deviation was estimated to be 0.43 mm (horizontal) × 0.17 mm (vertical) from the FWHM. Based on these values and assuming an elliptical beam, we estimate that 68% of the incident beam intensity fall in an area of 0.23 mm2. The number of photons was calculated based on the time required to measure one loop of core levels and the number of photons per loop, considering the corresponding slit and beam size for each sample. The fluence (total number of photons per cm2) was calculated by multiplying the number of photons with 0.68/0.0023 (see Table S3 in the ESI† to obtain all values per loop).
The exit slit was adjusted to 10 μm (low flux, LF = 1.2 × 1011 photons per second) for the MAPbBr3 and MAPbCl3 single crystals, and to 30 μm (high flux, HF = 3.6 × 1011 photons per second) for the more X-ray-stable MAPbI3 and CsPbBr3 compositions. FAPbBr3 was measured under both LF and HF. The flux values were theoretically estimated based on the used beamline settings as is described in the FlexPES beamline paper.30 These theoretical values are expected to be somewhat higher than the actual ones at the used photon energy, but give a good estimate, which is based on the beamline settings used in the experiment. The results presented for FAPbBr3 throughout the paper were measured under high flux, as this allows us to follow the degradation quicker and, therefore, improves comparability to the measurements of the other compositions. The core levels of FAPbBr3 measured using low flux can be found in the ESI† in Fig. S2. It should be noted that the exposure time for the measurement under high flux of photons was reduced to achieve the same photon count.
Results and discussion
The work presented here studied five distinct lead halide perovskite single crystal compositions (MAPbI3, MAPbBr3, MAPbCl3, FAPbBr3, and CsPbBr3), which can be categorized into two series. The first series consists of a constant cation (methylammonium) with varying halides (iodide, bromide, chloride) while the second series consists of a constant halide (bromide) with varying cations (methylammonium, formamidinium, cesium). Depending on the composition of the material, different degradation patterns were observed, including the formation of metallic lead (Pb0) and/or the degradation of the organic cation.Fig. 1 depicts the evolution of Pb 4f, N 1s, Cs 4d, and halide (I 4d, Br 3d, Cl 2p) core levels spectra for all the studied materials during irradiation. Fig. S3 in the ESI† provides additional core level spectra (C 1s and O 1s). All core level spectra presented in both figures were measured using a photon energy of 600 eV, intensity normalized to the total amount of Pb 4f (Pb0 + Pb2+), and energy calibrated against Pb 4f7/2 (Pb2+) at 138.54 eV binding energy. Fig. S4 in ESI† shows the same core level spectra as Fig. 1 but normalized to the perovskite contribution (Pb2+). All the measurements were carried out in loops for each single crystal under constant irradiation with the initial spectrum shown in blue and the final in orange.
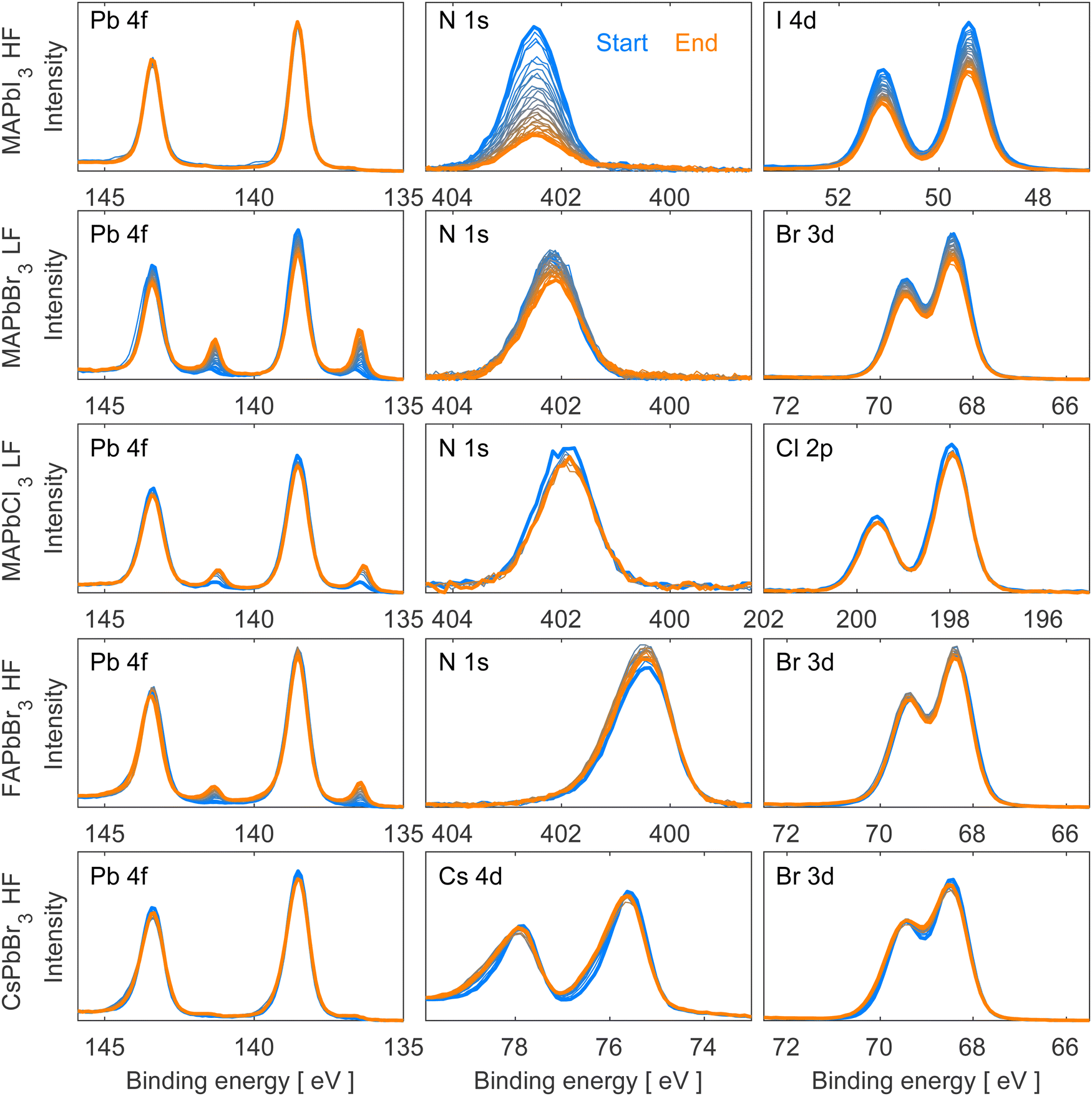 | ||
| Fig. 1 Pb 4f, N 1s/Cs 4d and I 4d/Br 3d/Cl 2p core level spectra of clean surfaces of MAPbI3, MAPbBr3, MAPbCl3, FAPbBr3 and CsPbBr3 single crystals, recorded using the FlexPES beamline (MAX IV) with a photon energy of 600 eV. The spectra are intensity normalized to the total amount of Pb 4f and energy calibrated against Pb 4f7/2 (Pb2+) = 138.54 eV binding energy. Low flux (LF = 1.2 × 1011 photons per second), high flux (HF = 3.6 × 1011 photons per second). | ||
The beam flux was adjusted depending on the exact crystal composition to avoid immediate beam degradation and enable degradation processes to happen within a reasonable timeframe. Since MAPbI3, FAPbBr3 and CsPbBr3 exhibit greater stability towards X-rays, they were exposed to a higher beam flux (HF = 3.6 × 1011 photons per second). Conversely, due to their lower stability towards X-rays, MAPbCl3 and MAPbBr3 were measured at a lower beam flux (LF = 1.2 × 1011 photons per second) to slow down the degradation processes.
We observe no oxygen contamination and a clear C 1s peak corresponding to the perovskite and no adventitious carbon, which usually appears lower binding energies (around 284.8 eV)31,32 for all measured single crystals except for FAPbBr3 and CsPbBr3 which both present a small contribution of adventitious carbon and oxygen (Fig. S3, ESI†). Despite not having a perfectly clean surface for these two crystals, the small amounts of oxygen and adventitious carbon do not change during the experiments, and therefore do not take part in the degradation process and should not interfere with our results.
As can be seen in Fig. 1 and Fig. S4 (ESI†), all measured single crystals present a similar shape of their Pb 4f core level corresponding to Pb2+ with the Pb 4f7/2 peak calibrated at 138.54 eV. A metallic lead (Pb0) contribution appears during X-ray irradiation at a binding energy of 137.0 eV33 and will be discussed in detail in the following paragraphs. All Br-based compounds also present a similar shape and position of Br 3d doublet associated with Br− (68.4 and 69.5 eV). The N 1s core level presents the same shape for all four hybrid materials with a clear difference in position for FA+-based (400.5 eV (Br)) and MA+-based (401.8 eV (Cl), 402.1 eV (Br) and 402.5 eV (I)) perovskites due to the different chemical environments of the elements in the organic cations. For MAPbI3 and MAPbCl3, the observation of a I 4d doublet with the I 4d5/2 at 49.4 eV and Cl 2p doublet with the Cl 2p3/2 at 197.9 eV confirms the presence of these anions. The detection of Cs 4d doublet with the Cs 4d5/2 at 75.5 eV confirms the presence of Cs+ in the CsPbBr3 compound.
Analyzing Fig. 1, MAPbI3 exhibited only small amounts of metallic lead during the experiment, while the organic cation (observed through nitrogen) and iodide intensities decreased significantly. In contrast, the other hybrid perovskite single crystals (MAPbBr3, MAPbCl3, and FAPbBr3) formed significant amounts of metallic lead under irradiation accompanied by a decrease of the organic cation and halide content in the case of MAPbBr3 and MAPbCl3. On the other hand, FAPbBr3 depicts only slight changes in the Br 3d spectra and an increase in the N 1s peak intensity, in agreement with literature.22 Lastly, the fully inorganic CsPbBr3 perovskite single crystal demonstrated minimal metallic lead formation and peak broadening of the Br 3d and the Cs 4d core level spectra. Similar results were previously reported by our group for CsPbBr3 thin films.23
To gain quantitative information about these changes and therefore the mechanisms behind them, all core levels were curve fitted. Notably, the N 1s and halide core level spectra of all crystal compositions except CsPbBr3 could be fitted well using the perovskite fit contributions. In other words, no additional non-perovskite contributions were found for those core levels for most compositions. Instead, we observed changes in peak intensities. For CsPbBr3, however, additional, non-perovskite peak contributions were found at higher binding energy for the Cs 4d and Br 3d core levels. Fits of the first and last measurement can be found in Fig. S5 and S6 in the ESI† and fitting details are described in the experimental section.
The trends of the metallic lead formation for the different studied perovskite crystal compositions and their dependence on the fluence (photons cm−2) were examined after careful curve fitting of all individual Pb 4f spectra. The contribution of metallic lead as a function of fluence (photons cm−2) is presented in Fig. 2, which clearly describes two trends. Firstly, focusing on the series with the same cation (MA+) at the A-site and varying X-site halides (I−, Br−, Cl−): a small amount of metallic lead was detected on the MAPbI3 crystal surface, which is consistent with the results presented in Fig. 1 and previous literature observations.34 About 1% of metallic lead was found after exposing the surface to a dose of 5 × 1017 photons cm−2. After this point the amount of metallic lead decreased somewhat to stabilize at below 1% despite using the higher flux (HF) and exposing the material to 22.5 × 1017 photons cm−2. However, the lower flux (LF) caused approximately 10% metallic lead on the MAPbBr3 surface after receiving 2.4 × 1017 photons cm−2 and 16% after receiving 5 × 1017 photons cm−2 and increased further until the end of the experiment (8.3 × 1017 photons cm−2). In comparison, MAPbCl3 formed 10% metallic lead after exposure to only half the fluence (1.2 × 1017 photons cm−2) compared to MAPbBr3.
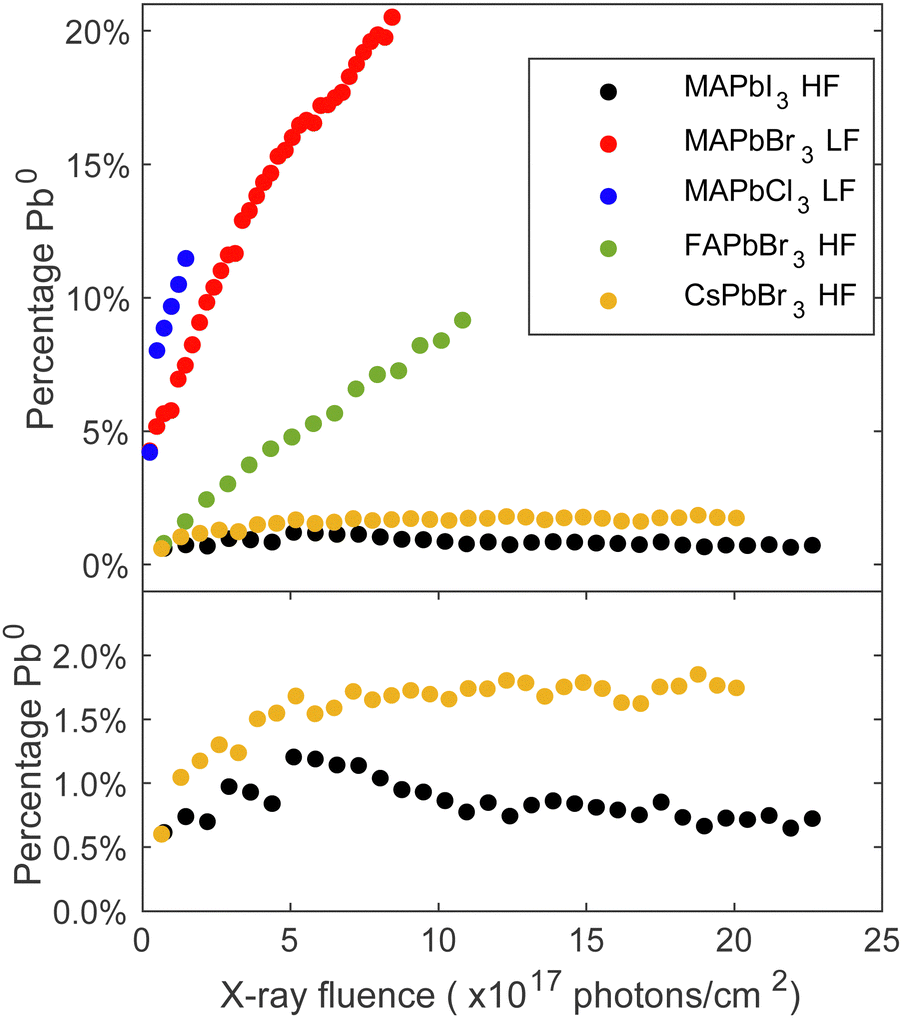 | ||
| Fig. 2 Percentage Pb0 (Pb0 intensity/total Pb intensity) on the surface of each material (MAPbI3, MAPbBr3, MAPbCl3, FAPbBr3, CsPbBr3) as a function of fluence (photons cm−2). Top panel: All compositions. Bottom panel: Zoom on MAPbI3 and CsPbBr3. | ||
In the series with a constant halide (Br−) and varying cations (Cs+, FA+, MA+), the MAPbBr3 crystal was least stable. FAPbBr3 formed 5% metallic lead after exposure to 5 × 1017 photons cm−2 using the higher flux (HF) and the fully inorganic CsPbBr3 demonstrated only a small amount of metallic lead formation. Approximately 1.6% metallic formed after receiving 5 × 1017 photons cm−2 using the higher flux (HF), and this amount did not increase much more under continued irradiation. This is expected as CsPbBr3 lacks the volatile organic cation.35 Other studies have reported the presence metallic lead in all these compositions under environmental stressors such as light or X-rays.22,27,34,36,37 However, most studies focus on polycrystalline thin films, where degradation is different and facilitated by the grain boundaries and vacancy formation. It is worth noting that exposing different spots on the same FAPbBr3 single crystal to an equal number of photons (e.g. 7.4 × 1017 photons cm−2) but with a different flux leads to different amounts of metallic lead formation: the spot measured with the high flux (HF) formed twice as much metallic lead as the spot measured with low flux (LF), as shown in Fig. S7 in the ESI.† Our previous study also presented a dependence on the flux and found that once Pb0 is detected, we observe an accelerated formation of Pb0.23 On the contrary, it is possible to prevent the formation of Pb0 by using a sufficiently low flux. Therefore, the choice of flux has a direct impact on the formation rate of Pb0 and, hence, on the degradation process of the single crystal surface. In the context of the FAPbBr3 crystal examined in this experiment, the crystal has the capacity to absorb a greater quantity of photons when they are distributed over a longer period of time. In summary, both the organic cation and the halide anion impact the degradation mechanism and the kinetics of forming metallic lead.
We will now continue to discuss the degradation mechanisms for each compound in detail starting with the MA+-series. To be consistent with the results presented in Fig. 1, all core level intensities are divided by the total lead intensity (Pb0 + Pb2+) and initial ratios of nitrogen and halide were set to those expected from the perovskite stoichiometry (1 MA+ cation, represented by the nitrogen, and 3 halides). Fig. 3 depicts the evolution of core level intensities of the MA+-series crystals as a function of fluence, showing the lead and nitrogen on the left axis (black) and the halide on the right axis (blue). The axes are scaled in a 1:3 (left:right) ratio for MAPbBr3 and MAPbCl3. This means that the intensities of the perovskite components are scaled in relation to their abundance in the pristine perovskite crystal. For MAPbI3 the axes are scaled in a 1:1 (left:right) ratio to be able to directly follow a radiolysis process expected for methylammonium iodide and how this develops during exposure. Overall, all materials, except MAPbI3, show a notable decrease in their Pb2+ intensities which stems from the formation of metallic lead and is consistent with the findings reported in Fig. 2. Furthermore, a general reduction in the halide and organic cation contents is observed for all compositions.
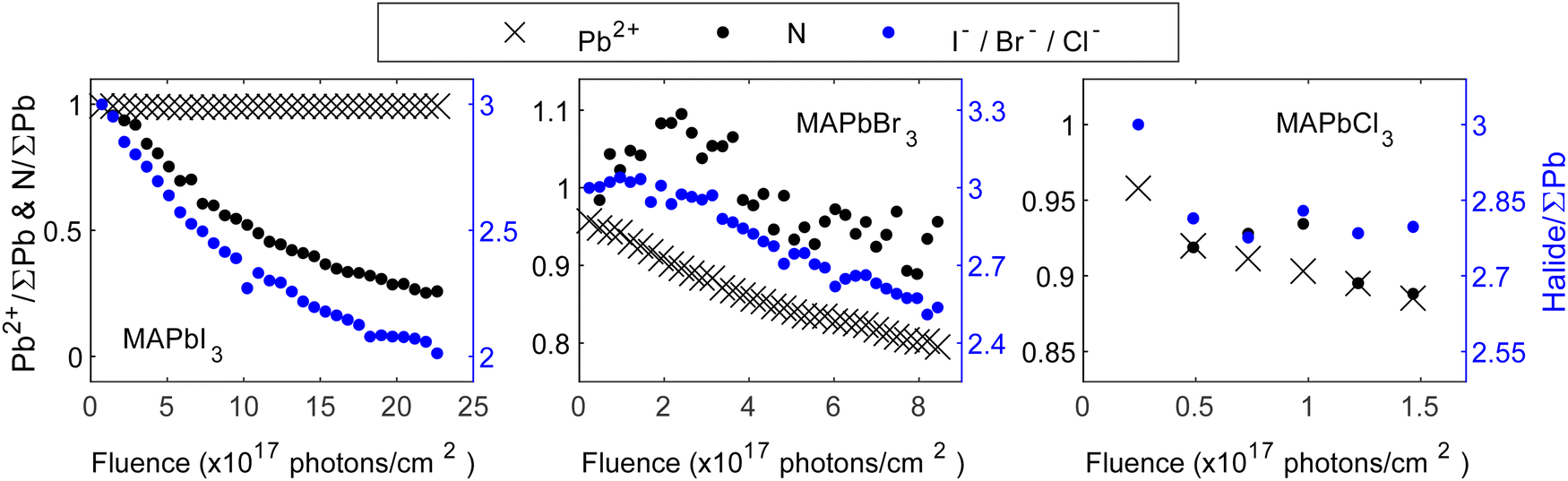 | ||
| Fig. 3 Core level intensity evolution for the crystal surfaces of MAPbI3 (left), MAPbBr3 (middle) and MAPbCl3 (right) as a function of the fluence (photons cm−2). Intensity normalized by the total amount of lead (Pb0 + Pb2+) and initial ratios set to those expected from the perovskite stoichiometry (1 nitrogen and 3 halides). The left axes (black) refer to Pb2+ and N, the right axes (blue) refer to the halide. | ||
Analyzing Fig. 3 in depth, different reaction mechanisms can be proposed. Starting with MAPbI3, a significant and rapid decrease in the intensities of its N 1s core level, accompanied by a decrease in its I 4d core level, is observed. Previous studies have reported the formation of MAI and PbI2, where PbI2 subsequently decays to metallic lead and iodine.20,38 Here, the Pb2+ signal remains almost stable throughout our measurements, indicating only a small amount of metallic lead formation. However, a possible degradation to MAI and PbI2 still holds and requires the simultaneous removal of one nitrogen and one iodide. With the scaling of the y-axis used here, this could have been observed through overlapping data points for N and I in Fig. 3. Considering the difference in IMFP of nitrogen and iodide (0.76 nm and 1.50 nm, respectively, further details in Table S1 in the ESI†), a stronger decrease of the N intensity would be expected if MAI is mainly removed in the surface region. However, here we observe that the I intensity decreases relatively more. This suggests that in addition to MAI removal from the perovskite surface, also some additional iodide is removed relative to the remaining perovskite components (Pb and organic cation). It is important to note that the PES measurements are carried out under UHV conditions. Hence, any gaseous degradation products are being quickly removed from the surface and any reversible reactions, as others have reported for MAPbI3, 21 cannot be observed in this experiment.
Overall, the results indicate the simultaneous disappearance of (at least) one iodide per organic cation from the surface of the single crystal. Therefore, our results align best with the hypothesis of MAI radiolysis, accompanied by the generation of PbI2:
| MAPbI3(s) → PbI2(s) + gaseous products | (1) |
Moving to the analysis of MAPbBr3, the Pb2+ intensity decreases throughout the experiment, and is accompanied by the formation of metallic lead, a result that has been reported frequently in literature.27–29 Furthermore, in both nitrogen and bromide an overall intensity decrease is observed, suggesting a partial removal of the organic cation and the halide from the substrate. It is important to note, that the observed decrease of the bromide and nitrogen intensities is delayed relative to the decrease of Pb2+, i.e., the cause of the initial decrease in the signals of the different components are different and thus originate from different reactions and kinetics. Furthermore, the N intensity initially increases followed by a decrease, and the Br intensity decreases more than the N intensity. This indicates that multiple processes are involved in the degradation, in contrast to the behavior observed for MAPbI3 where the organic cation and the halide disappear together. Ion migration has been frequently reported for lead halide perovskites and could explain some of the observed behavior.39–42 Specifically, the N intensity could increase initially due to ion migration and subsequently decrease due to radiolysis of MABr. The bromide intensity, on the other hand, may decrease faster than nitrogen due to a combination of MABr radiolysis and metallic lead formation. Overall, we propose that the degradation to MABr and Br228 is accompanied by ion migration of methylammonium ions from the bulk towards the surface as well as metallic lead formation:
| MAPbBr3 → Pb0(s) + MA+ + Br− + Br2(g) | (2.1) |
| Brbulk− → Brsurface− | (2.2) |
| MAbulk+ → MAsurface+ | (2.3) |
| MA+ + Br− → gaseous products | (2.4) |
In the case of MAPbCl3, the intensity decreases of Pb2+, nitrogen and chloride follow each other. Given the scaling of the y-axis, this means that we observe a simultaneous loss of all components in contrast to both MAPbI3 and MAPbBr3. Specifically, the formation of metallic lead and the loss of the organic cation and halide happen simultaneously, suggesting that radiolysis of MACl occurs simultaneously with metallic lead formation:
| MAPbCl3 → Pb0(s) + gaseous products | (3) |
In general, two different reactions have been discussed for removal of MAX from the perovskites in the scientific community:3,43 MAX can form NH3 and CH3X or CH3NH2 and HX. As an example, it was found that MAPbI3 forms CH3NH2 and HI at lower temperatures and MAI and NH3 at higher temperatures.44 However, from our experiments it is not possible to distinguish which gaseous products are formed.
After discussing the trends for MA-series we will now focus on the trends for remaining perovskites containing the Br− anion. Fig. 4 shows the core level intensities evolution of FAPbBr3 as a function of fluence (photons cm−2) where the intensity relation between the left:right axis is 1:3. Similar to Fig. 3, all core level intensities were normalized by the total amount of lead (Pb0 + Pb2+) and initial ratios were set to those expected from the perovskite stoichiometry (1 FA+ cation and 3 Br−). While the lead content decreases continuously, the bromide and nitrogen intensities initially increase, followed by a subsequent decrease, similar to the observations made for MAPbBr3. The bromide intensity returns close to its initial relative value of 3 Br−/∑Pb, while an excess of nitrogen remains at the surface. This suggests that the metallic lead formation is also accompanied by other reactions. Previous studies have reported self-healing effects in FAPbBr3 thin films.22 However, we observe a concentration increase beyond expected levels, followed by a decline and no recovery of metallic lead to Pb2+. In contrast, self-healing implies an initial decrease followed by restoration of the original composition. Therefore, ion migration from the bulk to the surface seems more likely, as it would lead to higher concentrations of FA+ and Br− at the surface but leave the bulk FABr-deficient. At the same time, the eventual decrease of nitrogen and bromide indicates a removal of the organic cation and bromide ions, most likely by radiolysis. Notably, the organic cation exhibits a stronger increase compared to the halide. Considering that the formation of metallic lead involves the loss of bromide ions, a decrease together with Pb2+ would be expected. However, this seems to be compensated by the bromide ion migration. Overall, similar reactions to MAPbBr3 seem likely:
| FAPbBr3 →FA+ + Br− + Pb0(s) + Br2(g) | (4.1) |
| Brbulk− → Brsurface− | (4.2) |
| FAbulk+ → FAsurface+ | (4.3) |
| FA+ + Br− → gaseous products | (4.4) |
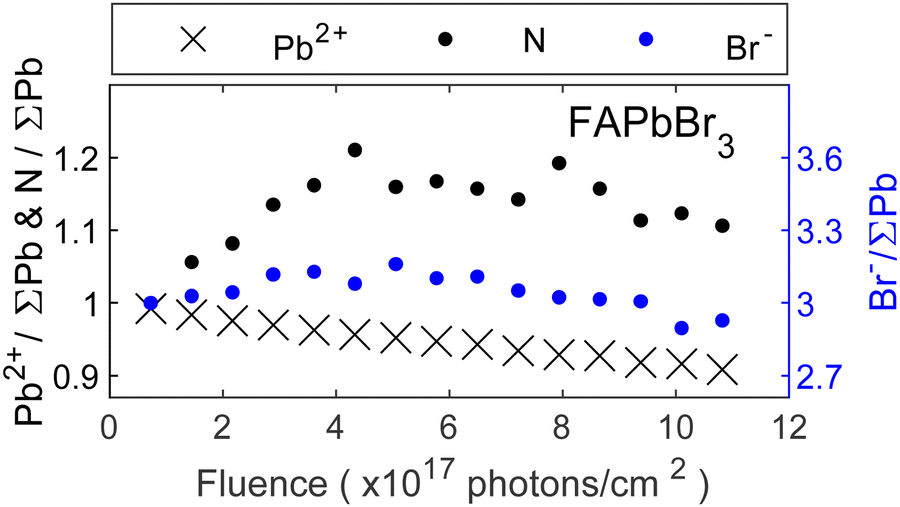 | ||
| Fig. 4 Core level intensity evolution for the FAPbBr3 crystal surface as a function of fluence (photons cm−2). Intensity normalized by the total amount of lead (Pb0 + Pb2+) and initial ratios set to those expected from the perovskite stoichiometry (1 cation represented by nitrogen and 3 bromide). | ||
However, the formation of metallic lead occurs at a slower rate compared to MAPbBr3 and ion migration leads to a temporary excess of FABr in the surface region.
Moving to the experiments on CsPbBr3, an in-depth analysis of the core level spectra via curve fitting showed that the degradation process involves the formation of a secondary species at higher binding energy in the Cs 4d and Br 3d core level spectra (fits can be seen in Fig. S5 and S6 in the ESI†). In this context, it is important to note that minor C 1s and O 1s contributions were present (see Fig. S3, ESI†). However, given their consistent intensity levels throughout the experiment and considering their low overall intensities, no significant impact on the presented results is expected. Fig. 5A depicts the evolution of core level intensities of total Cs 4d and total Br 3d normalized by total lead (Pb0 + Pb2+) as a function of fluence. The initial values were set to the expected CsPbBr3 perovskite stoichiometry (1 Cs+ and 3 Br−). Cesium intensities are represented by the left axis (black), while bromide is represented by the right axis (blue) with a linear 1:1 scaling between the two axes. Both, the total amount of Cs and Br increase together in a 1:1 ratio relative to the total Pb 4f intensities, suggesting that the excess of Cs and Br forms in the same reaction. As the intensity increase is observed relative to the total lead intensity, we suggest that ion migration of Br− and Cs+ is responsible.
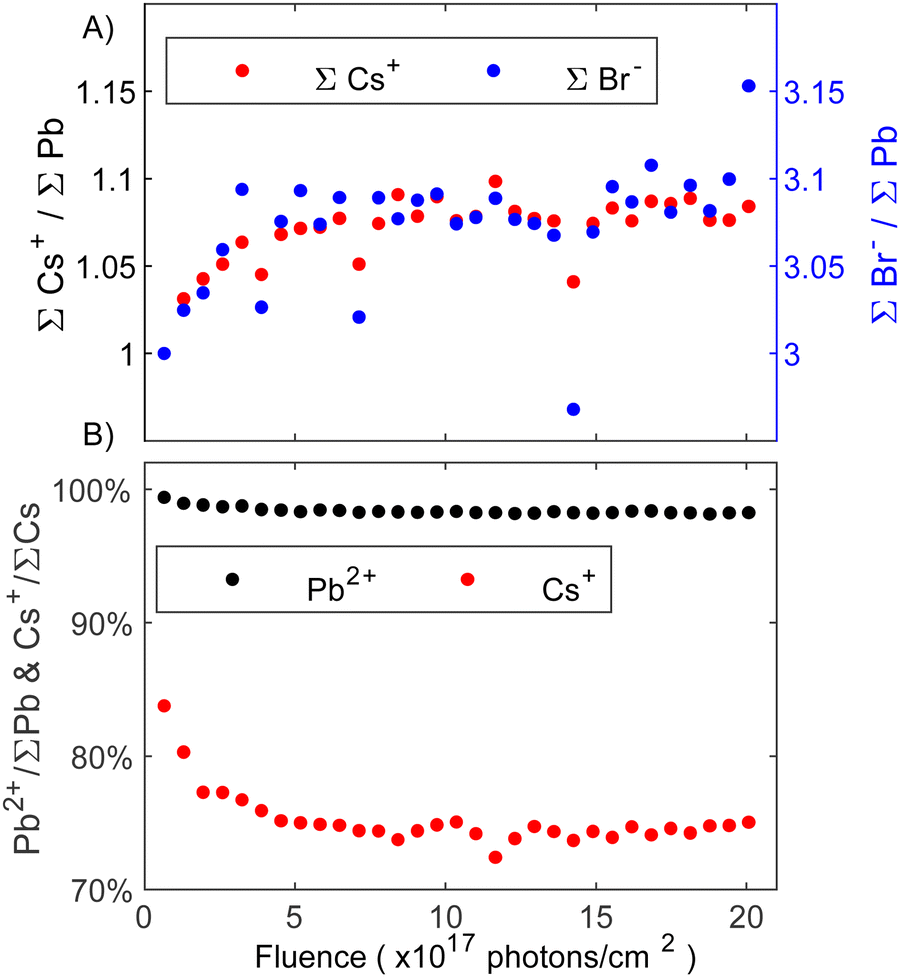 | ||
| Fig. 5 (A) Core level intensity evolution of total Cs+ (left, black axis) and total Br− (right, blue axis) of the CsPbBr3 surface as a function of fluence (photons cm−2). Intensity normalized by the total amount of lead (Pb0 + Pb2+) and initial ratios set to those expected from the perovskite stoichiometry (1 Cs+ and 3 Br−). The axes are scaled in a 1:1 ratio to one another. (B) Comparison of the percentage of decrease in perovskite Pb2+ and Cs+ contributions at the surface relative to total Pb 4f and total Cs 4d core level intensity, respectively. | ||
Fig. 5B shows the percentage of the perovskite Pb2+ and Cs+ core level intensities relative to the total amount of Pb and Cs, respectively. It can be seen in Fig. 5B that the perovskite Pb 4f contribution (Pb2+) presents an initial small decrease and then is almost constant throughout the entire experiment while the intensity of the perovskite Cs+ quickly decreases to 75% indicating a change in the surface composition. Considering that overall Cs+ and Br− increase by the same amount (Fig. 5A), the formation of CsBr seems likely. This aligns with our previous study on thin films where CsBr was identified as a degradation product with the CsBr core levels appearing at higher binding energies than the perovskite Cs and Br.23 Furthermore, PES experiments on (Cs0.06FA0.79MA0.15)Pb(I0.85Br0.15)3 perovskite thin films with varying amounts of KBr as a passivator similarly showed a secondary Br 3d contribution at higher binding energy, which was assigned to KBr.45 Despite different cations, one would expect similar trends in the binding energy shift for KBr and CsBr. We therefore assign the new Cs and Br species to CsBr. However, higher quantities of CsBr are formed significantly quicker on the single crystal surface investigated here than on the thin film surface studied previously. In this context it is important to note that small, non-perovskite Cs 4d and Br 3d contributions were observed from the start, which contrasts our previous study. This initial CsBr presence could explain why the degradation is accelerated, as the initial CsBr may act as a catalyst for further degradation. The CsBr layer stabilizes after receiving around 5 × 1017 photons cm−2 (Fig. 5B). Alongside CsBr, traces of metallic lead resulting from the degradation to PbBr2 on the surface were detected. Overall, the following degradation reactions proposed:
| CsPbBr3 → CsBr(s) + Pb0(s) + Br2(g) | (5.1) |
| Csbulk+ + Brbulk− → CsBrsurface | (5.2) |
Finally, our findings on the stability comparison of the MA+-series are summarized in Fig. 6. Considering the different degradation mechanism of each perovskite composition, the predominant reaction was used to show the percentage of remaining perovskite as a function of X-ray fluence. For MAPbI3, the percentage of N relative to total lead was used, while the percentage Pb2+ relative to total lead was used for MAPbBr3 and MAPbCl3. Given the complexity of the FAPbBr3 and CsPbBr3 degradation, selecting one core level as the main indicator of the remaining perovskite was not feasible. Consequently, these two compositions were excluded from this figure.
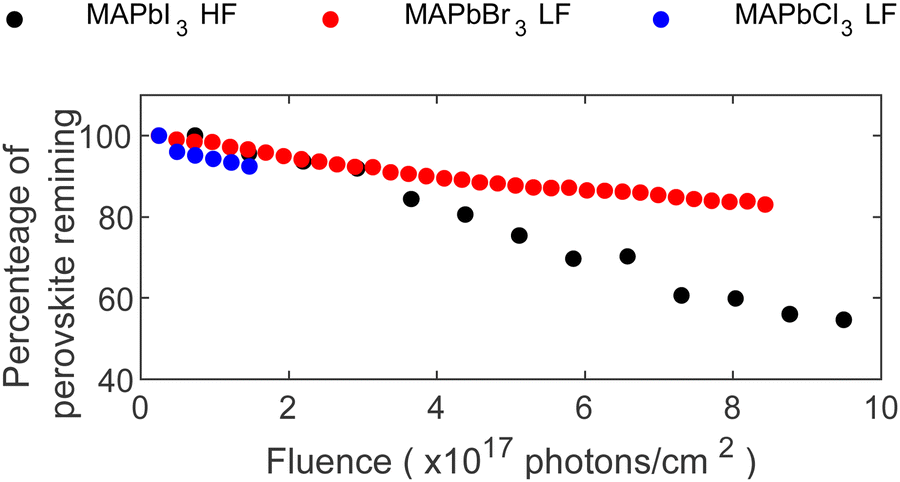 | ||
| Fig. 6 Percentage of perovskite remaining at the surface as function of X-ray fluence (photons cm−2). Determined by the percentage of N+/∑Pb in MAPbI3 and the percentage of Pb2+/∑Pb in MAPbBr3 and MAPbCl3. | ||
Comparing all investigated compositions in the MA+-series, MAPbCl3 shows the quickest degradation, and therefore lowest stability against X-rays, even under low flux conditions (LF). However, the experiment was stopped early in comparison due to the rapid accumulation of metallic lead on the surface, limiting the discussion of long-term behavior. The potential for longer experiments to study the evolution of MAPbCl3 remains open for exploration. Continuing with MAPbBr3, a similar but slower behavior as for MAPbCl3 under LF conditions can be found with the formation of metallic lead and a continuous decline in Pb2+ content. In comparison, MAPbBr3 retained 93% of its perovskite composition after exposure to 1.8 × 1017 photons cm−2 while MAPbCl3 retained only 89% after the same X-ray exposure.
In contrast, MAPbI3 behaved differently, i.e. formation of only little metallic lead despite exposure to higher photon flux (HF). Furthermore, its degradation mechanism was characterized by a consistent loss of the organic cation together with iodide, suggesting MAI radiolysis as a main degradation reaction. In contrast, MAPbBr3 involved multiple processes, including significant formation of Pb0 and ion migration. Comparable amounts of N and Pb2+ disappear for MAPbI3 and MAPbBr3, respectively, after receiving 1.8 × 1017 photons cm−2. Therefore, both compositions show similarly strong X-ray degradation but with different degradation paths. Radiolysis of MA+ is slowed down in MAPbBr3 compared to MAPbI3, while MAPbBr3 forms significantly larger amounts of Pb0. The difference in radiolysis could be related to an enhanced stability of MABr compared to MAI.46,47 Ion migration is in general observed in different perovskite compositions.39–41 McGovern et al. compared the halide migration ability of MAPbI3 and MAPbBr3 thin films and found similar activation energies for bromide migration in MAPbBr3 as for iodide ions in MAPbI3.47 Furthermore, they found that MA+ ion migration is hindered in MAPbBr3 but not in MAPbI3.47 In contrast, our experiments give direct evidence of ion migration in MAPbBr3 induced by X-ray irradiation of both Br− and MA+ through initial increase of both ion concentrations. On the other hand, ion migration is not directly observed for MAPbI3, as the intensity decrease of the MA+ and I− signals are most likely due to radiolysis. However, this does not exclude that ion migration also occurs in the MAPbI3, but if so, it is a slower process than radiolysis. Finally, the degradation of MAPbBr3 slowed down compared to MAPbI3 after receiving 3 × 1017 photons cm−2 resulting in a reduction to 80% Pb2+ for MAPbBr3 while the intensity of N in MAPbI3 decreased to 60% after receiving 8.2 × 1017 photons cm−2.
Conclusions
We successfully synthesized, in situ cleaved and characterized several lead halide perovskite single crystal compositions (MAPbI3, MAPbCl3, MAPbBr3, FAPbBr3, CsPbBr3). The study focused on their X-ray stability and different degradation mechanisms have been found. Distinct degradation pathways were identified for each material. In this context, MAPbI3 and CsPbBr3 form only small amounts of metallic lead, which stabilized during further X-ray radiation. MAPbCl3, MAPbBr3, FAPbBr3 formed substantial quantities of metallic lead, which continued to increase during the experiment. Additionally, our study revealed that while MAPbI3 and MAPbCl3 showed one main degradation pathway (radiolysis resulting in PbI2 and Pb0 formation, respectively), MAPbBr3, FAPbBr3 and CsPbBr3 underwent multiple parallel degradation processes. These included the formation of metallic lead, the formation of volatile and solid degradation products and ion migration. Specifically, the degradation of MAPbBr3, FAPbBr3 and CsPbBr3 involved the migration of both, the monovalent cation and the halide. Furthermore, our in-depth analysis revealed peak broadening in the cesium and bromide core levels of CsPbBr3, which was attributed to the formation of CsBr, which was potentially catalyzed by the presence of CsBr on the cleaved surface. Lastly, the MA+-series was compared to one another regarding their remaining perovskite after X-ray exposure. Using our experimental settings and exposure time, MAPbCl3 exhibited lowest stability against X-rays in the same fluence range compared to the other two crystals followed by MAPbI3 and MAPbBr3. Comparing MAPbI3 and MAPbBr3, both are similarly unstable under X-rays, but the dominating reactions are different. MAPbBr3 is more resistant to MA+ radiolysis but presents an easier formation of Pb0. MAPbI3, on the other hand, shows only little Pb0 formation and its degradation is dominated by MAI radiolysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Swedish Research Council (Grant no. VR 2018-04125, VR 2018-06465, VR 2018-04330 and 2022-03168), the Göran Gustafsson Foundation, the Swedish Energy Agency (Grant no. STEM P50626-1) and the Carl Tryggers Foundation (Grant no. CTS 18:59). This work was partially supported by the Wallenberg Initiative Materials Science for Sustainability (WISE) funded by the Knut and Alice Wallenberg Foundation. We acknowledge MAX IV Laboratory for time on Beamline FlexPES under Proposal 20220600. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. The authors also thank Alexei Preobrajenski, and Alexander Generalov for support during the FlexPES beamtime (proposal 20220600).References
- S. Porwal, D. Kumar, S. Ghosh, S. Kansal, S. Priya, A. Chandra and T. Singh, in Low-Dimensional Halide Perovskites, Elsevier, 2023, pp. 301–333 Search PubMed.
- X. Shen, K. Kang, Z. Yu, W. H. Jeong, H. Choi, S. H. Park, S. D. Stranks, H. J. Snaith, R. H. Friend and B. R. Lee, Joule, 2023, 7, 272–308 CrossRef CAS.
- J. Zhuang, J. Wang and F. Yan, Nano-Micro Lett., 2023, 15, 84 CrossRef CAS PubMed.
- Z.-Y. Chen, N.-Y. Huang and Q. Xu, Coord. Chem. Rev., 2023, 481, 215031 CrossRef CAS.
- S. Jia, Y. Xiao, M. Hu, X. He, N. Bu, N. Li, Y. Liu, Y. Zhang, J. Cui, X. Ren, K. Zhao, M. Liu, S. Wang, N. Yuan, J. Ding, Z. Yang and S. Liu, Adv. Mater. Technol., 2022, 7, 2100908 CrossRef CAS.
- W. Delmas, S. Erickson, J. Arteaga, M. Woodall, M. Scheibner, T. S. Krause, K. Crowley, K. T. VanSant, J. M. Luther, J. N. Williams, J. McNatt, T. J. Peshek, L. McMillon-Brown and S. Ghosh, Adv. Energy Mater., 2023, 13, 2203920 CrossRef CAS.
- X. He, Y. Deng, D. Ouyang, N. Zhang, J. Wang, A. A. Murthy, I. Spanopoulos, S. M. Islam, Q. Tu, G. Xing, Y. Li, V. P. Dravid and T. Zhai, Chem. Rev., 2023, 123, 1207–1261 CrossRef CAS PubMed.
- T. J. Jacobsson, S. Svanström, V. Andrei, J. P. H. Rivett, N. Kornienko, B. Philippe, U. B. Cappel, H. Rensmo, F. Deschler and G. Boschloo, J. Phys. Chem. C, 2018, 122, 13548–13557 CrossRef CAS.
- P. Schulz, D. Cahen and A. Kahn, Chem. Rev., 2019, 119, 3349–3417 CrossRef CAS PubMed.
- A. García-Fernández, B. Kammlander, S. Riva, D. Kühn, S. Svanström, H. Rensmo and U. B. Cappel, Inorg. Chem., 2023, 62, 15412–15420 CrossRef PubMed.
- B. Kammlander, S. Svanström, D. Kühn, F. O. L. Johansson, S. Sinha, H. Rensmo, A. G. Fernández and U. B. Cappel, Chem. Commun., 2022, 58, 13523–13526 RSC.
- S. Svanström, A. García-Fernández, T. J. Jacobsson, I. Bidermane, T. Leitner, T. Sloboda, G. J. Man, G. Boschloo, E. M. J. Johansson, H. Rensmo and U. B. Cappel, ACS Mater. Au, 2022, 2, 301–312 CrossRef PubMed.
- R. Larciprete, A. Agresti, S. Pescetelli, H. Pazniak, A. Liedl, P. Lacovig, D. Lizzit, E. Tosi, S. Lizzit and A. Di Carlo, Materials, 2021, 14, 3954 CrossRef CAS PubMed.
- J. Liang, X. Hu, C. Wang, C. Liang, C. Chen, M. Xiao, J. Li, C. Tao, G. Xing, R. Yu, W. Ke and G. Fang, Joule, 2022, 6, 816–833 CrossRef CAS.
- Y. Guan, C. Zhang, Z. Liu, Y. Zhao, A. Ren, J. Liang, F. Hu and Y. S. Zhao, Adv. Mater., 2022, 34, 2203201 CrossRef CAS PubMed.
- L. De Marco, G. Nasti, A. Abate and A. Rizzo, Solar RRL, 2022, 6, 2101085 CrossRef CAS.
- Z. Chen, B. Turedi, A. Y. Alsalloum, C. Yang, X. Zheng, I. Gereige, A. AlSaggaf, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2019, 4, 1258–1259 CrossRef CAS.
- J. Hieulle, D. Son, A. Jamshaid, X. Meng, C. Stecker, R. Ohmann, Z. Liu, L. K. Ono and Y. Qi, Adv. Funct. Mater., 2023, 33, 2211097 CrossRef CAS.
- V. V. Ozerova, N. A. Emelianov, D. P. Kiryukhin, P. P. Kushch, G. V. Shilov, G. A. Kichigina, S. M. Aldoshin, L. A. Frolova and P. A. Troshin, J. Phys. Chem. Lett., 2023, 14, 743–749 CrossRef CAS PubMed.
- W.-C. Lin, W.-C. Lo, J.-X. Li, Y.-K. Wang, J.-F. Tang and Z.-Y. Fong, npj Mater. Degrad., 2021, 5, 13 CrossRef CAS.
- A. G. Boldyreva, L. A. Frolova, I. S. Zhidkov, L. G. Gutsev, E. Z. Kurmaev, B. R. Ramachandran, V. G. Petrov, K. J. Stevenson, S. M. Aldoshin and P. A. Troshin, J. Phys. Chem. Lett., 2020, 11, 2630–2636 CrossRef CAS PubMed.
- V. Milotti, S. Cacovich, D. R. Ceratti, D. Ory, J. Barichello, F. Matteocci, A. Di Carlo, P. M. Sheverdyaeva, P. Schulz and P. Moras, Small Methods, 2023, 7, 2300222 CrossRef CAS PubMed.
- S. Svanström, A. García Fernández, T. Sloboda, T. J. Jacobsson, H. Rensmo and U. B. Cappel, Phys. Chem. Chem. Phys., 2021, 23, 12479–12489 RSC.
- A. García-Fernández, S. Svanström, C. M. Sterling, A. Gangan, A. Erbing, C. Kamal, T. Sloboda, B. Kammlander, G. J. Man, H. Rensmo, M. Odelius and U. B. Cappel, Small, 2022, 18, 2106450 CrossRef PubMed.
- M. Ghasemi, S. Yuan, J. Fan, B. Jia and X. Wen, J. Mater. Chem. A, 2023, 11, 3822–3848 RSC.
- K. Motoki, Y. Miyazawa, D. Kobayashi, M. Ikegami, T. Miyasaka, T. Yamamoto and K. Hirose, J. Appl. Phys., 2017, 121, 085501 CrossRef.
- G. Armaroli, L. Ferlauto, F. Lédée, M. Lini, A. Ciavatti, A. Kovtun, F. Borgatti, G. Calabrese, S. Milita, B. Fraboni and D. Cavalcoli, ACS Appl. Mater. Interfaces, 2021, 13, 58301–58308 CrossRef CAS PubMed.
- H. Syafutra, J.-H. Yun, Y. Yoshie, M. Lyu, S. N. Takeda, M. Nakamura, L. Wang and M.-C. Jung, Nanomaterials, 2020, 10, 1253 CrossRef CAS PubMed.
- C. Wang, B. R. Ecker, H. Wei, J. Huang and Y. Gao, J. Phys. Chem. C, 2018, 122, 3513–3522 CrossRef CAS.
- A. Preobrajenski, A. Generalov, G. Öhrwall, M. Tchaplyguine, H. Tarawneh, S. Appelfeller, E. Frampton and N. Walsh, J. Synchrotron Radiat., 2023, 30, 831–840 CrossRef CAS PubMed.
- H. Xie, X. Liu, L. Lyu, D. Niu, Q. Wang, J. Huang and Y. Gao, J. Phys. Chem. C, 2016, 120, 215–220 CrossRef CAS.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D’Haen, L. D’Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. De Angelis and H.-G. Boyen, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- B. Philippe, B.-W. Park, R. Lindblad, J. Oscarsson, S. Ahmadi, E. M. J. Johansson and H. Rensmo, Chem. Mater., 2015, 27, 1720–1731 CrossRef CAS.
- J. Liang, X. Hu, C. Wang, C. Liang, C. Chen, M. Xiao, J. Li, C. Tao, G. Xing, R. Yu, W. Ke and G. Fang, Joule, 2022, 6, 816–833 CrossRef CAS.
- S. Ullah, J. Wang, P. Yang, L. Liu, S.-E. Yang, T. Xia, H. Guo and Y. Chen, Mater. Adv., 2021, 2, 646–683 RSC.
- J. D. McGettrick, K. Hooper, A. Pockett, J. Baker, J. Troughton, M. Carnie and T. Watson, Mater. Lett., 2019, 251, 98–101 CrossRef CAS.
- X. Dai, C. Fei, P. Kandlakunta, L. Zhao, Z. Ni, L. R. Cao and J. Huang, IEEE Trans. Nucl. Sci., 2022, 69, 1850–1856 CAS.
- B. Philippe, B.-W. Park, R. Lindblad, J. Oscarsson, S. Ahmadi, E. M. J. Johansson and H. Rensmo, Chem. Mater., 2015, 27, 1720–1731 CrossRef CAS.
- E. J. Juarez-Perez, L. K. Ono, I. Uriarte, E. J. Cocinero and Y. Qi, ACS Appl. Mater. Interfaces, 2019, 11, 12586–12593 CrossRef CAS PubMed.
- E. Bi, Z. Song, C. Li, Z. Wu and Y. Yan, Trends Chem., 2021, 3, 575–588 CrossRef CAS.
- L. Chen, H. Wang, W. Zhang, F. Li, Z. Wang, X. Wang, Y. Shao and J. Shao, ACS Appl. Mater. Interfaces, 2022, 14, 10917–10926 CrossRef CAS PubMed.
- L. McGovern, I. Koschany, G. Grimaldi, L. A. Muscarella and B. Ehrler, J. Phys. Chem. Lett., 2021, 12, 2423–2428 CrossRef CAS PubMed.
- A. F. Akbulatov, L. A. Frolova, N. N. Dremova, I. Zhidkov, V. M. Martynenko, S. A. Tsarev, S. Yu. Luchkin, E. Z. Kurmaev, S. M. Aldoshin, K. J. Stevenson and P. A. Troshin, J. Phys. Chem. Lett., 2020, 11, 333–339 CrossRef CAS PubMed.
- J. A. McLeod and L. Liu, J. Phys. Chem. Lett., 2018, 9, 2411–2417 CrossRef CAS PubMed.
- Z. Andaji-Garmaroudi, M. Abdi-Jalebi, F. U. Kosasih, T. Doherty, S. Macpherson, A. R. Bowman, G. J. Man, U. B. Cappel, H. Rensmo, C. Ducati, R. H. Friend and S. D. Stranks, Adv. Energy Mater., 2020, 10, 2002676 CrossRef CAS.
- L. Shi, M. P. Bucknall, T. L. Young, M. Zhang, L. Hu, J. Bing, D. S. Lee, J. Kim, T. Wu, N. Takamure, D. R. McKenzie, S. Huang, M. A. Green and A. W. Y. Ho-Baillie, Science, 2020, 368, eaba2412 CrossRef CAS PubMed.
- L. McGovern, M. H. Futscher, L. A. Muscarella and B. Ehrler, J. Phys. Chem. Lett., 2020, 11, 7127–7132 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: [DETAILS]. See DOI: https://doi.org/10.1039/d3cp05061k |
| ‡ The authors contributed equally to the work. |
| This journal is © the Owner Societies 2024 |