
Radiolytic evaluation of a new technetium redox control reagent for advanced used nuclear fuel separations†
Anh N.
Dang
a,
Maya H.
Rogalski
a,
Corey D.
Pilgrim
 b,
Joseph R.
Wilbanks
b,
Dean R.
Peterman
b,
Jesse D.
Carrie
b,
Peter R.
Zalupski
b,
Stephen P.
Mezyk
*a and
Gregory P.
Horne
*b
b,
Joseph R.
Wilbanks
b,
Dean R.
Peterman
b,
Jesse D.
Carrie
b,
Peter R.
Zalupski
b,
Stephen P.
Mezyk
*a and
Gregory P.
Horne
*b
aDepartment of Chemistry and Biochemistry, California State University Long Beach, 1250 Bellflower Boulevard, Long Beach California, 90840-9507, USA. E-mail: stephen.mezyk@csulb.edu
bCenter for Radiation Chemistry Research, Idaho National Laboratory, 1955 N. Freemont Ave., P.O. Box 1625, Idaho Falls, ID 83415, USA. E-mail: gregory.holmbeck@inl.gov
First published on 15th January 2024
Abstract
Technetium is a problematic radioisotope for used nuclear fuel (UNF) and subsequent waste management owing to its high environmental mobility and coextraction in reprocessing technologies as the pertechnetate anion (TcO4−). Consequently, several strategies are under development to control the transport of this radioisotope. A proposed approach is to use diaminoguanidine (DAG) for TcO4− and transuranic ion redox control. Although the initial DAG molecule is ultimately consumed in the redox process, its susceptibility to radiolysis is currently unknown under envisioned UNF reprocessing conditions, which is a critical knowledge gap for evaluating its overall suitability for this role. To this end, we report the impacts of steady-state gamma irradiation on the rate of DAG radiolysis in water, aqueous 2.0 M nitric acid (HNO3), and in a biphasic solvent system composed of aqueous 2.0 M HNO3 in contact with 1.5 M N,N-di-(2-ethylhexyl)isobutyramide (DEHiBA) dissolved in n-dodecane. Additionally, we report chemical kinetics for the reaction of DAG with key transients arising from electron pulse radiolysis, specifically the hydrated electron (eaq−), hydrogen atom (H˙), and hydroxyl (˙OH) and nitrate (NO3˙) radicals. The DAG molecule exhibited significant reactivity with the ˙OH and NO3˙ radicals, indicating that oxidation would be the predominant degradation pathway in radiation environments. This is consistent with its role as a reducing agent. Steady-state gamma irradiations demonstrated that DAG is readily degraded within a few hundred kilogray, the rate of which was found to increase upon going from water to HNO3 containing solutions and solvents systems. This was attributed to a thermal reaction between DAG and the predominant HNO3 radiolysis product, nitrous acid (HNO2), k(DAG + HNO2) = 5480 ± 85 M−1 s−1. Although no evidence was found for the radiolysis of DAG altering the radiation chemistry of the contacted DEHiBA/n-dodecane phase in the investigated biphasic system, the utility of DAG as a redox control reagent will likely be limited by significant competition with its degradation by HNO2.
Introduction
About 6% of all uranium-235 fission events yield technetium-99 (99Tc),1,2 which for a typical Light Water Reactor (LWR) results in the production of ∼40 kg of 99Tc per year.3 This high fission yield, combined with its long half-life (τ1/2 = 211![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years), makes 99Tc a very important component of used nuclear fuel (UNF) and subsequent waste management.4 For example, at the Hanford site in Washington, USA, some 1500 kg of 99Tc is present in ∼53 million gallons of waste.5 Historically, some of the 177 waste tanks at the Hanford site have leaked and contaminated the local environment.6 These events are problematic for many reasons, but especially because of technetium's high mobility in all water environments,7 due to its most stable chemical form being the soluble pertechnetate anion (TcO4−).8
000 years), makes 99Tc a very important component of used nuclear fuel (UNF) and subsequent waste management.4 For example, at the Hanford site in Washington, USA, some 1500 kg of 99Tc is present in ∼53 million gallons of waste.5 Historically, some of the 177 waste tanks at the Hanford site have leaked and contaminated the local environment.6 These events are problematic for many reasons, but especially because of technetium's high mobility in all water environments,7 due to its most stable chemical form being the soluble pertechnetate anion (TcO4−).8
Technetium is similarly challenging in UNF reprocessing environments. The TcO4− ion is often coextracted as a counter ion, alongside nitrate ions (NO3−), when forming tributylphosphate (TBP) complexes of hexavalent uranium (UO22+) and tetravalent plutonium (Pu4+) under typical Plutonium Uranium Reduction Extraction (PUREX) process conditions:
| UO22+ + 2TBP + TcO4− + NO3− ⇌ [UO2(TcO4)(NO3)(TBP)2], | (1) |
| Pu4+ + 2TBP + TcO4− + 3NO3− ⇌ [Pu(TcO4)(NO3)3(TBP)2]. | (2) |
Multiple efforts to date have provided a quantitative foundational, understanding of technetium chemistry under UNF reprocessing conditions.9–25 However, the challenges of technetium management have been amplified by more recent efforts to simplify the traditional PUREX process, motivated by economics, plutonium proliferation concerns, and neptunium management.26–28 Some iterations of the PUREX process use hydrazine-stabilized ferrous and uranous nitrates for the reduction and separation of plutonium and neptunium (transuranics) from uranium-loaded organic phases.29 In addition, the hydrazine component acts as a nitrite ion (NO2−) scavenger and facilitates the reduction of the TcO4− ion to the non-extractable oxide (TcO2).26,30,31 Both are useful processes in the management of UNF. However, hydrazine also reacts with nitrous acid (HNO2), which in the presence of TBP results in the formation of explosive hydrazoic acid.29,32 Consequently, advanced PUREX formulations aim to use alternative redox control methods for transuranic and technetium management. Diaminoguanidine (DAG, Fig. 1(A)) has recently been identified as a potential candidate for this role in Advanced PUREX processes. Under moderately acidic conditions, DAG has been shown to facilitate the desired redox control of the aforementioned transuranic ions, in addition to the reduction of the TcO4− ion.33–35 This is not the case for other proposed substitutes, such as acetohydroxamic acid (AHA).36–42
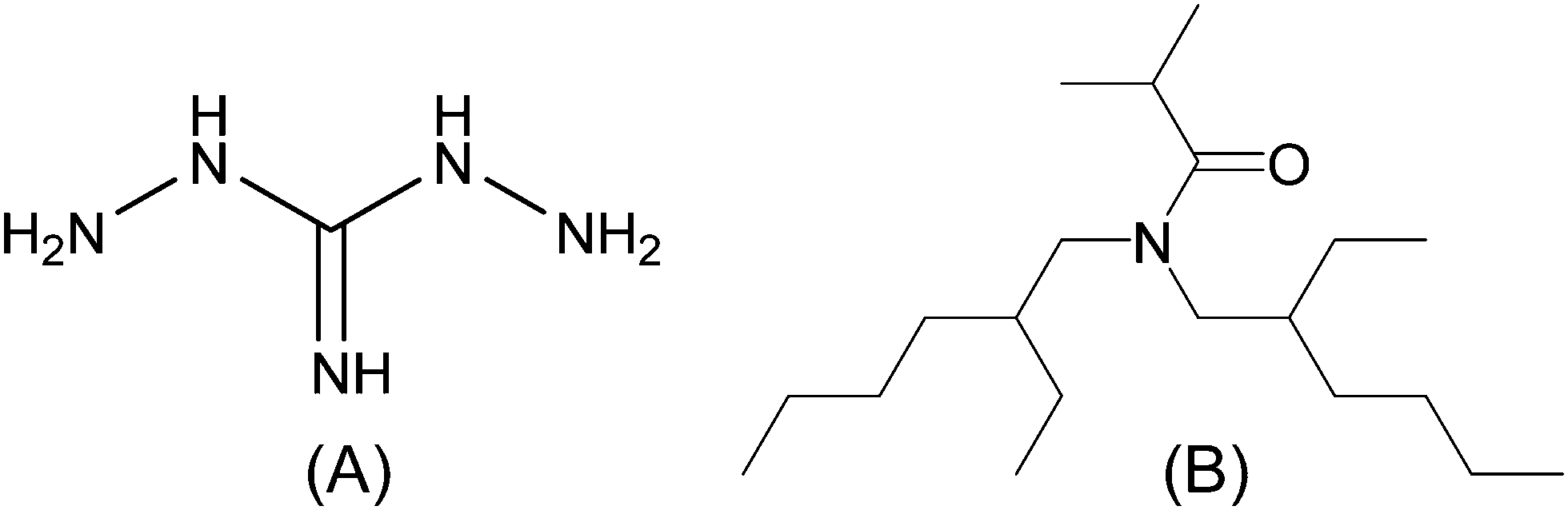 | ||
| Fig. 1 Molecular structure of diaminoguanidine (DAG, A) and N,N-di-(2-ethylhexyl)isobutyramide (DEHiBA, B). | ||
However, the radiation stability of DAG is currently unknown, and yet is a critical factor in determining the feasibility of any chemical employed in a UNF reprocessing flowsheet. The dissolved fuel's inherent ionizing radiation fields promote the destruction of useful process chemicals into a variety of degradation products that are often detrimental to process performance. In the case of DAG, which is expected to be ultimately consumed by its redox control reactions with TcO4− and transuranic ions, there are two major radiolytic concerns. The first involves the potential competition for DAG between the aforementioned metal ions and accumulated radiolysis products. Both the direct and indirect radiolysis of nitric acid (HNO3) affords the formation and accumulation of HNO2:43
NO3−/HNO3 ![[long arrow, wavy then straight]](https://www.rsc.org/images/entities/char_e0f6.gif) e−, NO3˙, NO2−/HNO2, O, Haq+, e−, NO3˙, NO2−/HNO2, O, Haq+, | (3) |
| NO3− + eaq−/H˙ → HNO2, | (4) |
The family of aminoguanidines has been shown to react with HNO2 under mild and strongly acidic conditions.44–48 Given the fact that HNO3 radiolysis leads to the accumulation of 10s of micromolar of HNO2 within only a few kGy,49 DAG may be preferentially consumed by HNO2, thereby inhibiting its redox control of TcO4− and transuranic ions.
The second radiolytic concern relates to whether the use of excess amounts of DAG affords the formation of hazardous degradation products, from the scavenging of radical and molecular HNO3 radiolysis products (eqn (3)). For example, explosive products and byproducts have been reported from syntheses involving the reaction of aminoguanidines with HNO2.44–50 In addition, degradation products with the capacity to migrate across the interface and negatively impact the longevity of Advanced PUREX process ligands, such as N,N-di-(2-ethylhexyl)isobutyramide (DEHiBA, Fig. 1(B)), could also be created by the HNO2 reaction.
Based on these concerns, and in support of the continued development of DAG-based technetium management strategies, we have investigated the radiation robustness of DAG. Here, we report the impacts of steady-state gamma irradiation on the radiolysis of DAG in water, aqueous 2.0 M HNO3, and in a solvent system composed of aqueous 2.0 M HNO3 in contact with 1.5 M DEHiBA dissolved in n-dodecane. Additionally, we report chemical kinetics for the reaction of DAG and a 1:1 perrhenate ion system, [DAG·ReO4], with key transients arising from the radiolysis of aqueous HNO3 solution, specifically the hydrated electron (eaq−), hydrogen atom (H˙), and hydroxyl (˙OH) and nitrate (NO3˙) radicals. We have also directly measured the thermal kinetics for the reaction of DAG with HNO2 in acidic aqueous media.
Methods
Chemicals
Acetonitrile (≥99.97%), diaminoguanidine hydrochloride (DAG, 98%), hexane (≥99%), iron(III) sulfate heptahydrate (FeSO4·7H2O, ReagentPlus®, ≥99%), n-dodecane (≥99% anhydrous), nitric acid (HNO3, ≥99.999% trace metals basis), parachlorobenzoic acid (pCBA, 99%), perchloric acid (HClO4, ≥99.999% trace metals basis), potassium thiocyanate (KSCN, ≥99.0% ACS Reagent Grade), sodium chloride (NaCl, 99.999% trace metals basis), sulfuric acid (H2SO4, 99.999%), and tertiary butanol (tBuOH, ≥99.5% anhydrous) were obtained from MilliporeSigma (Burlington, Massachusetts, USA). Potassium nitrite (KNO2, Reagent grade >99%) was supplied by Ward Scientific (Ontario, Canada). N,N-Di-(2-ethylhexyl)isobutyramide (DEHiBA, 99%) was sourced from Marshallton Research Laboratories Inc. (King, NC, USA). Potassium perrhenate (KReO4, 99% trace metals basis) was supplied by Beantown Chemical Corporation (Hudson, NH, USA). Dibasic phosphate (>99% ACS) was received from Thermo Fisher Scientific (Waltham, MA, USA). Unless otherwise specified, all chemicals were used without further purification. Compressed argon (Ar), nitrogen (N2), and nitrous oxide (N2O) gases were purchased from Airgas (Radnor, PA, USA) and Matheson (Irving, TX, USA) with purities ≥99.5%. Ultra-pure water (18.2 MΩ cm) was used to prepare all aqueous solutions.Time-resolved electron pulse irradiations
Chemical kinetics were measured for the reaction of DAG and a 1:1 [DAG·ReO4] system with radiation-induced transient aqueous radical species, specifically the eaq−, H˙ atom, and ˙OH and NO3˙ radicals. Reaction kinetics were measured using the Notre Dame Radiation Laboratory (NDRL) linear electron accelerator facility, details for which have been previously reported for both the electron accelerator and transient absorption detection system.51,52 Dosimetry was determined at the beginning of each day using N2O saturated solutions of 10 mM KSCN at λ = 475 nm (Gε = 5.2 × 10−4 m2 J−1),53 affording an average of 6 ± 1 (˙OH and eaq−) and 25 ± 2 Gy per pulse (H˙ and NO3˙). The DAG molecule was irradiated in aqueous solutions formulated to isolate specific radicals:54
• Hydrated electron (eaq−). Direct decay kinetics of the eaq− were observed at 720 nm using Ar or N2-saturated aqueous solutions containing 0.5 M tBuOH and 10 mM monobasic phosphate buffer at pH 6.95.
• Hydrogen atom (H˙). Growth kinetics of the transient pCBA-H adduct absorbance was observed at 360 nm using Ar or N2-saturated solutions containing 10 mM tBuOH, 1 mM HClO4, and 10 mM monobasic phosphate buffer at pH 3.06.
• Hydroxyl radical (˙OH). Transient growth kinetics of the oxidized DAG/1:1 [DAG·ReO4] species formed at 260 nm were observed using N2O saturated solutions containing 10 mM monobasic phosphate buffer at pH 7.01.
• Nitrate radical (NO3˙). Direct decay kinetics of the NO3˙ radical were observed at 640 nm using N2O-saturated aqueous solutions containing 5.0 M NaNO3/1 mM HClO4 at pH 3.01.
The 1:1 [DAG·ReO4] solutions were prepared by the dissolution of equivalent moles of ReO4− into the corresponding DAG solution, and then left for 24–36 hours.
The presented kinetic data were generated by averaging 10–15 individual measurements. Quoted errors for the derived second-order reaction rate coefficients (k) are a combination of measurement precision (∼4%) and initial (∼6%) and dilution (<1%) sample concentration errors.
Steady-state cobalt-60 gamma irradiations
Gamma irradiation of single and biphasic DAG solutions was achieved using a Foss Therapy Services Model 812 and a Nordion Gammacell 220E cobalt-60 irradiator at the Idaho National Laboratory (INL) Center for Radiation Chemistry Research, and a Shepherd 109-68R cobalt-60 irradiator at the NDRL. Single phase samples comprised of 50 mM DAG dissolved in either water or 2.0 M HNO3 solution. Biphasic samples comprised of 100 mM DAG dissolved in 2.0 M HNO3 solution contacted in a 1:1 aqueous-to-organic ratio with 1.5 M DEHiBA dissolved in n-dodecane solvent that had been pre-equilibrated thrice with 2.0 M HNO3 solution. Both sample solution formulations were irradiated in 20 mL screw-cap scintillation vials: 5 mL for single phase samples, and 10 mL for biphasic samples (5 mL of each phase). Although initially aerated, all samples were considered deaerated upon irradiation due to the radiolytic consumption of dissolved oxygen. Irradiations were performed in triplicate.
Dose rates were determined for each occupied irradiator sample position by chemical dosimetry using Fricke solution (1 mM FeSO4·7H2O and 1 mM NaCl in 0.4 M H2SO4),55 corrected for the radioactive decay of cobalt-60 (τ1/2 = 5.27 years) throughout the duration of the study. Furthermore, the gamma dose received by the organic phase was derived by accounting for the electron density difference of n-dodecane as compared to Fricke solution: (Zn-dodecane/An-dodecane)/(ZFricke/AFricke) × (ρn-dodecane/ρFricke) = 0.78,55 where Z, A, and ρ are the atomic number, mass number, and density, respectively. Previous work showed that the additional volume and height afforded by the 10 mL biphasic samples had a negligible effect (<2.5% higher) on the dose rate relative to that experienced by 5 mL samples.56
Aqueous phase analysis by liquid chromatography–mass spectrometry
Post irradiation, biphasic sample phases were separated. All aqueous phases were analyzed for the loss of DAG as a function of absorbed gamma dose and solution composition (water, 2.0 M HNO3, and 2.0 M HNO3:1.5 M DEHiBA/n-dodecane) at California State University Long Beach. The quantification of DAG was achieved by liquid chromatography–mass spectrometry (LCMS) using an Agilent (Santa Clara, CA, USA) 1260 Infinity II Liquid Chromatography system equipped with a binary solvent pump, an autosampler, and a column oven. Separation was achieved using an Agilent Poroshell 120 EC-C18 reversed-phase column (3.0 mm inner diameter × 50 mm length, 2.7 μm particle size). The sample injection volume was 1 μL and quantification was performed in triplicate. Analytes were detected with an Agilent 6470 triple quadrupole mass spectrometer, operating in the positive electrospray ionization mode with dynamic multiple reaction monitoring. Mass spectrometer parameters were as follows: 300 °C dry N2 gas temperature, 5 L min−1 dry N2 gas flow, 45 psi nebulizer pressure, 250 °C sheath N2 gas temperature, 11 L min−1 sheath N2 gas flow rate, 3500 V capillary voltage, and 500 V nozzle voltage.
For DAG samples initially dissolved in 2.0 M HNO3 and 2.0 M HNO3:1.5 M DEHiBA/n-dodecane, the mobile phase consisted of an isocratic mixture of 90% water and 10% acetonitrile with a flow rate of 0.30 mL min−1. The column temperature was maintained at 30 °C. DAG was quantified using a 5-point calibration curve (0.2–1.0 μM) made from DAG in 2.0 M HNO3. Samples were diluted up to 200000 times in 2.0 M HNO3.
For DAG samples dissolved only in water, the HNO3 method was modified to optimize the peak shape. The mobile phase consisted of an isocratic mixture of 70% water and 30% acetonitrile. The flow rate was 0.45 mL min−1 and the column temperature was 40 °C. A 5-point calibration curve made from DAG in water was used (20–100 nM). Samples were diluted by factors ranging up to 500000. A blank and a quality control (QC, 100 mM DAG in water) standard were injected after every five samples.
The degradation rates for the radiolysis of DAG in each solution and solvent system are expressed as dose constants (d, kGy−1) derived from fits between 0 and ∼30 kGy.57
Organic phase analysis by gas chromatography
Separated organic phases were analyzed at INL for the loss of DEHiBA as a function of absorbed gamma dose. Quantification of DEHiBA was achieved by gas chromatography (GC) using an Agilent 7890A gas chromatograph equipped with an autosampler and a flame ionization detector (FID). Sample organic phases were diluted by a factor of 1:20 in hexane. Each dilution replicate was injected four times using the following conditions: an Agilent Technologies HP-5 column (0.32 mm inner diameter × 30 m length × 0.25 μm film thickness) with an injector temperature of 300 °C; an oven temperature of 100 °C for 1 minute, ramped to 250 °C at 30 °C min−1. The split ratio was 40:1 with 2.7 mL min−1 flow through the column. The FID temperature was 350 °C. The dilution replicate injection order was randomized to help differentiate systematic instrument drift from real trends in the samples. The quadruple injections per dilution replicates were conducted sequentially to reduce any diluent evaporation. Quantification was performed using a calibration curve with standards prepared from neat DEHiBA dissolved in hexane. Five calibration points and a blank were used to construct calibration curves for each compound on each instrument. Each standard was injected four times and analyzed in order of increasing concentration.
QC standard concentrations at 50 mM DEHiBA/hexane were analyzed at the mid-point of the analysis and at the end to ensure the validity of the calibration curve throughout the entire measurement period. Each QC standard run was a separate aliquot of the QC standard in order to reduce the effects of diluent evaporation.
The radiation-induced degradation rate of DEHiBA is reported as a radiolytic yield (G-value, μmol J−1).
Stop-flow analysis for the reaction of DAG and HNO2
The reaction kinetics of DAG with HNO2 in acidic aqueous solutions was studied using a Hi-Tech Scientific (Bradford-on-Avon, UK) SF-61 DX2 double mixing stopped-flow spectrometer. Separate solutions of DAG (4–20 mM) and KNO2 (5–10 mM) in 2.0 M HNO3 were mixed at 25.0 °C in a 1:1 ratio. Upon mixing, the decay of HNO2 was directly monitored as a function of time (up to 10 seconds) at 350 nm. An ε350 of 18.7 ± 0.3 M−1 cm−1 was determined in this study, which is slightly lower than the literature value of ε354 = 22.1 ± 0.22 M−1 cm−1 in HCl solution at pH 1.5.58 A HNO2 concentration of 2.5 mM was chosen to obtain useful absorption changes. However, at this concentration it was difficult to maintain pseudo-first-order concentration kinetic conditions. Hence, the kinetics analyses were performed in two different ways depending on the initial concentration of DAG in 2.0 M HNO3. For the 5–10 mM DAG we assumed pseudo-first-order conditions and fitted single-exponential decay kinetics: | (5) |
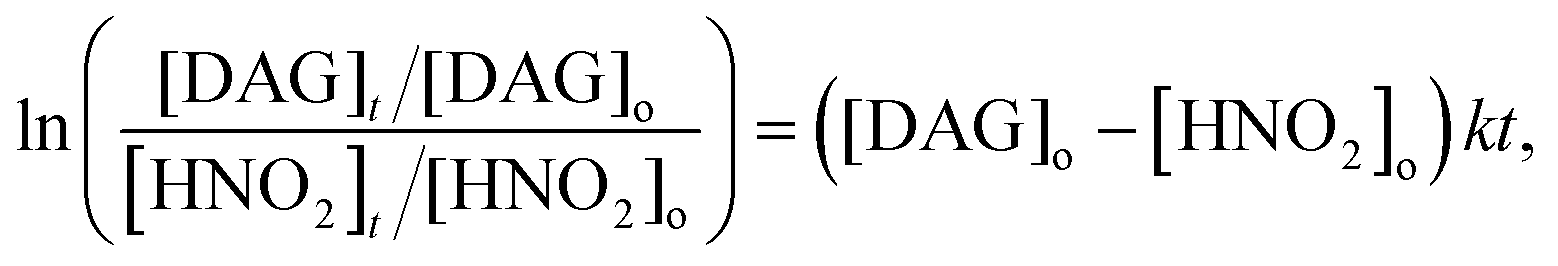 | (6) |
:1 reaction. The average second-order rate coefficient calculated in this manner had larger errors (∼10%) compared to the pseudo-first-order fitted k′ values. However, excellent agreement was obtained for the derived k value from both methods (within fitting error).
Results and discussion
Chemical kinetics
To elucidate the impact of ionizing radiation on DAG-based solvent systems, an understanding of its fundamental reaction kinetics with the expected suite of radiation-induced aqueous radicals is essential. Typical kinetic data are shown in Fig. 2 for the reaction of DAG and a 1:1 [DAG·ReO4] system with the oxidizing ˙OH radical, for which rate coefficients were determined.
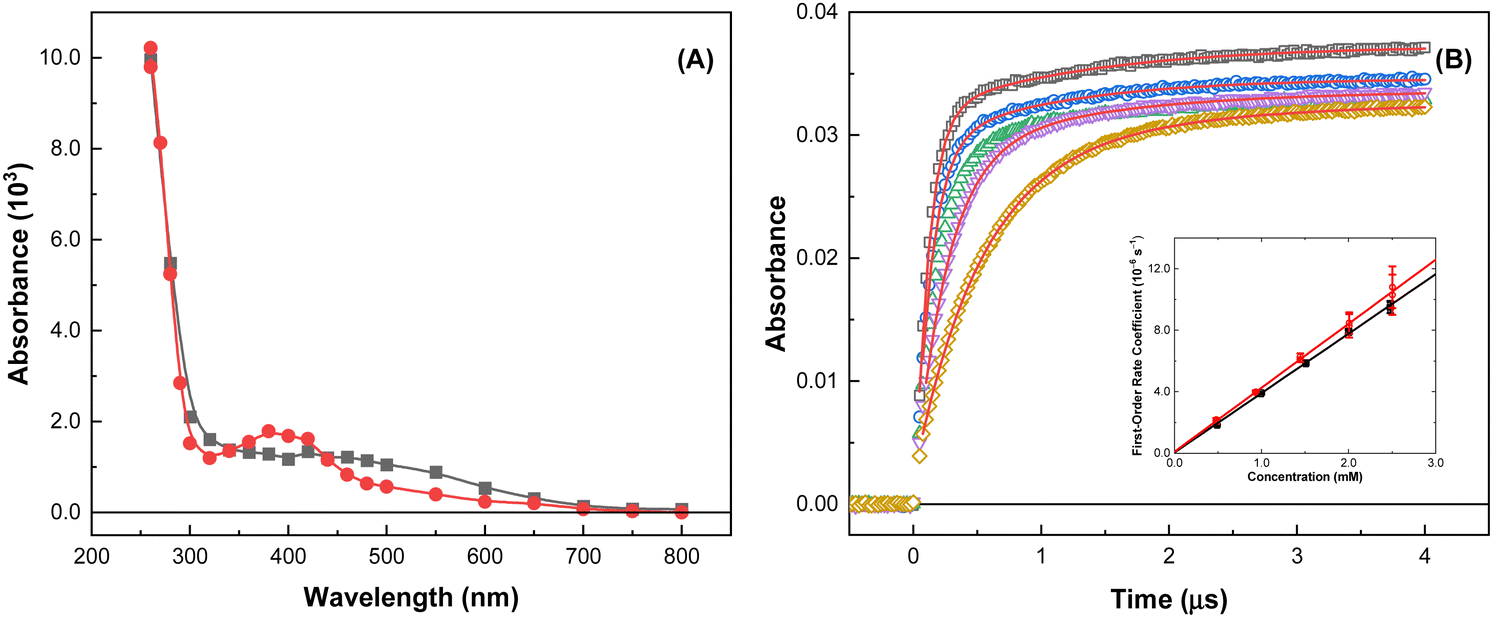 | ||
Fig. 2 (A) Transient absorption spectra from the reaction of the ˙OH radical with DAG from the electron pulse irradiation of 4.91 mM DAG in 2.0 M HNO3 ( ) and 2.50 mM of 1:1 [DAG·ReO4] in 2.0 M HNO3 ( ) and 2.50 mM of 1:1 [DAG·ReO4] in 2.0 M HNO3 ( ), respectively, at 22.7 ± 0.1 °C. Data obtained from limiting absorbance values for the faster exponential growth fits to measured kinetics. All data have had solution blank absorbances subtracted. (B) Transient kinetic growths measured at 260 nm for 4.91 ( ), respectively, at 22.7 ± 0.1 °C. Data obtained from limiting absorbance values for the faster exponential growth fits to measured kinetics. All data have had solution blank absorbances subtracted. (B) Transient kinetic growths measured at 260 nm for 4.91 ( ), 2.48 ( ), 2.48 ( ), 2.00 ( ), 2.00 ( ), 1.00 ( ), 1.00 ( ), and 0.49 ( ), and 0.49 ( ) mM DAG dissolved in 2.0 M HNO3 at 22.6 °C. Solid lines are double-exponential growth kinetics, corresponding to k′ = (9.77 ± 0.14) × 106, (7.96 ± 0.13) × 106, (3.97 ± 0.04) × 106, and (2.02 ± 0.13) × 106 s−1, respectively, for the faster exponential component. Inset: Second-order rate coefficient determination for the ˙OH radical reaction with DAG (■) and the 1:1 [DAG·ReO4] system ( ) mM DAG dissolved in 2.0 M HNO3 at 22.6 °C. Solid lines are double-exponential growth kinetics, corresponding to k′ = (9.77 ± 0.14) × 106, (7.96 ± 0.13) × 106, (3.97 ± 0.04) × 106, and (2.02 ± 0.13) × 106 s−1, respectively, for the faster exponential component. Inset: Second-order rate coefficient determination for the ˙OH radical reaction with DAG (■) and the 1:1 [DAG·ReO4] system ( ). Solid lines are weighted linear fits, corresponding to k(˙OH + DAG) = (3.87 ± 0.05) × 109 M−1 s−1 (R2 = 0.98) and k(˙OH + [DAG·ReO4]) = (4.15 ± 0.07) × 109 M−1 s−1 (R2 = 0.98). ). Solid lines are weighted linear fits, corresponding to k(˙OH + DAG) = (3.87 ± 0.05) × 109 M−1 s−1 (R2 = 0.98) and k(˙OH + [DAG·ReO4]) = (4.15 ± 0.07) × 109 M−1 s−1 (R2 = 0.98). | ||
The observed absorption growths exhibited double-exponential behavior:
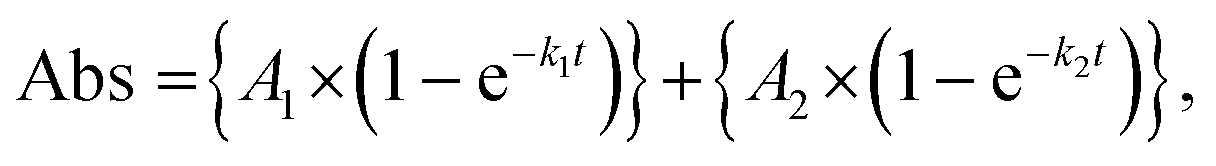 | (7) |
:1 [DAG·ReO4] with the eaq−, H˙ atom, and NO3˙ radical are given in the ESI,† and all the derived second-order rate coefficients (k) are summarized in Table 1.
:1 [DAG·ReO4] system
| Radical | Second-order rate coefficient (k, M−1 s−1) | |
|---|---|---|
| DAG | 1:1 [DAG·ReO4] |
|
| eaq− | (1.97 ± 0.06) × 108 | (1.62 ± 0.03) × 1010 |
| H˙ | (7.25 ± 0.23) × 106 | (3.19 ± 0.23) × 107 |
| ˙OH | (3.87 ± 0.05) × 109 | (4.15 ± 0.07) × 109 |
| NO3˙ | (2.19 ± 0.03) × 109 | (2.06 ± 0.03) × 109 |
Fast rate coefficients were measured for the reaction of only DAG with both the ˙OH and NO3˙ radicals, suggesting that this molecule is susceptible to oxidation, which is consistent with its proposed role as a TcO4− ion reductant. This was further supported by the slower (by orders of magnitude) reactivities measured for the reaction of only DAG with the reducing species from water radiolysis, i.e., the eaq− and H˙ atom. In addition, the high concentrations (molar) of HNO3 used under envisioned UNF reprocessing conditions would result in most of these reducing radicals being scavenged (eqn (4)), and thus unavailable for reaction with other solutes. Given these expected reprocessing conditions and the rate coefficient values given in Table 1, the radiolytic longevity of DAG is expected to be determined by its extent of reaction with the ˙OH and NO3˙ radicals, with their availability dictated by competition kinetics with the other constituents of a UNF reprocessing solvent system.
As the ReO4− ion is often used as a chemical surrogate for the TcO4− ion,60 the 1:1 [DAG·ReO4] system was chosen with the goal of investigating the chemistry of the rhenium product species (believed to be ReO2+) arising from the reduction of ReO4− by DAG. Insights into the radiation-induced chemical behavior of the ReO2+/TcO2+ species is also necessary for the continued development of DAG-based reprocessing flowsheets. However, in the presence of the ReO4− ion we found negligible impact on the reaction of DAG with the ˙OH and NO3˙ radicals (Table 1), suggesting that either little of the DAG had been consumed through the aforementioned ReO4− ion reduction process or that the resulting product ReO2+ species exhibited negligible reactivity with these two radicals. Confirmation of this chemistry was also provided by the measured rate for the reaction of the 1:1 [DAG·ReO4] system with the eaq−, which gave a coefficient that was almost identical to the single measurement reported for the ReO4− ion and similar to that reported for TcO4− ion:61–64
| ReO4− + eaq− → ReO42− k = 1.5 × 1010 M−1 s−1, | (8) |
| TcO4− + eaq− → TcO42− k = (1.3–2.5) × 1010 M−1 s−1. | (9) |
These kinetic measurements indicate that little to no ReO4− was reduced by DAG to the ReO2+ ion species within our 24–36-hour timeframe. As such, the measurements made with the 1:1 [DAG·ReO4] system only appear to represent the combined scavenging capacities of the two independent chemical species, and not the ReO2+ ion species. The measured kinetics for the H˙ atom for the 1:1 [DAG·ReO4] system also reflects only this radical's chemical reactivity with the ReO4− ion. This is an interesting finding in itself, as the corresponding reaction for the TcO4− ion has been reported to be fast:62
| TcO4− + H˙ → TcO42− + Haq+ k > 1 × 1010 M−1 s−1. | (10) |
Typically, H˙ atom reaction rate coefficients are several orders of magnitude slower than the corresponding eaq− reaction,54 which we find to be consistent for the ReO4− ion, but not for the TcO4− ion, suggesting an alternative reaction mechanism may be occurring.
Overall, these kinetic findings are consistent with observations reported by Zalupski et al.,34 who found that the addition of an aliquot of TcO4− (to a final concentration of 3 mM) to a 0.1 M solution of DAG resulted in an energetic reaction that afforded a reduced technetium product species (believed to be TcO2+), as identified by precipitation and a color change of solution. In dilute HNO3 (pH = 2), a black precipitate formed from the TcO2 species, while in 2.0 M HNO3, the initially colorless solution became a yellow hue, denoting the formation of the TcO2+ species. A much less energetic reaction was observed for the ReO4− ion in the presence of DAG, taking multiple days to achieve the same quantitative reduction. The reaction of DAG analogues, such as 1,1′-(ethane-1,2-diyl) diaminoguanidine, also showed similar differences in reactivity with the TcO4− and ReO4− ions.
Steady-state irradiations
The changes in DAG concentration upon its gamma irradiation in water, 2.0 M HNO3, and 2.0 M HNO3 contacted with 1.5 M DEHiBA/n-dodecane are shown in Fig. 3 and Fig. S4 (ESI†). | ||
Fig. 3 Normalized concentration of DAG as a function of absorbed dose from the gamma irradiation of: 50 mM DAG in water ( ); 50 mM DAG in 2.0 M HNO3 ( ); 50 mM DAG in 2.0 M HNO3 ( ); and 100 mM DAG in 2.0 M HNO3:1.5 M DEHiBA/n-dodecane ( ); and 100 mM DAG in 2.0 M HNO3:1.5 M DEHiBA/n-dodecane ( ) under ambient irradiator temperature conditions. Solid lines are first-order exponential fits to data for dose constant calculation. Non-normalized DAG concentration data given in Fig. S4 (ESI†). ) under ambient irradiator temperature conditions. Solid lines are first-order exponential fits to data for dose constant calculation. Non-normalized DAG concentration data given in Fig. S4 (ESI†). | ||
For all three investigated systems, the irradiation of DAG promoted its destruction, affording dose constants (d-values) of up to −31 × 10−3 kGy−1, as summarized in Table 2.
| Solvent system | d-value (10−3 kGy−1, R2 = 0.99) |
|---|---|
| H2O | −9.54 ± 0.31 |
| 2.0 M HNO3 | −30.70 ± 0.88 |
| 2.0 M HNO3:DEHiBA/n-dodecane | −24.60 ± 0.97 |
These degradation efficiencies are relatively high compared with other molecules considered for this redox-control role, such as AHA (d ∼ −(5.87 ± 0.61) × 10−3 kGy−1).56 Further, it is evident from Fig. 3 and the range of d-values in Table 2 that the rate of DAG radiolysis is very dependent upon solution composition, and that it was completely degraded within 100 kGy of absorbed gamma dose in systems containing 2.0 M HNO3. These observed changes in the rate of DAG radiolysis with solution composition are indicative of a reaction between DAG and HNO2, the dominant aqueous HNO3 radiolysis product. Here we measured a rate coefficient of k = 5480 ± 85 M−1 s−1, the kinetic data for which is shown in Fig. 4. This rate coefficient is consistent with the reactivity of DAG with HNO2 reported by Levering,45 where the addition of one equivalent of KNO2 to a solution of DAG in dilute HNO3 afforded an exothermic reaction accompanied by a sudden change in solution color from clear to bright orange and eventually yellow, highlighting the speed with which HNO2/NO2− (HNO2 ⇌ NO2− + Haq+, pKa = 3.2) reacts with the amine backbone of DAG.
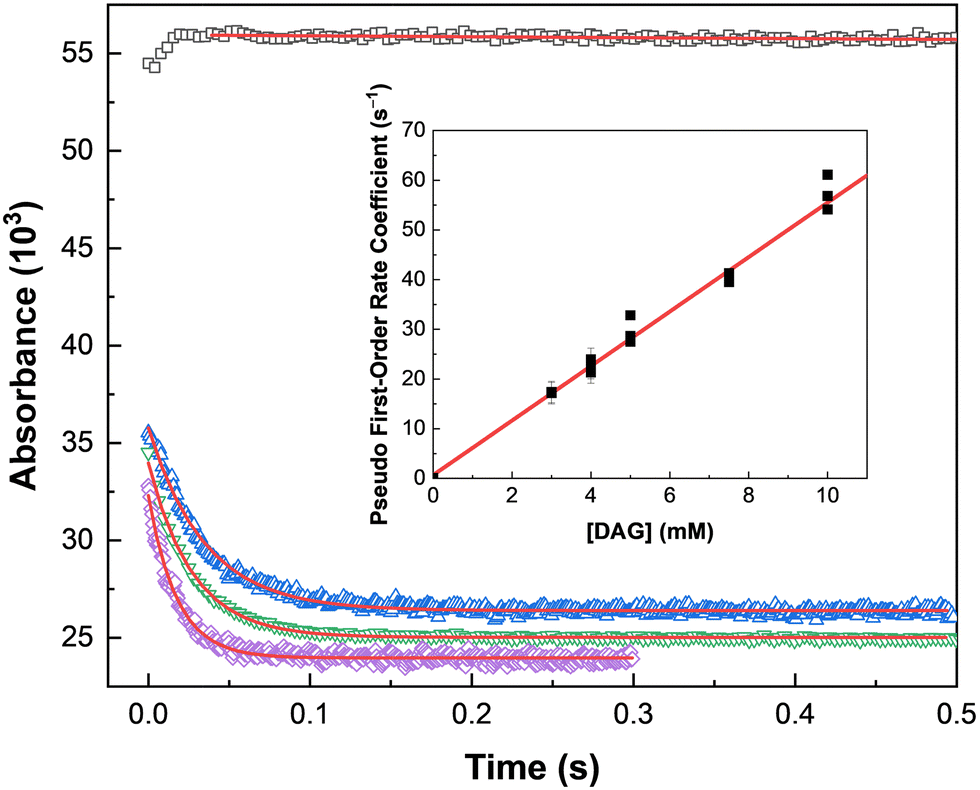 | ||
Fig. 4 Stopped-flow kinetic data for the reaction of DAG with HNO2 in 2.0 M HNO3 at 25.0 °C: 5.0 mM HNO2 self-reaction ( ); 5.0 mM DAG + 2.50 mM HNO2 ( ); 5.0 mM DAG + 2.50 mM HNO2 ( ); 7.5 mM DAG + 2.50 mM HNO2 ( ); 7.5 mM DAG + 2.50 mM HNO2 ( ); and 10.0 mM DAG + 2.50 mM HNO2 ( ); and 10.0 mM DAG + 2.50 mM HNO2 ( ). Solid lines are fitted pseudo-first-order exponential decays, corresponding to values of k′ = 0.04 ± 0.05, 22.29 ± 0.23, 39.51 ± 0.39, and 56.83 ± 0.69 s−1, respectively. Inset: Second-order determination for the reaction of DAG with HNO2 in 2.0 M HNO3. Solid line is a weighted linear fit, corresponding to k = 5480 ± 85 M−1 s−1, R2 = 0.99. ). Solid lines are fitted pseudo-first-order exponential decays, corresponding to values of k′ = 0.04 ± 0.05, 22.29 ± 0.23, 39.51 ± 0.39, and 56.83 ± 0.69 s−1, respectively. Inset: Second-order determination for the reaction of DAG with HNO2 in 2.0 M HNO3. Solid line is a weighted linear fit, corresponding to k = 5480 ± 85 M−1 s−1, R2 = 0.99. | ||
Although DAG was completely consumed within 160 kGy in the biphasic systems, higher gamma dose irradiations were also performed to determine whether the presence of DAG or its degradation products impacted the radiation chemistry of the organic phase. The concentration of DEHiBA as a function of absorbed gamma dose (corrected for the electron density of n-dodecane relative to Fricke solution) from the irradiated biphasic DAG solvent system is shown in Fig. 5. Within the investigated dose range, DEHiBA was found to degrade linearly with absorbed gamma dose, which is consistent with previous findings for the irradiation of DEHiBA under a variety of solvent system conditions.56,65
 | ||
| Fig. 5 Concentration of DEHiBA as a function of absorbed dose from the gamma irradiation of 100 mM DAG in 2.0 M HNO3 contacted with 1.5 M DEHiBA/n-dodecane under ambient irradiator temperature conditions. Solid line is a weighted linear fit to data, affording G(DEHiBA) = −0.55 ± 0.02 μmol J−1, R2 = 0.94. | ||
A linear fit to the data shown in Fig. 5 gave a G-value of −0.55 ± 0.02 μmol J−1 for the loss of DEHiBA, in excellent agreement with that reported by Drader et al. (−0.53 μmol J−1) for the gamma irradiation of 1 M DEHiBA/n-dodecane contacted with an aqueous HNO3 phase in the absence of DAG.65 Our data clearly show that the presence of DAG and its degradation products have little effect on interfacial radiation chemistry under the investigated conditions, which is consistent with findings for the irradiation of DEHiBA in contact with an AHA containing aqueous HNO3 phase.56 That said, the organic phases developed a green hue, the intensity of which increased with absorbed gamma dose. This was not observed in our previous DEHiBA irradiations in the absence of DAG.56
Conclusions
Foundational knowledge for the radiation robustness of DAG has been established in solutions and solvent systems of relevance to UNF reprocessing solvent systems. Reaction kinetic measurements for the suite of transient aqueous HNO3 radiolysis products (eaq−, H˙ atom, and ˙OH and NO3˙ radicals) with DAG indicate that the primary path for this molecules’ radiation-induced degradation is via fast reaction with the oxidizing ˙OH (k = (3.87 ± 0.05) × 109 M−1 s−1) and NO3˙ (k = (2.19 ± 0.03) × 109 M−1 s−1) radicals.The steady-state gamma irradiation of DAG afforded its complete destruction within a few hundred kilogray, with its observed loss in water being much slower than in the investigated 2.0 M HNO3 systems. This solution formulation dependence is attributed to an important thermal reaction between DAG and the major HNO3 radiolysis product, HNO2, which occurs with a relatively fast rate coefficient of k = 5480 ± 85 M−1 s−1 at 25.0 °C.
These steady-state and time-resolved findings suggest that the effectiveness of DAG as a TcO4− and transuranic ion redox control reagent will be limited by its competition with HNO2 and scavenging of oxidizing radicals (˙OH and NO3˙) from aqueous HNO3 radiolysis. That said, beyond exhibiting little capacity to interfere with organic phase radiation chemistry, it is currently unclear as to what the impact of DAG degradation products, from both radiolysis and reduction processes, have upon overall process performance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been funded by the U.S. Department of Energy (DOE) Assistant Secretary for Nuclear Energy, under the Material Recovery and Waste Form Development Campaign, DOE-Idaho Operations Office Contract DE-AC07-05ID14517. The NDRL is supported by the Division of Chemical Sciences, Geosciences and Biosciences, Basic Energy Sciences, Office of Science, US-DOE through award no. DE-FC02-04ER15533.References
- J. M. Siegel, J. Am. Chem. Soc., 1946, 68, 2411–2442 CrossRef CAS.
- A. L. Nichols, D. L. Aldama and M. Verdelli, Handbook of Nuclear Data for Safeguards, INDC International Nuclear Data Committee Report INDC(NDS)-0502, International Atomic Energy Agency, Vienna, 2007 Search PubMed.
- K. H. Lieser, Radiochim. Acta, 1993, 63, 5–8 CrossRef CAS.
- G. Choppin, J.-O. Liljenzin and J. Rydberg, Radiochemistry and Nuclear Chemistry, Butterworth-Heinemann, Woburn, Massachusetts, 2002, ch. 21, p. 593 Search PubMed.
- D. Banerjee, D. Kim, M. J. Schweiger, A. A. Druger and P. Thallapally, Chem. Soc. Rev., 2016, 45, 2724–2739 RSC.
- R. E. Gephart, A Short History of Hanford Waste Generation, Storage, and Release, Report PNNL-13605, Rev. 4, Pacific Northwest National Laboratory, Washington, 2003.
- J. P. Icenhower, W. J. Martin, N. P. Oafoku and J. M. Zachara, The geochemistry of technetium: A summary of the behavior of an artificial element in the natural environment, Report PNNL-18139, Pacific Northwest National Laboratory, 2008.
- E. Anders, The Radiochemistry of Technetium, National Academy of Sciences, National Research Council Report NAS-NS 3021, U.S. Atomic Energy Commission, 1960.
- F. Poineau, B. P. Burton-Pye, A. Maruk, G. Kirakosyan, I. Denden, D. B. Rego, E. V. Johnstone, A. P. Sattelberger, M. Fattahi, L. C. Francesconi, K. E. German and K. R. Czerwinski, Inorg. Chim. Acta, 2013, 398, 147–150 CrossRef CAS.
- M. H. Campbell, Anal. Chem., 1963, 35(13), 2052–2054 CrossRef CAS.
- X. Feng and C. Song, Solvent Extr. Ion Exch., 2001, 19(1), 51–60 CrossRef.
- N. C. Schroeder, M. Attrep Jr. and T. Marrero, Technetium and Iodine Separations in the UREX Process, LA-UR-01-6607, 2001.
- E. G. Orebaugh, Analysis of Technetium in SRP Uranium Product Streams, DPST-84-835, 1986.
- E. Vialard and M. Germain, Technetium Behaviour Control in the PUREX Process, Fr. Pat., FR8602443, 1992 Search PubMed.
- S. Tachimori, J. Nucl. Sci. Technol., 2012, 31(5), 456–462 CrossRef.
- G. D. Jarvinen, K. M. Long, G. S. Goff, W. H. Runde, E. J. Mousolf, K. R. Czerwinski, F. Poineau, D. R. McAlister and E. P. Horwitz, Solvent Extr. Ion Exch., 2013, 31(4), 416–429 CrossRef CAS.
- M. Ozawa, M. Ishida and Y. Sano, Radiochemistry, 2003, 45(3), 205–212 CrossRef.
- A. N. Mashkin, K. K. Korchenkin and N. A. Svetlakova, Radiochemistry, 2002, 44(1), 34–40 CrossRef.
- F. Liu, W.-F. Zheng, Y. Zhang, H. Wang and C.-X. Zhou, J. Radioanal. Nucl. Chem., 2013, 295, 1621–1625 CrossRef CAS.
- K.-W. Kim, S.-H. Kim, K.-C. Song, E.-H. Lee and S.-H. Yoo, J. Radioanal. Nucl. Chem., 2002, 253, 3–10 CrossRef CAS.
- E. Aneheim, C. Ekberg, A. Littley, E. Lofstrom-Engdahl and G. Skarnemark, J. Radioanal. Nucl. Chem., 2013, 296, 743–748 CrossRef CAS.
- P. Moeyaert, T. Dumas, D. Guillaumont, K. Kyashnina, C. Sorel, M. Miguirditchian, P. Moisy and J. F. Durfreche, Inorg. Chem., 2016, 55, 6511–6519 CrossRef CAS PubMed.
- K. Geroge, A. Masters, F. Livens, M. Sarsfield, R. Taylor and C. Sharrad, Hydrometallurgy, 2022, 211, 105892 CrossRef.
- H. Chen, M. Jobson, R. J. Taylor, D. A. Woodhead, A. J. Masters and C. Sharrad, Ind. Eng. Chem. Res., 2022, 61, 786–804 CrossRef CAS.
- H. Chen, M. Jobson, R. Taylor, M. Sarsfield, A. Masters, D. Woodhead and C. Sharrad, Prog. Nucl. Energy, 2023, 164, 104856 CrossRef CAS.
- R. S. Herbst and M. Nilsson, in Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment, ed. K. L. Nash and G. J. Lumetta, Woodhead Publishing Series in Energy, Cambridge, 2011, vol. II, ch. 6, pp. 141–175 Search PubMed.
- P. Baron, M. Lecomte, B. Boullis, N. Simon and D. Warin, Separation of the Long Lived Radionuclides: Current Status and Future R&D in France, Global, New Orleans, 2003.
- R. Taylor, G. Mathers and A. Banford, Prog. Nucl. Energy, 2023, 164, 104837 CrossRef CAS.
- P. Paviet-Hartmann, C. Riddle, K. Campbell and E. Mausolf, Overview of Reductants Utilized in Nuclear Fuel Reprocessing/Recycling, INL/CON-12-28006, United States, 2013.
- T. J. Kemp, A. M. Thyer and P. D. Wilson, J. Chem. Soc., Dalton Trans., 1993, 2601–2605 RSC.
- J. Garraway and P. Wilson, J. Less-Common Met., 1984, 97, 191–203 CrossRef CAS.
- E. K. Dukes and R. M. Wallace, Formation of hydrazoic acid from hydrazine in nitric acid solutions, DP-72B, United States, 1962.
- P. R. Zalupski, T. S. Grimes, C. D. Pilgrim and S. Jansone-Popova, Complete Initial Evaluation of Novel Complexants for Tc Holdback for Simplified Single Cycle Separations, Report INL/RPT-22-69248, Idaho National Laboratory, 2022.
- P. R. Zalupski, T. S. Grimes, C. D. Pilgrim, S. Jansone-Popova and M. Jopaul, Pertechnetate Recognition by Guanidinium-Based Compounds. Part I. Reagent Screening. Report INL/RPT-23-73272, Idaho National Laboratory, Idaho, 2023.
- P. R. Zalupski, T. S. Grimes, C. D. Pilgrim, S. Jansone-Popova and J. Mathew, Complete initial demonstration of guanidinium class of complexants for technetium with a goal of reducing the Tc distribution ratio to below 0.2 in the prototypic DEHiBA extraction system, Report INL/RPT-23-74656, Idaho National Laboratory, Idaho, 2023.
- J. E. Birkett, M. J. Carrott, O. D. Fox, C. J. Jones, C. J. Maher, C. V. Roube, R. J. Taylor and D. A. Woodhead, J. Nucl. Sci. Technol., 2007, 44, 337–343 CrossRef CAS.
- I. May, R. J. Taylor and G. Brown, J. Alloys Compd., 1998, 271–273, 650–653 CrossRef CAS.
- R. J. Taylor and I. May, Czech. J. Phys., 1999, 49, 617–621 CrossRef CAS.
- M. J. Carrott, O. D. Fox, C. J. Maher, C. Mason, R. J. Taylor, S. I. Sinkov and G. R. Choppin, Solvent Extr. Ion Exch., 2007, 25(6), 723–745 CrossRef CAS.
- M. J. Carrott, O. D. Fox, G. LeGurun, C. J. Jones, C. Mason, R. J. Taylor, F. P. L. Andrieux and C. Boxall, Radiochim. Acta, 2008, 96(6), 333–343 CrossRef CAS.
- R. J. Taylor, S. I. Sinkov, G. R. Choppin and I. May, Solvent Extr. Ion Exch., 2008, 26(1), 41–61 CrossRef CAS.
- P. Tkac, B. Matteson, J. Bruso and A. Paulenova, J. Radioanal. Nucl. Chem., 2008, 277(1), 31–36 CrossRef CAS.
- S. P. Mezyk and D. M. Bartels, J. Phys. Chem. A, 1997, 101, 6233–6237 CrossRef CAS.
- E. Lieber and G. B. L. Smith, Chem. Rev., 1939, 25(2), 213–271 CrossRef CAS.
- D. R. Levering, The reaction of diaminoguanidine with nitrous acid, Doctoral Dissertation, Illinois Institute of Technology, 1950.
- E. Lieber, E. Sherman, R. A. Henry and J. Cohen, J. Am. Chem. Soc., 1951, 73, 2327–2329 CrossRef CAS.
- E. Lieber and D. R. Levering, J. Am. Chem. Soc., 1951, 73, 1313–1317 CrossRef CAS.
- J. Wang, M. Cai, F. Zhao and K. Xu, Molecules, 2019, 24(19), 3616 CrossRef CAS.
- G. P. Horne, C. R. Gregson, H. E. Sims, R. M. Orr, R. J. Taylor and S. M. Pimblott, J. Phys. Chem. B, 2017, 121(4), 883–889 CrossRef CAS PubMed.
- T. M. Klapötke and J. Stierstorfer, J. Am. Chem. Soc., 2009, 131, 1122–1134 CrossRef PubMed.
- K. Whitman, S. Lyons, R. Miller, D. Nett, P. Treas, A. Zante, R. W. Fessenden, M. D. Thomas and Y. Wang, Proceedings of the ’95 Particle Accelerator Conference and International Conference on High Energy Accelerators, IEEE, New Jersey, 1996.
- G. L. Hug, Y. Wang, C. Schöneich, P. Y. Jiang and R. W. Fessenden, Radiat. Phys. Chem., 1999, 54, 559–566 CrossRef CAS.
- G. V. Buxton and C. R. Stuart, J. Chem. Soc., Faraday Trans., 1995, 91, 279–281 RSC.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- J. W. T. Spinks and R. J. Woods, An Introduction to Radiation Chemistry, Wiley Press, New York, 1990 Search PubMed.
- R. E. Umpleby, J. K. Conrad, J. R. Wilbanks, K. D. Schaller and G. P. Horne, Radiat. Phys. Chem., 2023, 207, 110799 CrossRef CAS.
- B. J. Mincher and R. D. Curry, Appl. Radiat. Isot., 2000, 52, 189–193 CrossRef CAS PubMed.
- G. McKay, B. Sjelin, M. Chagnon, K. P. Ishida and S. P. Mezyk, Chemosphere, 2013, 92, 1417–1422 CrossRef CAS PubMed.
- P. Atkins and J. de Paula, Physical Chemistry, W.H. Freeman and Co., New York, 7th edn, 2001 Search PubMed.
- R. J. Drout, K. Otake, A. J. Howarth, T. Islamoglu, L. Zhu, C. Xiao, S. Wang and O. K. Farha, Chem. Mater., 2018, 30(4), 1277–1284 CrossRef CAS.
- K. Libson, J. C. Sullivan, W. A. Mulac, S. Gordon and E. Deutsch, Inorg. Chem., 1989, 28, 375–377 CrossRef CAS.
- L. Heller-Grossman, S. Abrashkin, A. Shafferman, M. A. Davis and R. A. Taube, Int. J. Appl. Radiat. Isot., 1981, 32, 501–506 CrossRef CAS.
- E. Deutsch, W. R. Heineman, R. Hurst, J. C. Sullivan, W. A. Mulac and S. Gordon, J. Chem. Soc., Chem. Commun., 1978, 1038–1040 RSC.
- A. K. Pikaev, S. V. Kryuchkov, A. F. Kuzina and V. I. Spitsyn, Dokl. Phys. Chem., 1977, 236, 992–995 Search PubMed.
- J. Drader, G. Saint-Louis, J. M. Muller, M.-C. Charbonnel, P. Guilbaud, L. Berthon, K. M. Roscioli-Johnson, C. A. Zarzana, C. Rae, G. S. Groenewold, B. J. Mincher, S. P. Mezyk, K. McCann, S. G. Boyes and J. Braley, Solvent Extr. Ion Exch., 2017, 35, 480–495 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. Contains additional chemical kinetic measurement data and non-normalized DAG concentrations vs. absorbed gamma dose data. See DOI: https://doi.org/10.1039/d3cp04987f |
| This journal is © the Owner Societies 2024 |