
Adsorption and dehydrogenation of ammonia on Ru55, Cu55 and Ru@Cu54 nanoclusters: role of single atom alloy catalyst
D.
Chattaraj
 *a and
C.
Majumder
b
*a and
C.
Majumder
b
aProduct Development Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, India. E-mail: debchem@barc.gov.in; Fax: +91 22 2550 5151; Tel: +91 22 2559 6446
bChemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, India
First published on 12th December 2023
Abstract
Hydrogen production by the catalytic decomposition of ammonia (NH3) is an important process for several important applications, which include energy production and environment-related issues. The role of single Ru-atom substitution in a Cu55 nanocluster (NC) has been illustrated using the NH3 decomposition reaction as a model system. The structural stability of Ru@Cu54 NC has been evaluated using Ru55 and Cu55 NCs for comparison. Ru@Cu54 prefers an icosahedron structure (Ih), like Ru55 and Cu55 NCs, with almost comparable average binding energies of −5.55 eV per atom. The adsorption of NHx (x = 0–3) on different adsorption sites of the icosahedron Ru@Cu54 NC has also been studied and the corresponding adsorption energies have been estimated. The site-preference investigation suggested that NH3 prefers to adsorb vertically to the Ru@Cu54. The stable geometries of the N and H atoms on the high symmetry adsorption sites of Ru@Cu54 NC have been studied. Although the N atom favours top and hollow sites, the H atom prefers to stay in the Ru–Cu bridge site along with the hollow sites. The adsorption energy of N on the Ru@Cu54 NC fcc site is found to be −5.42 eV, which is very close to the optimal value (−5.81 eV) of the ammonia decomposition volcano curve. The reaction energies for stepwise H atom elimination from an adsorbed NH3 molecule have been estimated. Finally, NH3 adsorption and decomposition on Ru@Cu54 have been illustrated in terms of electronic structure analysis. The energetics calculations for the dehydrogenation of NH3 suggest that Ru@Cu54 NC can be a suitable catalyst.
Introduction
In recent times, the research and development of molecular hydrogen generation reactions have become an active area of scientific exploration.1,2 An economy based on hydrogen fuel is expected to reduce dependency on fossil fuels and reduce environmental pollution. Hence, it is now obvious that hydrogen is going to be the fuel for on-board vehicular applications and portable devices in coming decades.3,4 Conventional hydrogen production processes, such as partial oxidation, steam reforming of hydrocarbons, etc., are associated with the emission of COx (x = 1, 2) as by-products. As NH3 is produced in large scale worldwide with an existing huge distribution infrastructure, hydrogen production by NH3 decomposition could counter the above-mentioned problems. Ammonia, with its 17.8% hydrogen carrying capacity, has been found to be a potential source for COx (x = 1, 2) free hydrogen production through its decomposition.5 The decomposition of NH3 can produce high-purity hydrogen for fuel cells and has good practical applications.The fundamental reaction mechanism of NH3 decomposition on catalytic surfaces has become very important to design novel catalysts with higher chemical reactivity and selectivity. Ru and Ir, among the Pt group metals, are regarded as the most active catalysts for the decomposition of NH3.6,7 Mainer et al. reported a combined experimental and theoretical study in which they demonstrated NH3 adsorption and dehydrogenation on a Ru (0001) surface.8 However, the high cost and limited availability of Ru and Ir catalysts created the necessity of finding a suitable catalyst for this purpose. Less-expensive and easily available alternatives such as Ni-, Co-, Fe-, Cu-, and Pd-based catalysts posses’ good catalytic properties for NH3 adsorption and decomposition. A variety of such metal surfaces, like Ni,9–12 Co,13 Fe,13,14 Cu,15,16 Pd17–19etc., have been studied extensively as catalysts for this purpose.
Cu is regarded as a good catalyst for the purpose of gas-molecule adsorption and decomposition on its surface. The interaction of NH3 with Cu surfaces has been investigated by several authors, both experimentally and theoretically.20–24 Bartels et al. investigated NH3 molecule adsorption on a Cu(111) surface and inferred that multiple electronic excitation of the ammonia-substrate bond can lead to the desorption of the molecule from the substrate.20 Using scanned-energy-mode photoelectron diffraction, Baumgartel et al. studied the local structure of a Cu(111) surface with ammonia adsorbed and found that the NH3 molecule occupies the top site with a Cu–N bond length of 2.09 Å.21 In addition to experimental studies, several computational studies based on quantum mechanical principles on NH3 dehydrogenation process on a pure Cu surface are also available.22–24 Jiang et al. reported the adsorption configuration and dissociation mechanism of NH3 on Pd(111) and Cu(111) surfaces.22 It was revealed that the adsorption energy trend of NHx (x = 0–3) is NH3 < NH2 < NH < N and that the ammonia dissociation over Pd-based and Cu-based catalysts is a structure-sensitive reaction. In addition, Robinson and Woodruff23 and Xing24 have studied the complete dissociation of NH3 on a Cu(111) surface.
As per the study of Jiang et al.,25 NH dissociation is the most energy-intensive step in NH3 dehydrogenation on clean Cu(111) surfaces, making it the rate-determining step. Subsequently, effort has been made to improve the catalytic activity of Cu by alloying it with other transition metals such as Pd, Ni, etc.26,27 Jiang et al. systematically investigated NH3 dehydrogenation on Pd–Cu(111) and Cu–Pd(111) surfaces using density functional theory (DFT) with the aim of exploring the effect of the surface composition of the catalysts and the role of the dopant metal on the catalytic activity of NH3 dehydrogenation.26 They showed that as compared to Pd(111) and Cu(111) surfaces, the synergistic effect exists in different layers of the catalyst surfaces, which may help to design an optimal catalyst for ammonia dissociation by doping suitable atoms. The main limitation of the large energy barrier for NH dissociation has not been reduced significantly or eliminated despite all these efforts.
So far, most of the NH3 dehydrogenation studies are limited to metal or bimetallic surfaces, and very few are available on nanodomains. In the nano regime, the step-by-step NH3 dehydrogenation process has been computationally studied on 13-atom Cu-, Ni- and Cu-doped Ni clusters by Chen et al.27 They reported that adsorption energy of N on the Ni12Cu cluster is very close to the optimum value of the volcano curve, making it a suitable catalyst for NH3 dehydrogenation. The heat of reaction for the NH3 dehydrogenation process for Ni12Cu is found to be in between those of Ni13 and Cu13 clusters. X. Chen et al. investigated the NH3 decomposition reaction on three types of M@Ni core–shell nanoparticles (M = Fe, Ru, Ir) with 13 core M atoms and 42 shell Ni atoms. Regarding NH3 decomposition, they found that the Ru@Ni core–shell nanocluster possesses catalytic performance comparable to that of single metal Ru, but the catalytic activity of Fe@Ni and Ir@Ni core–shell NCs was found to be unsatisfactory as compared to the active metal Ru.28 In addition to these studies, Lanzani et al. studied the bonding and dissociation of NH3 and its fragments on a nanosized icosahedral Fe55 cluster using spin-polarized DFT.29 They suggested that the catalytic activity of iron surfaces towards ammonia-like molecules is enhanced when the metal is in the nanostructured phase.
Both Ru and Cu catalysts are widely known for their use as catalysts in various catalytic applications. The catalytic activity of these catalysts increases as we move from the bulk to the surface to the nano domain due to the increase in surface area, which are obvious in surface catalytic reactions. Nanoclusters have drawn considerable attention from researchers in the fields of physics, chemistry, materials science and especially catalysis, as they play an important role in bridging the gap between isolated atoms and bulk material.30,31
There are few catalytic studies on Ru55 nanoclusters, which may be because their limited availability and very high cost makes them a poor choice in catalysis. One of the important applications of Ru55 nanoclusters as a catalyst is described in ‘Ru55 nanoparticle catalyze the dissociation of H2O monomer and dimer to produce hydrogen: A comparative DFT study’.32 Here, the authors reported the H2O monomer and dimer dissociation followed by H2 release on a Ru55 nanocluster using DFT calculations. Su et al. used a Ru55 nanocluster to enhance the hydrogen evolution reaction (HER) performance as compared to the conventionally used catalysis.33 They used a Ru55 nanocluster catalyst deposited on O-doped graphene decorated with single metal atoms (e.g., Fe, Co and Ni) for HER. Consistent with theoretical predictions, these hybrid catalysts show outstanding HER performance, much superior to other reported electro-catalysts, such as the state-of-the-art Pt/C catalyst. Ajaml et al. reported the relationship between particle size, intermediate structure and energies of water reduction to produce hydrogen (H2) on Rux (x = 6, 13 and 55) clusters on a g-CN support using DFT calculations as well as by experiment.34 Ungerer et al. investigated the electron distribution and magnetic properties of Ru fcc nano-dots, both in icosahedral (13 and 55 atoms) and cubic shape (13 and 63 atoms).35
Similarly, the Cu55 nanocluster is also known for its several applications as a catalyst. Among other applications, it is used in acetylene hydrogenation, methanol and ethanol formation from syngas, conversion of CO2 to hydrocarbon fuels, CO oxidation, and as a degradant agent for SF6 molecules.36–42 Zhao et al. reported the effect of the size of the Cu nanocluster on the selectivity and activity of acetylene-selective hydrogenation to ethylene using DFT calculations.36 They have shown that on the Cu55 cluster, C2H2 is easily hydrogenated to form C2H4via a CHCH2 intermediate, and C2H4 prefers desorption over its hydrogenation, suggesting that the Cu55 cluster exhibits a good selectivity towards C2H4 formation. The catalytic activity of catalysts greatly depends on their local structure. The local structure changes of the Cu55 nanocluster during heating were investigated by Lin et al. using MD simulation.37 They reported how the local structure changes accompanying the atom packing affect the internal energy with increasing temperature by calculating the radial distribution function. Zhang et al. reported the formation of C2 oxygenates from syngas on Cu clusters of different sizes (Cu13, Cu38 and Cu55) and the size effect of Cu clusters on the catalytic performance of C2 oxygenates in order to search for a novel Cu-based catalyst with improved activity and selectivity towards C2 oxygenates.38 Lim et al. studied the conversion of CO2 into hydrocarbon fuels (CH4, CO and HCOOH) on defective-graphene-supported Cu nanoparticles (Cu55 clusters) using a first-principles method.39
However, in the above paragraphs, we mentioned that the use of Ru55 or Cu55 alone is not effective for the dehydrogenation of NH3 to H2 and N2, although a combination of these two elements can be useful for this purpose. Hence, the single-atom catalyst Ru@Cu54 has been chosen for the study of NH3 dehydrogenation.
To the best of our knowledge, earlier studies on the adsorption and dehydrogenation of ammonia have mainly focused on pure Ru and Cu metal surfaces. Limited systematic studies have been carried out to understand the effect of dopant metals on the dehydrogenation of ammonia. Also, few reports are available in the literature on the finite size effect in this dehydrogenation process. Therefore, it is of interest to probe the effect of the dopant, structure and size of bimetallic single-atom alloy catalysts on the adsorption and dehydrogenation of ammonia. In the present study, an effort has been made to incorporate the effect of the Ru atom by using it to replace a Cu atom in Cu55 NC and study the NH3 dehydrogenation mechanism process using the DFT method, with the aim of clarifying the favourable reaction mechanisms and the effect of the single Ru atom on the Cu surface. To determine the exact mechanism of NH3 decomposition on this surface, the elementary steps have been calculated, along with the adsorption energy (Eads) and reaction energy (ΔE). The regeneration of the catalyst in terms of nitrogen removal from the NCs has also been investigated for its reuse. This study will help to provide a fundamental understanding of the structural, energetic and catalytic properties of the Ru@Cu54 catalyst for NH3 dehydrogenation and explain why this catalyst can be useful for the production of H2 molecules via NH3 decomposition.
Computational details
All calculations were performed within the spin-polarized density functional theory using the plane wave-pseudopotential approach as implemented in the Vienna ab initio simulation package (VASP).43–45 The electron–ion interaction and the exchange correlation energy were described under the projector-augmented wave (PAW) method and the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE), respectively.46–48 The energy cut-off for the plane wave basis set for assuming a good energy convergence was fixed at 500 eV. We considered a large number of initial geometries for the clusters with intermediates and found the lowest-energy structures. A large number of spin states were also considered for all these configurations. Ionic optimization was carried out using the conjugate gradient scheme, and the forces on each ion were minimized to 5 meV Å−1.49,50 A suitable simulation box of size 20 × 20 × 20 Å was considered for all the calculations. The k-point sampling in the Brillouin zone (BZ) was treated with the Monkhorst–Pack scheme using the gamma point.51 The total energies of each relaxed structure using the linear tetrahedron method with Blochl corrections was subsequently calculated in order to eliminate any broadening-related uncertainty in the energies.52 The average binding energy (Eb) of the Ru@Cu54 NC was estimated from the total energy calculations using eqn (1)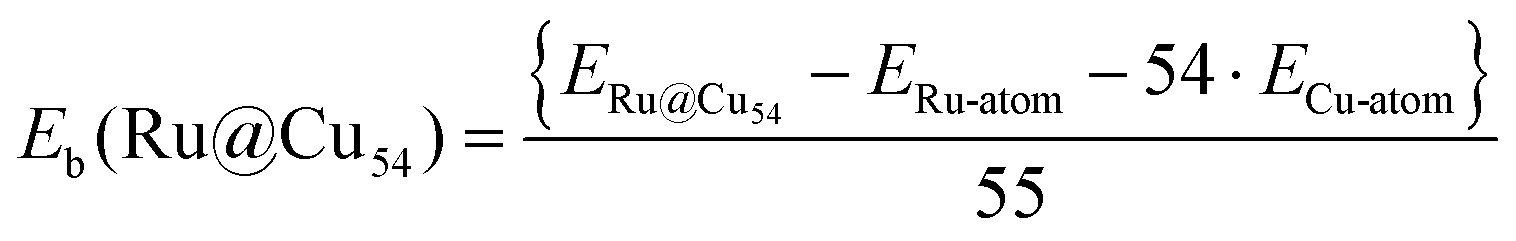 | (1) |
The adsorption energies of the adsorbate on Ru@Cu54 NC, Eads, was calculated using eqn (2)
| Eads = Eadsorbate+cluster − (Eadsorbate + Ecluster) | (2) |
Results and discussion
Structural stability of metal clusters
Ru has been regarded as a very good catalyst for ammonia (NH3) decomposition, as Ru lies in the optimum position of the volcano curve that depicts the choice of materials for NH3 decomposition. However, Ru has limitations due to its higher cost and low availability. On the other hand, Cu is a versatile catalyst used in different catalytic processes. Like Ru, Cu also has the limitation of a higher energy requirement during the last NH dissociation, which makes this the rate determining step. To rationalize the cost effectiveness of materials and reduce energy expenses, a combination of Cu and Ru atoms has been considered. Nanoclusters may possess better catalytic activity as compared to their bulk counterparts. The stability of 55-atom Cu55 and Ru55 nanoclusters (NCs), along with a Cu55 species with a single Ru atom substitution (Ru@Cu54), has been investigated using DFT. It is found that, like Cu55 and Ru55 NCs, the substituted Ru@Cu54 species possesses an icosahedron (Ih) structure in its ground state, as shown in Fig. 1(a–c). The cohesive energies and interatomic bond distances of the Cu55, Ru@Cu54 and Ru55 NCs are summarized in Table 1. From the table, it can be seen that Ru@Cu54 has more or less the same cohesive energy as the Cu55 and Ru55 NCs. Hence, stability-wise, Ru@Cu54 does not differ much from the 55-atom NCs of its constituent elements. The spin moments of these NCs are also listed in Table 1. We calculated the stability of the Ru@Cu54 nanocluster with Ru atom at the top and edge positions. The configuration with Ru at the top position was found to be 0.12 eV more stable compared to that at the edge position in terms of average binding energy. Hence, the Ru@Cu54 nanocluster with the Ru atom at the top position was used throughout our entire study. We assumed that the Ru atom should be on the surface of the Ru@Cu54 nanocluster, as it would facilitate NH3 adsorption followed by its dehydrogenation through direct interaction with the NH3 molecule. Investigation of the structural stability with the Ru atom at the inner position of Ru@Cu54 nanocluster is of no use here, as there would be no direct interaction between the Ru atom and NH3 molecule. Hence, the structural stability of the Ru@Cu54 nanocluster with Ru as an inner atom has not been studied here.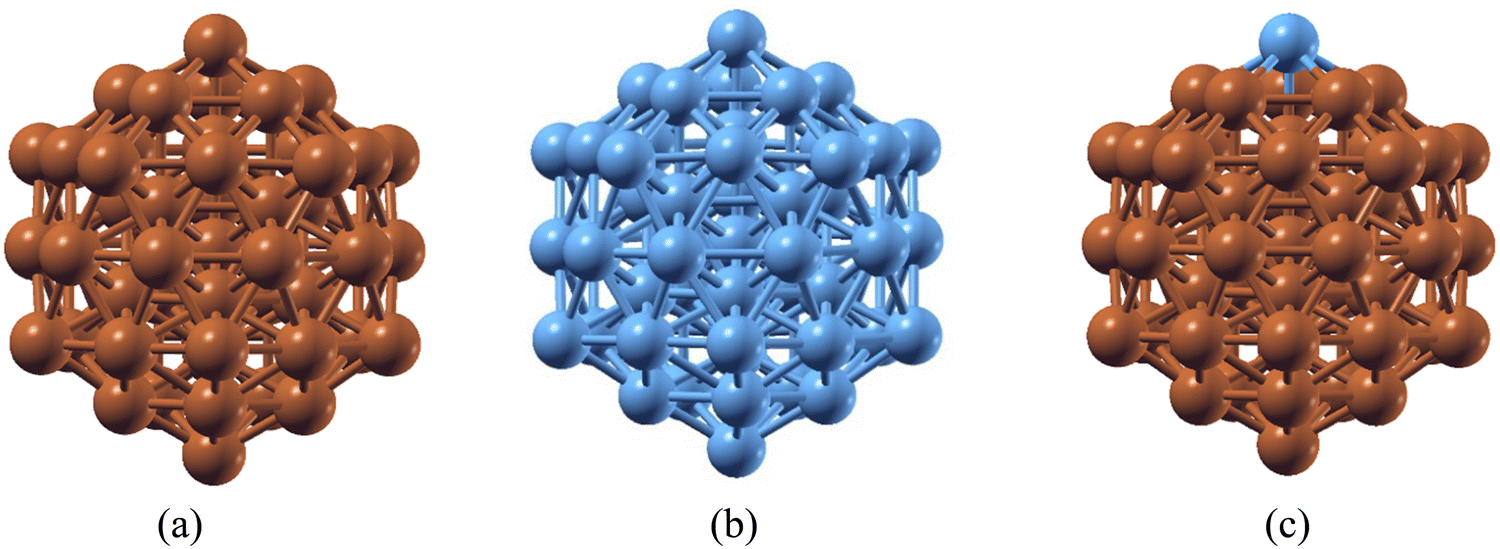 | ||
| Fig. 1 Optimized structures of the (a) Cu55, (b) Ru55 and (c) Ru@Cu54 nanoclusters. (Atom colours: Sky blue = Ru, Brown = Cu). | ||
| Species | Average binding energy | Average bond length (Å) | Spin moment (μB) | ||
|---|---|---|---|---|---|
| Cu–Cu | Cu–Ru | Ru–Ru | |||
| Cu55 | −5.59 | 2.67 | — | — | 14 |
| Ru@Cu54 | −5.55 | 2.68 | 2.79 | — | 14 |
| Ru55 | −5.54 | — | — | 2.70 | 13 |
It has been reported that the N atom adsorption energy is one of the important factors for the NH3 dehydrogenation catalytic reaction. In order to determine the catalytic activity of the Ru@Cu54 NC for NH3 dehydrogenation, the adsorption energy of the N intermediate was compared with the optimum value of the famous volcano curve (−5.81 eV), and it was found that the adsorption energy of N on the Ru@Cu54 NC fcc site (−5.42 eV) (see Table 2) is very close to that value. In a similar way, the N-adsorption energy of the Cu@Ru54 NC was also calculated and found to be −1.85 eV (see Table 2), which is far from the optimum value of the volcano curve (−5.81 eV). Hence, Cu@Ru54 was not considered further for NH3 dehydrogenation.
| Species | E ads | d M–N | d N–H | <H–N–H |
|---|---|---|---|---|
| Ru55–NH3 | −1.05 | 2.78 | 1.03 | 107.58 |
| Cu@Ru54–NH3 | −0.87 | 2.04 | 1.03 | 107.31 |
| Cu55–NH3 | −0.84 | 2.06 | 1.03 | 107.35 |
| Ru@Cu54–NH3 | −0.85 | 2.23 | 1.02 | 107.7 |
| Ru55–N | −5.03 | 1.67 | ||
| Cu@Ru54–N | −1.85 | 1.75 | ||
| Cu55–N | −4.36 | 1.84 | ||
| Ru@Cu54–N | −5.42 | 1.65 |
Adsorption of reaction intermediates
After establishing the equilibrium structures of Ru@Cu54, the adsorption of an NH3 monomer, NHx (x = 0–2) and H atoms on it were studied. The Ru@Cu54 cluster has twenty faces made up of Cu and Ru atoms. For the NH3, NHx (x = 0–2), N and H species, there are six types of adsorption sites, one top site t1, one edge site t2, two bridge sites b1 and b2, and two hollow sites h1(fcc) and h2(hcp). The optimized structures of the Ru@Cu54 NC with adsorbed NH3, NHx (x = 0–2), H, 2H, H2, 2N and N2 are shown in Fig. 2(a–o). The adsorption energy and geometric parameters are summarized in Tables 2 and 3.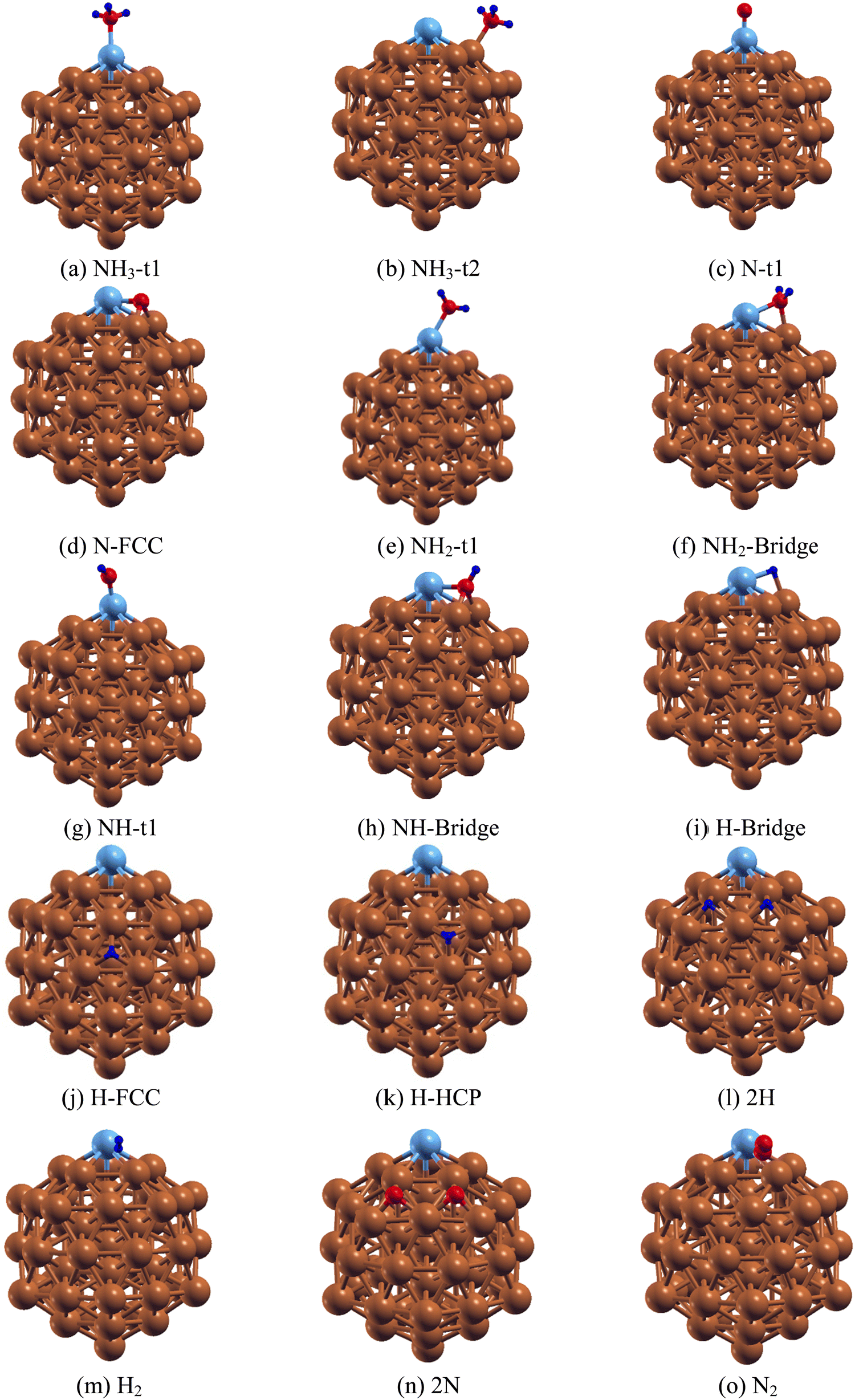 | ||
| Fig. 2 (a)–(o) Optimized structures of different fragments of NH3 on Ru@Cu54 nanoclusters: (a) NH3-t1 (one NH3 located at the t1 site of Ru@Cu54), (b) NH3-t2, (c) N-t1, (d) N-FCC, (e) NH2-t1, (f) NH2-bridge, (g) NH-t1, (h) NH-bridge, (i) H-bridge, (j) H-FCC, (k) H-HCP, (l) 2H, (m) H2, (n) 2N and (o) N2. (Atom colours: Sky blue = Ru, Brown = Cu, Red = N and Blue = H). | ||
| Species | Adsorption site | E ads | D M–N/M–H (M = Ru, Cu) | d N–H | <H–N–H |
|---|---|---|---|---|---|
| NH3 | Top(t1) | −0.85 | 2.23 (Ru–N) | 1.02 | 107.7 |
| Top(t2) | −0.48 | 2.12 (Cu–N) | 1.02 | 107.7 | |
| NH2 | Top(t1) | −4.55 | 1.90 (Ru–N) | 1.02 | 111.23 |
| Bridge(B) | −4.48 | 2.08 (Ru–N) | 1.02 | 111.5 | |
| 2.0 (Cu–N) | |||||
| NH | Top(t1) | −4.05 | 1.77 | 1.03 | — |
| Bridge(B) | −4.34 | 1.93 (Ru–N) | 1.03 | — | |
| 1.93 (Cu–N) | |||||
| N | Top(t1) | −5.38 | 1.65 (Ru–N) | — | — |
| FCC | −5.42 | 1.72 (Ru–N) | — | — | |
| 1.92 (Cu–N) | |||||
| H | Bridge(B) | −2.70 | 1.70 (Ru–H) | — | — |
| 1.79 (Cu–H) | |||||
| FCC | −2.54 | 1.80 (Cu–H) | — | — | |
| HCP | −2.41 | 1.75 (Cu–H) | — | — |
For the adsorption of NH3 molecule on Ru@Cu54, calculation shows that the NH3 molecule is preferentially adsorbed at the top site t1 and edge site t2 with binding energies of −0.85 and −0.48 eV, respectively, through the formation of Ru–N (for t1) and Cu–N (for t2) bonds (shown in Fig. 2(a) and (b)). Here, the Ru–N and N–H bond distances were found to be 2.23 and 1.02 Å for NH3 adsorbed at the t1 site, whereas the Cu–N and N–H bond distances were found to be 2.12 and 1.02 Å for the t2 site, respectively. Upon NH3 adsorption at the t1 site, the H–N–H bond angle was calculated to be 107.7°, which is comparable to the value obtained in the case of NH3 adsorption on the Ru(110) surface.12 The obtained geometric parameters, such as the Ru–N and N–H bond lengths and the H–N–H bond angle of NH3 adsorbed on Ru@Cu54, which are listed in Table 2, are comparable with those in the pure Ru55 NC.
For the generation of H2 molecules, NH3 should be dissociated into N and H atoms on the Ru@Cu54 surface. Thus, it is important to determine the stable positions of the intermediate N, H, NH2 and NH species on Ru@Cu54 NC. First, we investigated the stability of the N atom at different sites of Ru@Cu54. For those adsorptions, six initial configurations were considered. In these six configurations, the N atom positions were at top site t1, edge site t2, the two bridge sites b1 and b2 and the two three-atom hollow sites h1(fcc) and h2 (hcp). Among these, two sites, i.e., the top (t1) and three-atom hollow fcc site, were found to be stable sites for N atom adsorption, as shown in Fig. 2(c and d). The adsorption energy of the reaction intermediate N on the Ru@Cu54 fcc site is found to be −5.42 eV (Table 3), which is very close to the optimal value (−5.81 eV) of famous ammonia decomposition volcano curve.53
With respect to NH2 adsorption on the 55-atom Ru@Cu54 NC, NH2 can be adsorbed on both the Ru-top and Ru–Cu bridge site, as shown in Fig. 2(e) and (f). From Table 3, it is seen that NH2 at the top site (−4.55 eV) on Ru@Cu54 was found to be more stable than that at the bridge site (−4.48 eV). For pure Ru55, the NH2 adsorption energy is −5.04 eV for the bridge configuration, whereas it is −4.67 eV for the top (t1) site. For NH2 adsorbed at the t1 site on Ru@Cu54 NC, the Ru–N and N–H bond lengths were found to be 1.90 Å and 1.02 Å, respectively, while the H–N–H cone bond angle is 111.23°. Similarly, for the bridge site, the Ru–N and Cu–N bond lengths were optimized to be 2.08 and 2.00 Å, respectively, with N–H bond lengths of 1.02 Å and an H–N–H bond angle of 111.5°.
For NH adsorption on Ru@Cu54, two types of stable adsorption positions (top and bridge sites) were examined. The adsorption of NH on the top and bridge sites of Ru@Cu54 NC is shown in Fig. 2(g) and (h), respectively. NH at the Ru–Cu bridge site is found to be more stable as compared to that on the top site by 0.29 eV. The NH adsorption energy on the Ru@Cu54 NC bridge was calculated to be −4.34 eV (Table 3). In the bridge configuration, the Ru–N, Cu–N and N–H bond lengths were found to be 1.93, 1.93 and 1.03 Å, respectively.
To determine the stability of the H atom on Ru@Cu54, the H atom was placed in all the probable sites, i.e., the top, edge, and Ru–Cu and Cu–Cu bridge sites, as well as the fcc and hcp three-atom hollow sites. Even after placing the H atom at the t1 and t2 sites, it was found that it went to the Ru–Cu bridge position after optimization. The stable structures with the H atom at the bridge, FCC and HCP sites of Ru@Cu54 NC are shown in Fig. 2(i–k), and the corresponding adsorption energies are summarized in Table 3. From Table 3, it can be seen that the H atom of the Ru–Cu bridge site has higher stability compared to those at the hollow sites. The Ru–H and Cu–H bond lengths are found to be 1.70 and 1.79 Å, respectively, for the bridge configuration shown in Fig. 2(i).
The release of hydrogen molecules from the nanoclusters depends on the binding of H atoms on them. To determine the hydrogen release behaviour, the H atom stability in presence of an N atom on Ru@Cu54, as well as on Cu55 and Ru55, was studied. The binding energy of the H atom on Cu55–N, Ru@Cu54–N, Ru55–N was calculated to be −2.63, −2.65 and −2.81 eV, respectively. From the H atom binding energy, it can be stated that hydrogen release from Ru@Cu54 NC will be almost the same as that from Cu55 NC, but easier than that from Ru55.
After the dehydrogenation of NH3 into N and H atoms, the associative recombination of two H atoms followed by the desorption of the H2 molecule is an important step for the production of hydrogen. Desorption of the H2 molecule will depend on the diffusion of H atoms on the surface, which in turn is proportional to the limiting strength of the H atom on the surface. For the formation of H2, the most stable co-adsorption configuration was taken as the initial state, in which two H atoms adsorb on adjacent fcc sites, as shown in Fig. 2(l), for Ru@Cu54. For Cu55 and Ru55, the co-adsorption of two H atoms also occurs on adjacent fcc sites. The potential energy diagram for H2 desorption from the Ru55, Ru@Cu54 and Cu55 NCs are shown in Fig. 3. In the first step of the reaction, two H atoms adsorbed on the stable fcc sites combine to form an H2 molecule (Fig. 2(m)) with a reaction enthalpy of 0.25, 0.30, 0.54 eV for Cu55, Ru@Cu54, and Ru55, respectively. In the second step of the reaction, the adsorbed H2 molecule desorbs from the catalyst surface with a reaction energy of 0.77, 0.82 and 1.03 eV for Cu55, Ru@Cu54, and Ru55, respectively. From Fig. 3, it can be seen that for Ru@Cu54, the enthalpy of the reaction falls between those of the Cu55 and Ru55 NCs. It can be stated that substitution of one Ru atom in the Cu55 nanocluster will be useful for the production of H2 molecules.
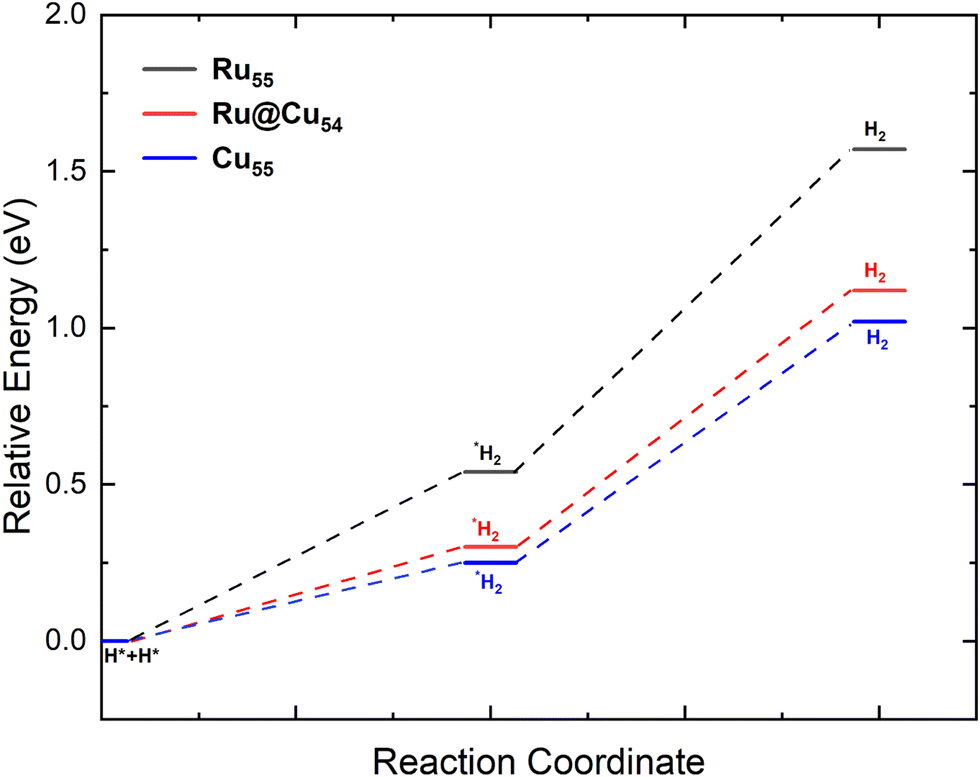 | ||
| Fig. 3 Potential-energy diagram of H2 desorption processes on the Ru55, Ru@Cu54 and Cu55 nanoclusters. | ||
Thermochemistry of NH3 decomposition
In the earlier section, it was mentioned that the adsorption energy of the intermediate N on Ru@Cu54 NC is very close to the optimal value of the volcano curve. To further understand the NH3 decomposition behaviour on Ru@Cu54, the potential energy diagram of NH3 decomposition on this NC has been calculated. The potential energy diagram of NH3 decomposition on the Ru55, Cu55 and Ru@Cu54 NCs is shown in Fig. 4. The most stable adsorption configuration of Ru@Cu54 NC, which is icosahedral, has been chosen as the initial state. We have considered the local interactions between Ru and Cu atoms like other bimetallic systems.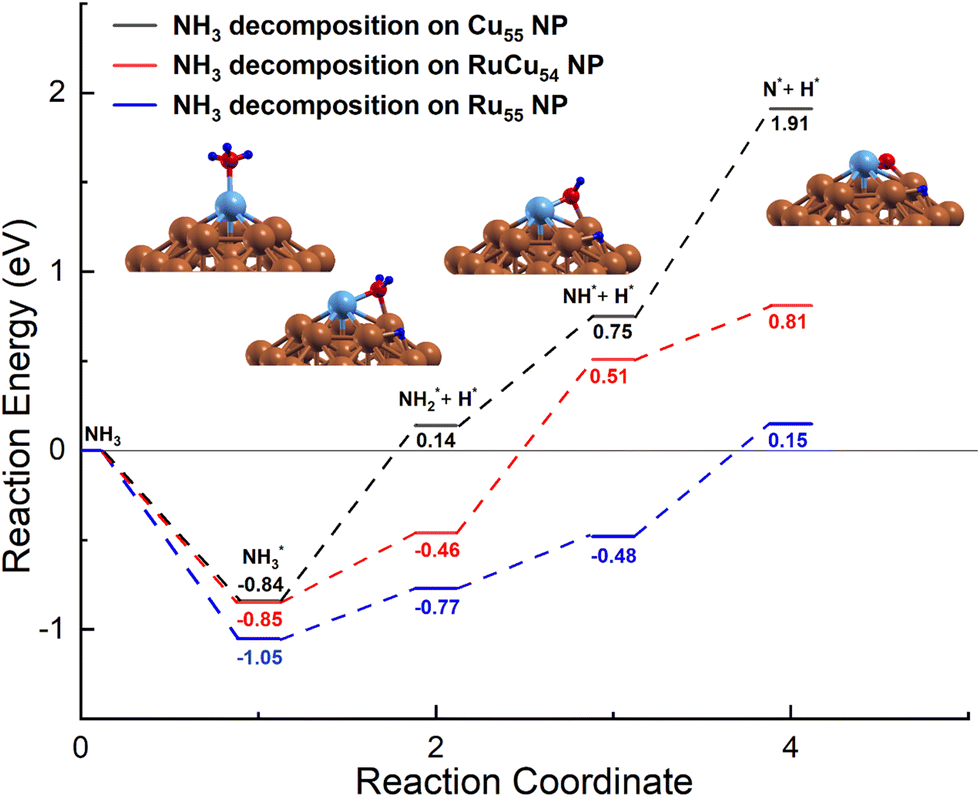 | ||
| Fig. 4 Potential-energy diagram of NH3 decomposition on the Cu55, Ru@Cu54 and Ru55 nanoclusters. (Atomic colours: Sky blue = Ru, Brown = Cu, Red = N and Blue = H). | ||
In the first step of dehydrogenation, NH3 adsorbs on the top site on Ru@Cu54 with an adsorption energy of −0.85 eV and becomes dissociated into NH2 and H intermediates with the breakage of one of the three N–H bonds. The NH2 intermediate remains in the Ru–Cu bridge site, and the H atom moves to the hcp hollow site, as shown in Fig. 4. The dehydrogenation reaction is an endothermic process with a reaction energy of 0.39 eV on Ru@Cu54. The reaction energy for the same dehydrogenation process on Cu55 is 0.98 eV, which is higher than that on Ru@Cu54. For the second step of dehydrogenation on Ru@Cu54, the NH2 adsorbed at the bridge site is considered; it becomes dissociated into NH and H fragments. Although NH remains on the bridge site, the H atom moves to the hcp hollow site, as shown in Fig. 4. The corresponding reaction energy is 0.97 eV, while the value for Cu55 is 0.61 eV. Similarly, for the third step of dehydrogenation, NH sits at the bridge site initially and is then dissociated into N (fcc) and H fragments (hcp), as shown in Fig. 4. This process is endothermic with a reaction energy of 0.30 eV on the Ru@Cu54 NC. The same process of dehydrogenation is also endothermic on the Cu55 NC with a comparatively higher reaction energy of 1.16 eV. This suggests that the dehydrogenation of the NH intermediate is the rate-determining step for Cu55 due to the high positive reaction heat (Fig. 4). The lower reaction energy of the last step of dehydrogenation for Ru@Cu54 makes it a better catalyst for NH3 decomposition compared to Cu55.
By comparing the data obtained in this work, the results show that the overall reaction energy for NH3 dehydrogenation on Ru@Cu54 NC (1.66 eV) is comparable to that on Ru55 NC (1.2 eV), but lower as compared to that on pure Cu55 NC (2.75 eV), which is consistent with the order of N adsorption energies. As expected, the Ru@Cu54 NC is thermodynamically feasible for the reaction of NH3 decomposition and can act as a better catalyst than pure Cu55 NC for this purpose.
In catalysis, the activation energy is a very important parameter, and is the one of the characteristics of the activity of catalysts. Recently, it has been established that for the dissociative chemisorption of a number of molecules, the activation energy depends linearly on the adsorption energy.54,55 The Brønsted–Evans–Polanyi (BEP) relation has been found to be applicable for this type of surface reactions. In the NH3 dehydrogenation process, Duan et al. showed that activation energy for the associative desorption of an N2 molecule and the N-adsorption energy follow a linear relationship for the Fe(110), Co(111), Ni(111) and Cu(111) surfaces.13 Here, the N-adsorption energy for the Cu55, Ru55 and Ru@Cu54 NCs was calculated to be −4.36, −5.03 and −5.42 eV, respectively. From this, we can conclude that the activation energy for the associative desorption of an N2 molecule on Ru@Cu54 NC will be within the range of Ru55.
Regeneration of catalyst
Catalyst regeneration is a very important step in catalysis for reuse and economic viability. Hence, N removal is necessary after the release of a hydrogen molecule from Ru@Cu54. The stability of the two adsorbed N atoms located at different fcc and hcp sites was investigated. Two N atoms were found to be more stable at adjacent fcc sites, as shown in Fig. 2(n), and the corresponding adsorption energy was found to be −5.48 eV per N atom. In the next step, the two N atoms are combined into an N2 molecule vertically on the catalytic surface (Fig. 2(o)) with a reaction enthalpy of 0.12, 0.35, and 0.75 eV for Cu55, Ru@Cu54, and Ru55, respectively. N2 then desorbs from the catalyst surface with a reaction energy of 0.43, 0.76, and 0.63 eV for Cu55, Ru@Cu54, and Ru55, respectively. The distance of N2 molecule from the nearest Ru/Cu atom was found to be 1.91, 1.90, and 1.94 Å for Ru@Cu54, Cu55 and Ru55, respectively. The potential energy diagram and the minimum energy path for the conversion of N2(g) from the two adsorbed N atoms is shown in Fig. 5. From Fig. 5, it can be seen that N2 removal from Ru@Cu54 will be easier, as the enthalpy of the reaction lies between those of Cu55 and Ru55.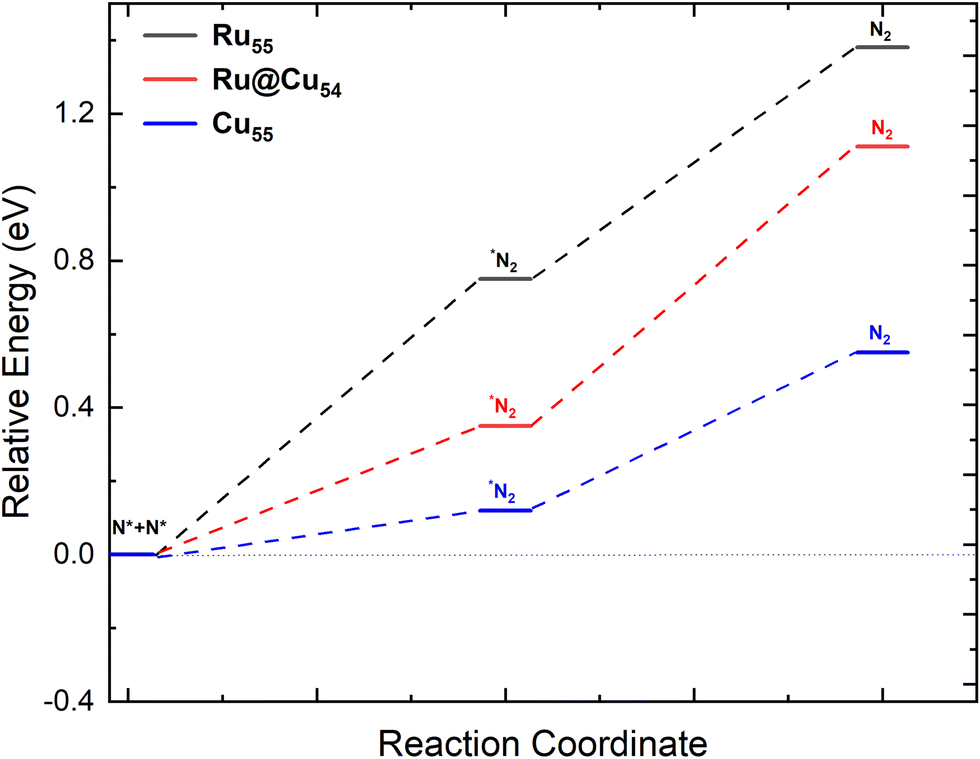 | ||
| Fig. 5 Potential-energy diagram of N2 desorption processes on the Ru55, Ru@Cu54 and Cu55 nanoclusters. | ||
Electronic structure analysis
Since the N atom adsorption energy is one of the important factors in the catalytic NH3 dehydrogenation reaction, in order to interpret the N atom adsorption energy difference, the projected density of states must be calculated for the nanoclusters under study. The interaction between the adsorbate and substrate has been studied by performing the density of states spectra and d-band calculations for the Cu55, Ru@Cu54 and Ru55 nanoclusters, which are shown in Fig. 6. From the figure, it can be seen that the d-bands are more populated near the Fermi level for Cu55 and Ru@Cu54 as compared to Ru55. This facilitates the N atom adsorption on Ru@Cu54, as for metals, a d-band centre close to the Fermi energy can facilitate a stronger interaction between the adsorbate and substrate.56 The calculated d-band centres of Ru55, Ru@Cu54 and Cu55 nanoclusters were found to be −2.85, −2.82 and −3.96 eV, respectively. As the d-band centre of Ru@Cu54 is nearest to the Fermi level, the N atom adsorption energy will be higher for Ru@Cu54 as compared to Ru55 and Cu55. In the previous section, it was reported that the N atom adsorption energy is the highest for Ru@Cu54 among the three nanoclusters.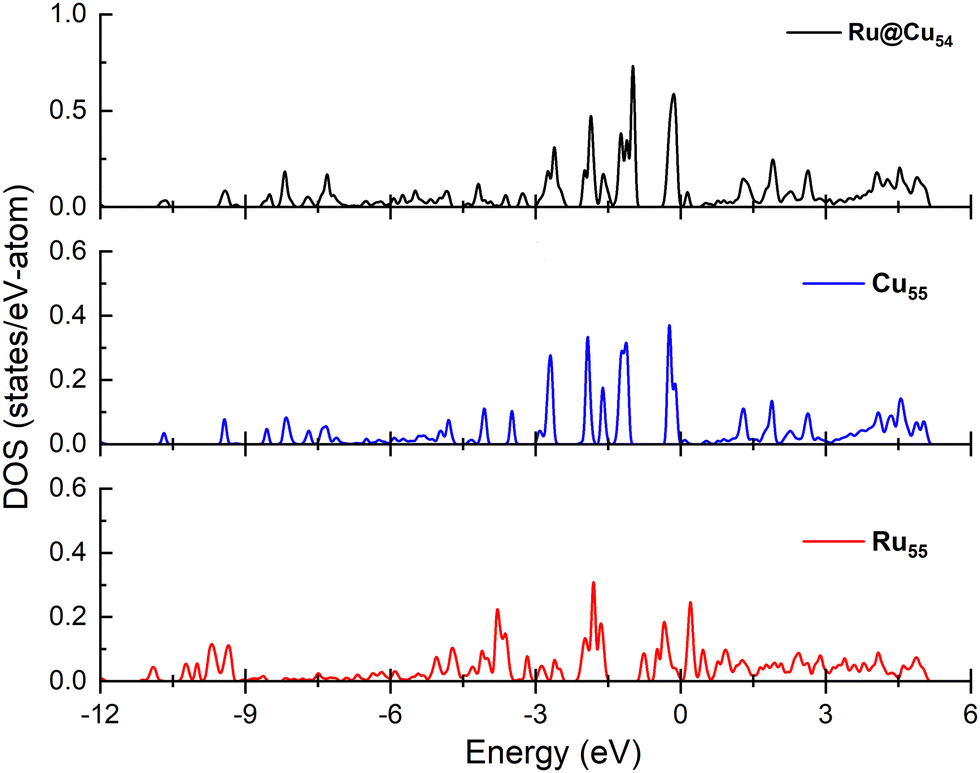 | ||
| Fig. 6 Density of states projected onto the d-band of the Ru@Cu54, Cu55 and Ru55 nanoclusters. | ||
Conclusion
A systematic study of ammonia adsorption and dehydrogenation on small Ru@Cu54 nanoclusters has been carried out using DFT calculations. The most stable adsorption structures for the NH3, NH2, NH, N, and H species were determined, indicating that NH3, NH2 and NH prefer to adsorb on the top site while N and H atoms preferentially adsorb on the fcc and bridge sites, respectively. In order to determine the catalytic activity of the Ru@Cu54 NC for NH3 dehydrogenation, the adsorption energy of the N intermediate was compared with the optimum value of the famous volcano curve (−5.81 eV), and it was found that the adsorption energy of N on the Ru@Cu54 NC fcc site (−5.42 eV) is very close to that value. The overall reaction energy for stepwise dehydrogenation on Ru@Cu54 is found to be lower than that of Cu55. Also, the energy required for the last step of the dehydrogenation of NH3 in the Cu55 NC is reduced abruptly when an apex Cu atom is replaced by Ru. The reduced energy requirement for NH3 dehydrogenation in the case of Ru@Cu54 compared to that of Cu55 can make it a better catalyst for this purpose. Also, Ru@Cu54 is found to be advantageous for catalyst regeneration by the removal of residual N atoms. Additionally, the d-band centre calculation of the three nanoclusters also confirms the calculated N atom adsorption energy trend.Data availability
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to ethical restrictions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to Dr P. K. Mohapatra, Associate Director, Radiochemistry and Isotope Group, Bhabha Atomic Research Centre (BARC) and Dr S. C. Parida, Head, Product Development Division, BARC for their interest and encouragement during progress of this work. The authors are also thankful to the members of the Computer Division, BARC for their kind cooperation during the work.References
- L. Schlapbach, Nature, 2009, 460, 809–811 CrossRef CAS PubMed.
- T. N. Veziroglu, Chem. Ind., 1999, 53, 383–393 Search PubMed.
- M. Momirlan and T. N. Veziroglu, Sustainable Energy Rev., 2002, 6(1–2), 141–179 CrossRef CAS.
- P. Parthasarathy and K. S. Narayanan, Renew. Energy, 2014, 66, 570–579 CrossRef CAS.
- R. Z. Sørensen, L. J. E. Nielsen, S. Jensen, O. Hansen, T. Johannessen, U. Quaade and C. H. Christensen, Catal. Commun., 2005, 6, 229–232 CrossRef.
- J. Zhang, H. Y. Xu, X. L. Q. J. Jin and W. Z. Li, Catal. Commun., 2006, 7, 148–152 CrossRef CAS.
- S. F. Yin, B. Q. Xu, X. P. Zhou and C. T. Au, Appl. Catal., A, 2004, 277, 1–9 CrossRef CAS.
- S. Maier, I. Stass, J. I. Cerda and J. Salmeron, J. Phys. Chem. C, 2012, 116(48), 25395–25400 CrossRef CAS.
- E. Salli, V. Hanninen and L. Halonen, J. Phys. Chem. C, 2010, 114, 4550–4556 CrossRef CAS.
- S. Stolbov and T. S. Rahman, J. Chem. Phys., 2005, 123, 204716 CrossRef PubMed.
- M. Grunze, P. A. Dowben and C. R. Brundle, Surf. Sci., 1983, 128, 311–324 CrossRef CAS.
- X. Duan, G. Qian, C. Fan, Y. Zhu, X. Zhou, D. Chen and W. Yuan, Surf. Sci., 2012, 606, 549–553 CrossRef CAS.
- X. Duan, J. Ji, G. Qian, C. Fan, Y. Zhu, X. Zhou, D. Chen and W. Yuan, J. Mol. Catal. A: Chem., 2012, 357, 81–86 CrossRef CAS.
- R. J. Lin, F. Y. Li and H. L. Chen, J. Phys. Chem. C, 2011, 115, 521–528 CrossRef CAS.
- J. Robinson and D. P. Woodruff, Surf. Sci., 2002, 498, 203–211 CrossRef CAS.
- B. Xing and G. C. Wang, Phys. Chem. Chem. Phys., 2014, 16, 2621–2629 RSC.
- J. A. Herron, S. Tonelli and M. Mavrikakis, Surf. Sci., 2012, 606, 1670–1679 CrossRef CAS.
- G. Novell-Leruth, A. Valcarcel, J. Perez-Ramirez and J. M. Ricart, J. Phys. Chem. C, 2007, 111, 860–868 CrossRef CAS.
- Z. Jiang, Q. Pan, M. Li, T. Yan and T. Fang, Appl. Surf. Sci., 2014, 292, 494–499 CrossRef CAS.
- L. Bartels, M. Wolf, T. Klamroth, P. Saalfrank, A. Kuhnle, G. Meyer and K. H. Rieder, Chem. Phys. Lett., 1999, 313, 544–552 CrossRef CAS.
- P. Baumgartel, R. Lindsay, T. Giessel, O. Schaff, A. M. Bradshaw and D. P. Woodruff, J. Phys. Chem. B, 2000, 104, 3044–3049 CrossRef CAS.
- Z. Jiang, P. Qin and T. Fang, Chem. Phys., 2014, 445, 59–67 CrossRef CAS.
- J. Robinson and D. P. Woodruff, Surf. Sci., 2002, 498, 203–211 CrossRef CAS.
- B. Xing and G.-C. Wang, Phys. Chem. Chem. Phys., 2014, 16, 2621–2629 RSC.
- Z. Jiang, P. Qin and T. Fang, Chem. Phys., 2014, 445, 59–67 CrossRef CAS.
- Z. Jiang, P. Qin and T. Fang, Appl. Surf. Sci., 2016, 371, 337–342 CrossRef CAS.
- S. Chen, X. Chen and H. Zhang, J. Mat. Sci., 2017, 52, 3162–3168 CrossRef CAS.
- X. Chen, J. Zhou, S. Chen and H. Zhang, J. Nanopart. Res., 2018, 20, 148–156 CrossRef.
- G. Lanzani and K. Laasonen, Int. J. Hydrogen Energy, 2010, 35, 6571–6577 CrossRef CAS.
- W. Eberhardt, Surf. Sci., 2002, 500, 242–270 CrossRef CAS.
- F. Baletto and R. Ferrando, Rev. Mod. Phys., 2005, 77, 371–423 CrossRef CAS.
- P. Cheng, Y. Yang, N. Ahmed, S. Zhang and S. Huang, Int. J. Hydrogen Energy, 2016, 41, 3844–3855 CrossRef CAS.
- P. Su, W. Pei, X. Wamg, Y. Ma, Q. Jiang, J. Liang, S. Zhao, J. Zhao, J. Liu and G. Q. Lu, Angew. Chem., 2021, 133(29), 16180–16186 CrossRef.
- S. Ajmal, H. T. D. Bui, V. Q. Bui, T. Yang, X. Shao, A. Kumar, S. G. Kim and H. Lee, Chem. Eng. J., 2022, 429, 132282 CrossRef CAS.
- M. J. Ungerer and N. H. de Leeuw, Nanomaterials, 2023, 13, 1118–1131 CrossRef CAS PubMed.
- B. Zhao, R. Zhang, Z. Huang and B. Wang, Appl. Catal., A, 2017, 546, 111–121 CrossRef CAS.
- Z. Lin, Z. Cai-Bei and Q. Yang, Chin. Phys., 2007, 16, 77–82 CrossRef CAS.
- R. Zhang, M. Peng, T. Duan and B. Wang, Appl. Surf. Sci., 2017, 407, 282–296 CrossRef CAS.
- D. H. Lim, J. H. Jo, D. Y. Shin, J. Wilcox, H. C. Ham and S. W. Nam, Nanoscale, 2014, 6, 5087–5092 RSC.
- L. Ma and J. Akola, Phys. Chem. Chem. Phys., 2019, 21, 11351–11358 RSC.
- S. M. Sintilo, A. B. Hernandez, A. A. P. Cid, W. I. Hernandez and M. S. Villanueva, ACS Omega, 2022, 7, 34401–34411 CrossRef PubMed.
- M. B. Gawande, A. Goswami, F. X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116(6), 3722–3811 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- W. Kohn and L. Sham, Phys. Rev. A, 1965, 140, 1133–1138 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- R. P. Feynman, Phys. Rev., 1939, 56, 340–343 CrossRef CAS.
- H. Hellman, Introduction to Quantum Chemistry; Deuticke, Leipzig, 1937.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- P. E. Blöchl and O. Jepsen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef PubMed.
- D. A. Hangsen, D. G. Vlachos and J. G. Chen, Nat. Chem., 2010, 2(6), 484–489 CrossRef PubMed.
- A. Logadottir, T. H. Rod, J. K. Nørskov, B. Hammer, S. Dahl and C. J. H. Jacobsen, J. Catal., 2001, 197, 229–231 CrossRef CAS.
- T. Bligaard, J. K. Nørskov, S. Dahl, J. Matthiesen, C. H. Christensen and J. Sehested, J. Catal., 2004, 224, 206–217 CrossRef CAS.
- B. Hammer and J. K. Norskov, Adv. Catal., 2000, 45, 71–129 CAS.
| This journal is © the Owner Societies 2024 |