
ReaxFF molecular dynamics of graphene oxide/NaCl aqueous solution interfaces†
Rokhsareh
Akbarzadeh
 a and
Milan
Předota
*b
a and
Milan
Předota
*b
aDepartment of Physics, Faculty of Science, University of South Bohemia, Branišovská 1760, 370 05 České Budějovice, Czech Republic
bDepartment of Physics, Faculty of Science, University of South Bohemia, Branišovská 1760, 370 05 České Budějovice, Czech Republic. E-mail: predota@prf.jcu.cz
First published on 4th December 2023
Abstract
In this work, the interaction of NaCl aqueous solution with graphene (G), graphene oxide (GO), and graphite oxide (GTO) is studied using the ReaxFF module of Amsterdam Modeling Suite (AMS) software. We consider four models using the NaCl aqueous solution, containing a graphene sheet (G), a single sheet of GO with epoxide and hydroxyl groups on its surface, 4 layers of GO to model GTO, and a bulk NaCl solution as a reference. The structural and dynamical properties of G, GO, and GTO were quantified by analyzing the functional groups, radial distribution functions, density profiles and diffusivities of water and ions. Due to the reactive force field, the systems underwent spontaneous modification of surface functional groups during the first 750 ps after which the structure stabilizes (the energy stabilizes in less than 400 ps). Pristine graphene in contact with the NaCl solution formed hydroxyl groups on the edges, i.e., converted to partially reduced graphene oxide. The epoxy groups (Oe) on the initial GO were rather unstable, leading to a reduction of their number, however, there was an increase in the number of hydroxyl groups (Oh), mainly at the edges. The interactions of NaCl with the carbon-based sheets are rather weak, including GO and GTO which are decorated with numerous functional groups. Diffusion coefficients of water agree with the available data, but discrepancies in Na+ and Cl− diffusivity compared to other references underscore the need for further development in the dynamic parameters of the reactive force field used. In essence, our research provides specific data previously unreported, laying a foundation for advancing water desalination system design. The study's novelty lies in its realistic approach to graphene/graphene oxide modification, comprehensive characterization, and the application of the reactive force field to explore the graphene oxide-NaCl aqueous interface, contributing to the development of a practical membrane system for water desalination.
Introduction
During the last decade, graphene (G) and graphene oxide (GO) have been attracting growing interest in the field of water purification due to their outstanding performances. Many researchers suggested the graphene-based material as a unique material in the field of membrane development that could be used in the reverse osmosis system (RO) and more recently in capacitive deionization (CDI).1–5 Graphene oxide is one of the graphene derivatives, and unlike graphene, GO exhibits good hydrophilicity, which is because of oxygen-containing functional groups such as hydroxyl, epoxy, and carboxyl groups in its plane and at the edges. There have been numerous studies on graphene or graphene oxide interfaces with water, yet an atomistic understanding of the water, ions, and GO interface, important for the development of GO for the water purification industry, is lacking.6–8On one hand, the synthesis of graphene is cumbersome, and, on the other hand, GO is a precursor to produce G. In addition, GO as a rising material among 2D materials has become the centre of attraction because of its simple and low-cost synthesis. Since graphene is costly and challenging to synthesize, attention has been given to its derivatives such as GO. Another advantage of GO is that it is dispersible in water and other solvents.9 Some of the unique properties of graphene can only be accessible if it is functionalized with organic groups such as hydroxyl, carboxyl, or amino. For instance, graphene family materials will acquire dispersibility and colloidal stability in aqueous solutions when their surfaces are functionalized. Moreover, some derivatives of graphene, such as graphene oxide (GO) and reduced graphene oxide (rGO), have abundant functional groups in their structures which help them interact with other molecules and disperse easily.10
GO is a monolayered graphene with a high oxygen content and it can be obtained from graphite oxide (GTO). Based on a report by Rowley-Neale and colleagues, GO has a C/O ratio of less than 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and closer to 2:1.11 However, Mouhat and his colleagues reported a ratio of 4:1 for C/O.12 GO is the oxygenated derivative of graphene covered with hydroxyl and epoxy groups on the basal plane as well as carboxyl groups at the edges.13–15 To be able to utilize GO as a water purification real system, a clear understanding of the effect of GO on water and salt diffusion is vital to find the permeation and transport of water and ion species. In this work, molecular dynamics (MD) simulations were conducted to reveal the interaction of wrinkled graphene and GO sheets with water and NaCl ions. The graphene and GO sheets exhibit a corrugated structure in contact with water, and this corrugation in the case of GO increases due to the functional groups.15 However, researchers already showed that crumbling could enhance the electrochemical behaviour of graphene, increasing the current density,16 which could be an advantage in some applications.
:1 and closer to 2:1.11 However, Mouhat and his colleagues reported a ratio of 4:1 for C/O.12 GO is the oxygenated derivative of graphene covered with hydroxyl and epoxy groups on the basal plane as well as carboxyl groups at the edges.13–15 To be able to utilize GO as a water purification real system, a clear understanding of the effect of GO on water and salt diffusion is vital to find the permeation and transport of water and ion species. In this work, molecular dynamics (MD) simulations were conducted to reveal the interaction of wrinkled graphene and GO sheets with water and NaCl ions. The graphene and GO sheets exhibit a corrugated structure in contact with water, and this corrugation in the case of GO increases due to the functional groups.15 However, researchers already showed that crumbling could enhance the electrochemical behaviour of graphene, increasing the current density,16 which could be an advantage in some applications.
This research provides information about the structural and dynamic properties of wrinkled G and GO in systems consisting of water molecules, Na+, and Cl− ions. The mean square displacement (MSD) of confined water, the radial distribution function (RDF) between atoms of the functional groups and atoms of water molecules and Na+ and Cl− ions, and G and GO were obtained along with the density profiles with respect to carbon atoms. This finding is valuable in solid–liquid interface design such as selective transport and membrane design and application.
Methodology and molecular dynamics simulation details
Construction of G and GO structures
In this study, G, GO, and GTO sheets were constructed using Amsterdam Modeling Suite (AMS) software. The rectangular hexagonal honeycomb structure of the graphene slab with the lattice vector (a × b) size of 29.5 × 23.5 Å was created in the ReaxFF environment available in AMS. To model GO, this slab was modified by surface hydroxyl and epoxy functional groups. The distribution of these groups resulted from the analysis of the experimental and simulation data of the GO structure reported by Mouhat and his colleagues.12 Among the known different types of GO structure models, the one proposed by Lerf and colleagues14 which is formed by hydroxyl and epoxide groups distributed on the basal plane of graphene17 is considered to be the best to describe the experimental results. Therefore, in this study, the hydroxyl and epoxy groups were selected to create GO. The ratio of O/C was selected as 1/4, while the number of hydroxyl groups was twice the number of epoxy groups attached on the basal plane of the graphene sheet as reported by many researchers.12,13,15 This arrangement of negatively charged oxygen layers could prevent a nucleophilic attack on carbon atoms, explaining the relative chemical inactivity of the epoxide groups in GO.14 Geometry optimization was conducted using a universal force field (UFF) prior to the MD simulation. The optimization resulted in wrinkling of GO due to the presence of functional groups attached to the carbons (see Fig. 1), which is in good agreement with the findings of researchers14 who reported that the carbon grid is nearly flat in graphene, but after the attachment of OH groups, it distorts, resulting in the wrinkling of the GO sheet. In graphene's structure hexagonal network, the C–C distance is 1.42 Å, while the mean values for the C–C bonded to hydroxyl and epoxy are 1.52 and 1.65 Å, respectively. Utilizing ReaxFF within the AMS suite, the atomic charges are allowed to fluctuate dynamically throughout the course of the simulations. Table 1 shows the Lennard-Jones (LJ) input parameters used in our simulations and the resulting average charges of atoms. The evolution of these charges over time is shown in Fig. S3 (ESI†). | ||
| Fig. 1 The hexagonal honeycomb structure of graphene (a), the structure of graphene oxide top view (b) and side view (c) with epoxy and hydroxyl functional groups randomly attached to the surface of the graphene layer. Carbon, oxygen, and hydrogen atoms are represented by grey, red, and white, respectively. | ||
| Atom type | ε [kJ mol−1] | σ [Å] | Average charge q [e] |
|---|---|---|---|
| C | 0.0105 | 3.851 | 0.012 |
| Ow | 0.060 | 3.500 | −0.720 |
| Oh | 0.060 | 3.500 | −0.545 |
| Oe | 0.060 | 3.500 | −0.440 |
| Oc | 0.060 | 3.500 | −0.409 |
| Hw | 0.044 | 2.886 | 0.366 |
| Na | 0.030 | 2.983 | 1.022 |
| Cl | 0.227 | 3.947 | −0.730 |
Construction of G, GO, and GTO systems
Four systems of H2O–NaCl (Bulk), H2O–NaCl with a single graphene layer (G), H2O–NaCl with a single GO layer (GO), and H2O–NaCl with four layers of GO to represent graphite-oxide (GTO) were built in the ReaxFF environment of AMS software, and their solid–liquid interface behaviour in the NaCl aqueous solution was investigated. Real graphite is composed of stacked graphene sheets with an interlayer spacing of 3.4 Å and similarly, graphite oxide is composed of stacked graphene oxide (GO) sheets but with a larger interlayer spacing which has been reported between 7.54 and 12 Å.18,19 In this study, the initial interlayer spacing of 10 Å for the GTO system was selected. The simulation box was designed with dimensions of 40 × 40 × 40 Å for bulk, G, and GO, while the simulation box for GTO with four layers of GO was set to 40 × 40 × 70 Å. These box sizes allowed at least 10.5 Å and 16.5 Å gaps between the periodic replicas of the G or GO sheets in the x and y dimensions, respectively. Numbers of water molecules and other atoms were selected based on the volume of the simulation box, as shown in Table 2. Randomly distributed water molecules were replaced by Na+ and Cl− ions to provide a NaCl concentration of 0.5 M. The snapshots of the initial simulation boxes are shown in Fig. 2.| System | Bulk | G | GO | GTO |
|---|---|---|---|---|
| Initial no. of C atoms and Oh/Oe/Oc/Od | 0 | 264, 0/0/0/0/0 | 264, 44/22/0/0 | 1056, 176/88/0/0 |
| No. of C, Oh/Oe/Oc/Od after stabilization | 0 | 264, 22/0/2/0 | 264, 59/6/2/1 | 1056, 233/47/4/2 |
| No. of H2O (initial/after stabilization) | 2100/2078 | 1900/1849 | 1850/1825 | 2700/2642 |
| No. of NaCl pairs | 30 | 30 | 30 | 44 |
| Box size (Å) | 40 × 40 × 40 | 40 × 40 × 40 | 40 × 40 × 40 | 40 × 40 × 70 |
| Average density (g cm−3) | 1.027 | 1.030 | 1.035 | 1.012 |
 | ||
| Fig. 2 Snapshots of initial simulation boxes for G (a), GO (b), and GTO (c). | ||
The TiOCHNCl.ff force field, incorporated into the ReaxFF library of Amsterdam Modeling Suite (AMS) and containing the force field parameters for all the elements within our system, was chosen for the present simulations. This force field was originally developed and trained by Kim et al.20 using several previously available force fields21–27 and its applicability and reliability have been affirmed by multiple studies.28–31 The Berendsen thermostat controlled the temperature at 300 K with a relaxation time of 100 fs. During the 1 ns equilibration run, the NPT ensemble set the pressure to 1 atm using the Berendsen barostat. The production run of 1 ns was carried out in the NVT ensemble. We employed a time step of 0.25 femtoseconds (fs) to maintain a balance between capturing accurate dynamics and ensuring computational feasibility. This choice of time step aligns with recommendations for ReaxFF simulations, which are typically run using a 0.25 fs time step for the purpose of ensuring energy conservation and charge accuracy. It is reported that ReaxFF simulations tend to be significantly slower by factors ranging from tens to hundreds, compared to simulations using traditional non-reactive force fields, as demonstrated in previous studies.32,33
Results and discussion
Structure of surfaces in contact with the aqueous solution
It has been reported that water can disrupt the GO structure and create unstable hydrogen bonds that destabilize the interlayer bonding.34 Therefore, it is worth studying the structure of GO exposed to water.Fig. 3 shows the snapshots of final configurations of G, GO, and GTO in solution after 1 ns production run. The orientation of the G sheet changes over time due to interactions with the aqueous solution, which also causes slight wrinkling. For GO and GTO, the wrinkling effect increases due to the presence of functional groups which could even cause it to self-fold or scroll over time,35 a phenomenon not observed in our relatively small structures. Wrinkling is a common phenomenon in 2D materials because of their low out-of-plane stiffness.36 The bond breaking and binding between functional groups and other species during interaction could be an additional cause of this behaviour. Protolytic reactions of water were identified along with the creation of bonds forming NaOH and Na(OH)2 (see Tables S2 and S3, ESI† for details). Table 2 and Fig. S1 (ESI†) show that the originally pure graphene sheet was converted to partially reduced GO as the carbonyl and hydroxyl groups (and one diol) formed at the edge of the graphene sheet during equilibration, but then its composition remained unchanged during the production run (attachment and detachment of only 2 OH groups happened afterward). The number of functional groups on G, GO, and GTO surfaces, given in Table 2, shows newly formed hydroxyl groups at the edges of G, GO, and GTO, and carbonyl and diol groups formed at the corners of the GO sheet (Fig. S2, ESI†). The number of epoxy groups on GO reduced to 6, which shows an instability of epoxy groups after interaction with water under ambient conditions in the current system. The total number of functional groups of GTO reached 280 and the number of hydroxyl groups increased to 233, while the epoxy groups’ population dropped to 47 after equilibration. The number of water molecules changed not only due to their adsorption and desorption leading to functional group formations and detachments but varied even after the stabilization of the graphene and graphene oxide structure due to their protolysis and reactions with ions, forming intermediates such as NaOH and H3O+ (see Tables S2 and S3, ESI†). The hydroxyl and epoxy groups on the GO surface facilitate both physical (physisorption) and chemical (chemisorption) interactions with water. These interactions are essential to consider when studying the behavior of GO in different environments and for applications where water adsorption and desorption play a significant role. Understanding the balance between these two types of interactions is crucial for tailoring the properties of graphene oxide for specific purposes.
 | ||
| Fig. 3 Snapshots of G (a), GO (b), and GTO (c) systems after 1 ns production run. | ||
Fig. 4 shows the evolution of the three interlayer distances between the four GO layers of GTO based on the z-distance of central carbon of each sheet from the other sheet (4 sheets and 3 interlayers). As also shown in Fig. 3c, all interlayer distances in GTO increased during the 1 ns equilibration period from the initial 10 Å to an average of 12.16 Å, and varied differently during the production run, ultimately growing to an average of 12.8 Å. This might be in part related to newly formed hydroxyl groups on the surface or edges of the GTO sheets and due to the hydration mechanism as reported.18,37 Determining the separation distance between two wrinkled graphene sheets poses a complex challenge and the representation in the provided figure is a partial estimate specifically focussed on the central carbon region rather than on the entire sheet.
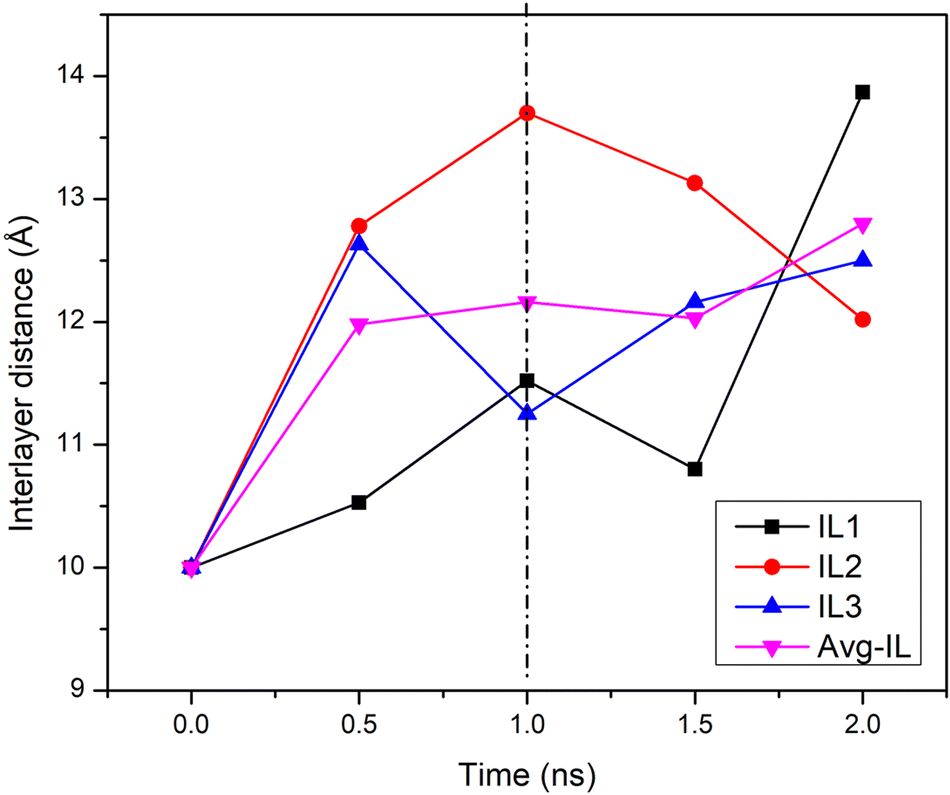 | ||
| Fig. 4 The evolution of the three interlayer (IL) distances between the central carbons of neighbouring graphene sheets in GTO during the 2 ns simulation. The vertical dashed line distinguishes the equilibration and production run. | ||
While many studies report that structure stabilization occurs approximately at the same time as the energy optimization, we found that energy optimization occurs at ∼400 ps, but the structure gets stabilized after ∼750 ps. These two distinct stages are detailed as follows: (i) Energy optimization: During the initial ∼400 ps, our observations revealed a systematic trend in the total energy of the system, influenced by solvation, adsorption, surface optimization, and reactions altering the nature and number of functional groups. Subsequently, beyond this period, the energy of the system exhibited fluctuations around a consistent value. (ii) Structural stabilization: Over an additional duration of up to 350 ps, we observed limited reactions, including 2 instances, where the identity of functional groups changed. Following this time frame, only occasional reactions involving surface functional groups were detected.
We acknowledge that with the application of ReaxFF, both forward and backward equilibrium reactions are expected even during the productive phase. These were observed numerously in the case of reactions of ions with water (see Table S3, ESI†) but not in the case of surface functional groups. The time evolution of the total surface charge of a graphene or graphene oxide sheet including functional groups at different times, given in Table S1 (ESI†), indicates that the resulting surface charges are about −7 e for G and −11.5 e for a single GO sheet.
Radial distribution functions
In this section, the radial distribution functions (RDFs) of either water molecules, Cl or Na ions with respect to carbon atoms of G, GO, or GTO are discussed and compared to those in a bulk system to have an insight into the local structure and the distribution of species around carbon surfaces.As shown in Fig. 5a, the Ow–Ow distribution has well-defined peaks at 2.8, 4.4, and 6.8 Å in agreement with experimental and simulation results reported elsewhere,38–40 confirming that the ReaxFF forcefield reproduces the water structure well.
 | ||
| Fig. 5 Radial distribution functions of the OW–OW (a) and Na–Cl pairs (b) in the four studied systems. | ||
The analysis of Na–Cl radial distribution functions (RDFs) compared to the bulk system reveals a reduction in the main peak at 4.8 Å in the presence of graphene (G), graphene oxide (GO), and least prominently in the case of the partially reduced graphene oxide (GTO) (Fig. 5b).
Note that the ReaxFF force field employed in our simulations does not predict contact ion pairs below 3 Å. This behaviour contrasts significantly with models incorporating full charges, which predict a notable presence of NaCl pairs within this range. However, ReaxFF aligns more closely with the performance of models utilizing scaled charges, predicting only a small amount of contact NaCl pairs. Additional details and a comprehensive comparison with other force fields can be found in the ESI,† Fig. S4.
Fig. 6 shows the RDFs of Na–O, Cl–O, and Cl–H in the four systems, distinguishing O and H atoms of water and surface groups to compare their interactions. In Fig. 6a, there are two main Na–Ow peaks at 2.35 Å (first hydration shell) and 4.7 Å (second hydration shell), respectively. The intensity of main peaks of Na–Ow in the case of GO and GTO is reduced, but a shoulder at a closer position of around 1.9 Å indicates intramolecular bonding of Na–Ow, leading to the formation of sodium hydroxide species, in agreement with the data reported by Kim and colleagues, who assigned this shoulder to the formation of NaOH.20 We have checked that the oxygen forming these bonds with sodium originates exclusively from water molecules and not from epoxy or surface hydroxyl groups. Oxygens in hydroxyls interact weakly, which is true for the peak at 4.7 Å and even more for the first peak, which is very low and shifted to further distances. This is not surprising considering that Oh is fully coordinated and the hydroxyl group is terminated by positive hydrogen. In contrast, epoxy oxygens present on GO and GTO interfaces preferably interact with Na+, with the peak at 2.6–2.7 Å, indicating the formation of Na–Oe contact pairs.
 | ||
| Fig. 6 The radial distribution function of Na–O (a), Cl–O (b), and Cl–H (c) in the four systems. The inserrts zoom the first peak region. | ||
The RDF of Cl–O shows in Fig. 6b a contact peak at 3.05 Å which is strong due to hydration of Cl− by water molecules, weak for oxygens of hydroxyls, Oh, and absent for epoxy oxygens, Oe. The second peak at 4.6 Å is prominent for water oxygen Ow, smaller than unity for Oh and very small for Oe. The interaction of Cl− with epoxy oxygens is weak at all distances.
The Cl–H RDFs show in Fig. 6c similar trends as those observed for Cl–Ow and Cl–Oh. The pronounced first peak centred at 2.05 Å stems from contact pairs with water hydrogens Hw and to a smaller extent with hydroxyl hydrogens Hh of GTO. In the simulation of graphene (see also Table S2, ESI†), a shoulder at distances less than 2 Å could indicate H–Cl bond formation. The second Cl–Hw peak is located at 3.5 Å. The positions of the Cl–Hw peaks agree with the data reported elsewhere for water interaction with sodium chloride.41
Overall, the RDFs indicate weak interaction of epoxy oxygens with ions compared to Ow and Oh.
The RDFs of various O–H pairs provide information on hydrogen bonding between water and surface groups (Fig. 7). The first peak at 1 Å corresponds to intramolecular O–H covalent bonds. Note that we observe this peak not just for water atoms, i.e., Ow–Hw, but also observe instances when a water molecule forms a bond with hydroxyl species, forming a limited number of Oh–Hw bonds in G and GO systems and significant GTO systems. The RDF peak positions of the O–H pair agree with the data reported in an earlier study for the water/GO system.42,43 The RDF data that we obtained for the bulk system at 0.99 and 1.75 Å agree with the reported data and the position of the first peak is even closer to the experimental result of 0.97 Å.18,43,44 The hydrogen bonds of hydroxyl hydrogen Hh to water oxygens Ow are stronger compared to those of hydroxyl or epoxy oxygens to water hydrogens Hw, which leads to a sharper second peak of Ow–Hh at a shorter distance of 1.75 Å in agreement with the reported data.38,45 Oppositely, the Oh–Hw and Oe–Hw peaks at 2.0–2.2 Å indicate weak hydrogen bonds.18,46 The bulk hydrogen bonds Ow–Hw are found between these two types. The third peak of RDFs around 3.2 Å for both Ow–Hh and Oh–Hw represents the intermolecular hydrogen-bonding network of water molecules.
 | ||
| Fig. 7 The RDF of oxygen–hydrogen pair of water molecules (w) and hydroxyl groups (h) in G and GO systems. | ||
Density profiles
To characterize the proximity of species to non-planar carbon-based sheets, we determined using our code for each O, H, Na+, or Cl− atom its closest distance d from any carbon atom to plot the density profiles of atoms as a function of distance d from the carbon sheet. These density profiles also include contributions from atoms interacting with the edges of carbon sheets (if the edge C atom is the closest C atom), but Fig. 3 shows that most interactions occur above/below the surfaces of the sheets. These density profiles are a much better measure of the interaction of species with the carbon sheets than RDFs with respect to C atoms, which would lead to broad distributions of distances starting from the closest approach due to the large number of C atoms. All the density profiles decay to zero around 20 Å because in our simulation boxes basically all atoms are within 20 Å of some carbon atoms.Fig. 8 presents the density profiles of Na, Cl, O, and H atoms at the interface with G, GO, and GTO. As discussed above, even though the G surface has been prepared as pristine graphene without any hydroxyl or epoxy groups, the first oxygen peak in Fig. 8a at about 1.5 Å indicates that some water molecules dissociated during the simulation. This dissociation resulted in a covalently bonded oxygen with a carbon atom at the G surface, leading to the spontaneous formation of hydroxyl and carbonyl groups while no epoxy groups were formed. In the case of GO and GTO, oxygens of hydroxyl as well as epoxy groups (giving rise to the shoulder as close as 1.2 Å) form the first peak. The second peak at 3 Å corresponding to the interaction of carbon atoms with oxygens of water molecules reveals that the presence of 4 sheets enhances the chances of a water molecule being closer to GO compared to G due to the presence of functional groups. However, it is significantly higher for GTO because the presence of 4 sheets enhances the chances of a water molecule to be closer to one of the sheets. The latter fact also explains the decreasing trend of GTO density profiles at larger distances. The hydrogens of hydroxyls are found at 2.0–2.2 Å, followed by hydrogens from water molecules. Sodium approaches all surfaces closer and adsorbs in bigger amounts than chloride ions. Its peak is at 4–4.2 Å from the closest carbons, and even closer in the case of GO. Up to about 8 Å from the surface, there is a surplus of Na+ to Cl−, while Cl− ions are pushed farther from the carbon surfaces, found in excess in the 10–18 Å distances from carbon atoms. We ascribe this polarization of the interface to stronger interactions of Na+ with surface Oh or Oe oxygens and/or water molecules forming the layer at 3 Å compared to interactions of Cl− with surface or water hydrogens. The role of surface oxygens in this charge displacement is supported by the fact that the difference in Na+ and Cl− densities is smallest for the G surface and largest for the GO surface. In the case of GTO, the adsorption of Na+ is strongest, but due to geometric reasons, it quickly decays at distances larger than 5 Å, i.e., about half the distance between GO sheets.
 | ||
| Fig. 8 Density profiles of atomic species as a function of the shortest distance from any C atom of the G (a), GO (b) and GTO sheets (c). The peak's height of O distribution for GTO is 14. | ||
Diffusion of water and ions
To characterize the effect of carbon-based sheets on the dynamics of aqueous solutions, we calculated the diffusivity of water and ions from the slope of the time evolution of the mean square displacement (MSD), using the equations:| MSD(t) = 〈[ri(t) − ri(0)]2〉 | (1) |
 | (2) |
The computed water diffusion coefficient in the system composed of pure water, DW = 2.29 × 10−9 m2 s−1, agrees with the experimental value of DW = 2.299 × 10−9 m2 s−1 reported by others.47,48 The computed water diffusion reduces to DW = 2.03 × 10−9 m2 s−1 in the bulk system composed of only water and 0.5 NaCl. The diffusivity of water remains nearly the same in the G system but reduces significantly in the GO and GTO systems, which means that the mobility of water molecules and salt ions decreases due to the interactions with the hydroxyl and epoxy functional groups, including the formation of hydrogen bonds. The diffusion is lowest for GTO, where we have 4 layers of GO, and a bigger part of the box volume is located near the surfaces as opposed to the bulk solution. The effect of the temporary trapping within GTO sheets also cannot be neglected.
Table 3 compares the results obtained in this study with the selected experimental and simulation results. The diffusivity of water agrees well with the experimental and simulated data. However, even considering the decrease of diffusivity with salt concentration, the diffusivity of Cl− obtained using ReaxFF strongly disagrees with the experimental value. ReaxFF predicts the diffusivity of Cl− to be smaller compared to that of Na+, while the opposite is true in reality. This discrepancy is observed already for bulk 0.5 M aqueous solution (Water + NaCl system shown in Table 3) and propagates to result in systems containing surfaces. We link the low diffusivity of Cl− to its very structured first shell, as detailed in the ESI.†
| System | Density [g cm−3] | Simulation/experiment | Salt concentration [m] | D W [10−9 m2 s−1] | D Na [10−9 m2 s−1] | D Cl [10−9 m2 s−1] | Ref. |
|---|---|---|---|---|---|---|---|
| Water + NaCl | 1.027 | Simulation | 0.5 | 2.03 | 0.85 | 0.50 | This work |
| Water + NaCl + G | 1.030 | Simulation | 0.5 | 2.05 | 0.83 | 0.51 | This work |
| Water + NaCl + GO | 1.035 | Simulation | 0.5 | 1.80 | 0.63 | 0.45 | This work |
| Water + NaCl + GTO | 1.012 | Simulation | 0.5 | 1.60 | 0.62 | 0.46 | This work |
| Water + NaCl | 1.018 | Simulation | 0.5 | 1.85 | — | — | 49 |
| Water + GO | — | Experiment | 0 | 0.96–2.14 | — | — | 18 |
| Water + NaCl | 1.017 | Simulation | 0.5 | 2.24 | 0.70 | 1.33 | 44 |
| Water + GO | — | Simulation | 0 | 1.98 | — | — | 50 |
| Water | — | Experiment | 0 | 2.3 | — | — | 51 |
| Water + Na | 0.997 | Simulation | — | 2.24 | — | — | 48 |
| Water | 1.06 | Experiment | 0 | 2.30 | — | — | 47 |
| Water | 0.997 | Experiment | 0 | 2.30 | 1.33 | 2.03 | 52 |
Conclusions
This study presents a pioneering investigation of graphene, graphene oxide, and graphite oxide interfaces with NaCl aqueous solution employing the reactive force field using the ReaxFF module of Amsterdam Modeling Suite. We found that during the 1 ns equilibration simulation, pristine graphene was modified spontaneously by the formation of a limited number of functional groups at the edges of the graphene sheet and in fact, it converted to a partially reduced graphene oxide. Changes to the initial setup of functional groups at GO and GTO sheets were also observed, particularly at their edges, but in all cases, the structures stabilized in less than 1 ns, allowing detailed analysis of surface species and their interactions during the production runs of 1 ns.Each system was analysed using radial distribution functions and density profiles and the diffusivities were calculated from the mean square displacement. The results were compared with the other reported experimental and simulation results. Except for diffusivity, there is good agreement of the obtained simulation results with other experimental and computational results which is encouraging for further MD simulation using ReaxFF to predict the structural and dynamic properties of graphene-based systems and their applications. Specific interactions of ions and water molecules with surface oxygens and hydrogens were analysed because they provide insights into the mechanisms of interactions and chemical reactions occurring at surfaces and interfaces. The epoxy oxygen Oe was found to interact weakly, while the hydroxyl hydrogens Hh interacted strongly compared to water hydrogens Hw. We have communicated the observed discrepancy in Cl− diffusivity with the developers of Amsterdam Modeling Suite, who acknowledged that the current reactive force field may not have been explicitly trained for this property. This underscores the need for further development and optimization of the dynamic properties of ions within the reactive force field to enhance their predictive capabilities and application range. Despite this discrepancy, the ReaxFF TiOCHNCl.ff force field remains a valuable tool for capturing interactions within complex systems.
The reported findings contribute to the evolving understanding of graphene-based materials and should be considered in the development of GO membranes for water remediation and desalination.
Abbreviations
| G | Graphene |
| GO | Graphene oxide |
| GTO | Graphite oxide |
| Ow | Oxygen of water |
| Oh | Oxygen of hydroxyl |
| Oe | Oxygen of epoxy |
| Oc | Oxygen of carbonyl |
| Od | Oxygen of diol |
Author contributions
RA has carried out data curation (all the simulations), investigation, and visualization; MP carried out funding acquisition, project administration, resources, software (code providing the density profiles), and supervision. Both authors were involved in the conceptualization, formal analysis, methodology, validation, and manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Czech Science Foundation project 21-27338S.References
- J. Han, T. Yan, J. Shen, L. Shi, J. Zhang and D. Zhang, Capacitive Deionization of Saline Water by Using MoS2-Graphene Hybrid Electrodes with High Volumetric Adsorption Capacity, Environ. Sci. Technol., 2019, 53, 12668–12676 CrossRef CAS PubMed.
- J. Martinez, M. Colán, R. Catillón, J. Huamán, R. Paria, L. Sánchez and J. M. Rodríguez, Desalination Using the Capacitive Deionization Technology with Graphite/AC Electrodes: Effect of the Flow Rate and Electrode Thickness, Membranes, 2022, 12, 717 CrossRef CAS PubMed.
- M. Shahbabaei and D. Kim, Exploring fast water permeation through aquaporin-mimicking membranes, Phys. Chem. Chem. Phys., 2020, 22, 1333–1348 RSC.
- M. Shahbabaei and D. Kim, Molecular Dynamics Simulation of Water Transport Mechanisms through Nanoporous Boron Nitride and Graphene Multilayers, J. Phys. Chem. B, 2017, 121, 4137–4144 CrossRef CAS.
- C. Shao, Y. Zhao and L. Qu, ChemNanoMat, 2020, 6, 1028–1048 CrossRef CAS.
- D. Konatham, J. Yu, T. A. Ho and A. Striolo, Simulation insights for graphene-based water desalination membranes, Langmuir, 2013, 29, 11884–11897 CrossRef CAS PubMed.
- C. D. Williams and M. Lísal, Coarse grained models of graphene and graphene oxide for use in aqueous solution, 2D Mater., 2020, 7, 025025 CrossRef CAS.
- L. Zhu, X. Guo, Y. Chen, Z. Chen, Y. Lan, Y. Hong and W. Lan, Graphene Oxide Composite Membranes for Water Purification, ACS Appl. Nano Mater., 2022, 5, 3643–3653 CrossRef CAS.
- Y. Liu, in IOP Conference Series: Earth and Environmental Science, Institute of Physics Publishing, 2017, vol. 94.
- S. S. M. Sastry, S. Panjikar and R. S. Raman, Nanotechnol., Sci. Appl., 2021, 14, 197–220 CrossRef PubMed.
- S. J. Rowley-Neale, E. P. Randviir, A. S. Abo Dena and C. E. Banks, Appl. Mater. Today, 2018, 10, 218–226 CrossRef.
- F. Mouhat, F. X. Coudert and M. L. Bocquet, Structure and chemistry of graphene oxide in liquid water from first principles, Nat. Commun., 2020, 11, 1566 CrossRef CAS.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392–2415 CrossRef CAS.
- A. Lerf, H. He, M. Forster and J. Klinowski, Structure of Graphite Oxide Revisited, 1998 Search PubMed.
- X. Shen, X. Lin, N. Yousefi, J. Jia and J. K. Kim, Wrinkling in graphene sheets and graphene oxide papers, Carbon, 2014, 66, 84–92 CrossRef CAS.
- D. Dong, W. Zhang, A. Barnett, J. Lu, A. C. T. van Duin, V. Molinero and D. Bedrov, Multiscale modeling of structure, transport and reactivity in alkaline fuel cell membranes: Combined coarse-grained, atomistic and reactive molecular dynamics simulations, Polymers , 2018, 10, 1289 CrossRef PubMed.
- A. F. Fonseca, H. Zhang and K. Cho, Formation energy of graphene oxide structures: A molecular dynamics study on distortion and thermal effects, Carbon, 2015, 84, 365–374 CrossRef CAS.
- L. Liu, R. Zhang, Y. Liu, W. Tan and G. Zhu, Insight into hydrogen bonds and characterization of interlayer spacing of hydrated graphene oxide, J. Mol. Model., 2018, 24, 137 CrossRef PubMed.
- S. N. Tripathi, G. S. S. Rao, A. B. Mathur and R. Jasra, RSC Adv., 2017, 7, 23615–23632 RSC.
- S. Y. Kim, A. C. T. Van Duin and J. D. Kubicki, Molecular dynamics simulations of the interactions between TiO2 nanoparticles and water with Na+ and Cl−, methanol, and formic acid using a reactive force field, J. Mater. Res., 2013, 28, 513–520 CrossRef CAS.
- S. Y. Kim, N. Kumar, P. Persson, J. Sofo, A. C. T. Van Duin and J. D. Kubicki, Development of a ReaxFF reactive force field for titanium dioxide/water systems, Langmuir, 2013, 29, 7838–7846 CrossRef CAS PubMed.
- O. Rahaman, A. C. T. Van Duin, W. A. Goddard and D. J. Doren, Development of a ReaxFF reactive force field for glycine and application to solvent effect and tautomerization, J. Phys. Chem. B, 2011, 115, 249–261 CrossRef CAS PubMed.
- O. Rahaman, A. C. T. Van Duin, V. S. Bryantsev, J. E. Mueller, S. D. Solares, W. A. Goddard and D. J. Doren, Development of a ReaxFF reactive force field for aqueous chloride and copper chloride, J. Phys. Chem. A, 2010, 114, 3556–3568 CrossRef CAS PubMed.
- S. Monti, A. C. T. Van Duin, S. Y. Kim and V. Barone, Exploration of the conformational and reactive dynamics of glycine and diglycine on TiO2: Computational investigations in the gas phase and in solution, J. Phys. Chem. C, 2012, 116, 5141–5150 CrossRef CAS.
- H. Manzano, F. J. Ulm, A. van Duin, R. Pellenq, F. Marinelli and S. Moeni, Water polarization and dissociation in confined nanopores: Mechanism, dipole distribution, and impact on the substrate properties, J. Am. Chem. Soc., 2012, 134, 2208–2215 CrossRef CAS PubMed.
- H. Manzano, R. J. M. Pellenq, F. J. Ulm, M. J. Buehler and A. C. T. Van Duin, Hydration of calcium oxide surface predicted by reactive force field molecular dynamics, Langmuir, 2012, 28, 4187–4197 CrossRef CAS.
- K. Chenoweth, A. C. T. Van Duin and W. A. Goddard, ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation, J. Phys. Chem. A, 2008, 112, 1040–1053 CrossRef CAS PubMed.
- S. Huygh, A. Bogaerts, A. C. T. Van Duin and E. C. Neyts, Development of a ReaxFF reactive force field for intrinsic point defects in titanium dioxide, Comput. Mater. Sci., 2014, 95, 579–591 CrossRef CAS.
- A. Włodarczyk, M. Uchroński, A. Podsiadły-Paszkowska, J. Irek and B. M. Szyja, Mixing ReaxFF parameters for transition metal oxides using force-matching method, J. Mol. Model., 2022, 28, 8 CrossRef.
- L. Huang, K. Gubbins, L. Li, X. Lu and K. E. Gubbins, Water on Titanium Dioxide Surface: A Revisit by Reactive Molecular Dynamics Simulations, Langmuir, 2014, 30, 14832–14840 CrossRef CAS.
- S. Y. Kim, Development of ReaxFF reactive force field for titanium dioxide/water systems and its application to etching, nanoparticles with organic solvents and ion adsorptions on nanocrystalline surfaces, PhD thesis, The Pennsylvania State University, 2013.
- I. Leven and T. Head-Gordon, Inertial Extended-Lagrangian Scheme for Solving Charge Equilibration Models, Phys. Chem. Chem. Phys., 2019, 21, 18652–18659 RSC.
- A. Rahnamoun, C. Kaymak, M. Manathunga, A. W. Götz, A. C. T. Van Duin, K. M. Merz and H. M. Aktulga, ReaxFF/AMBER for Reactive Molecular Dynamics Simulations, J. Chem. Theory Comput. , 2020, 16, 7645–7654 CrossRef CAS PubMed.
- L. E. Paniagua-Guerra, M. Terrones and B. Ramos-Alvarado, Effects of Moisture and Synthesis-Derived Contaminants on the Mechanical Properties of Graphene Oxide: A Molecular Dynamics Investigation, ACS Appl. Mater. Interfaces, 2022, 14, 54924–54935 CrossRef CAS PubMed.
- S. Deng and V. Berry, Materials Today, 2016, 19, 197–212 CrossRef CAS.
- X. Zeng, B. Bin Zhu, W. Qiu, W. L. Li, X. H. Zheng and B. Xu, Xinxing Tan Cailiao/New Carbon Mater., 2022, 37, 290–302 CrossRef CAS.
- B. Rezania, N. Severin, A. V. Talyzin and J. P. Rabe, Hydration of bilayered graphene oxide, Nano Lett., 2014, 14, 3993–3998 CrossRef CAS.
- A. K. Soper, The radial distribution functions of water and ice from 220 to 673 K and at pressures up to 400 MPa, 2000.
- J. Yu, Q. Zheng, D. Hou, J. Zhang, S. Li, Z. Jin, P. Wang, B. Yin and X. Wang, Insights on the capillary transport mechanism in the sustainable cement hydrate impregnated with graphene oxide and epoxy composite, Composites, Part B, 2019, 173, 106907 CrossRef CAS.
- A. C. T. Van Duin and S. R. Larter, Molecular dynamics investigation into the adsorption of organic compounds on kaolinite surfaces, 2001.
- H. Shen, T. Hao, J. Wen, R. R. Tan and F. S. Zhang, Properties of pure water and sodium chloride solutions at high temperatures and pressures: A simulation study, Mol. Simul., 2015, 41, 1488–1494 CrossRef CAS.
- E. K. Goharshadi, G. Akhlamadi and S. J. Mahdizadeh, Investigation of graphene oxide nanosheets dispersion in water based on solubility parameters: a molecular dynamics simulation study, RSC Adv., 2015, 5, 106421–106430 RSC.
- A. Nicolaï, P. Zhu, B. G. Sumpter and V. Meunier, Molecular dynamics simulations of graphene oxide frameworks, J. Chem. Theory Comput., 2013, 9, 4890–4900 CrossRef.
- A. Ghaffari and A. Rahbar-Kelishami, MD simulation and evaluation of the self-diffusion coefficients in aqueous NaCl solutions at different temperatures and concentrations, J. Mol. Liq., 2013, 187, 238–245 CrossRef CAS.
- M. Zokaie and M. Foroutan, Comparative study on confinement effects of graphene and graphene oxide on structure and dynamics of water, RSC Adv., 2015, 5, 39330–39341 RSC.
- L. Liu, J. Zhang, J. Zhao and F. Liu, Mechanical properties of graphene oxides, Nanoscale, 2012, 4, 5910–5916 RSC.
- K. Krynicki, C. D. Green and D. W. Sawyer, Pressure and temperature dependence of self-diffusion in water, Faraday Discuss. Chem. Soc., 1978, 66, 199–208 RSC.
- P. Cicu, P. Demontis, S. Spanu, G. B. Suffritti and A. Tilocca, Electric-field-dependent empirical potentials for molecules and crystals: A first application to flexible water molecule adsorbed in zeolites, J. Chem. Phys., 2000, 112, 8267–8278 CrossRef CAS.
- S. Bouazizi and S. Nasr, Effect of solvent composition on the structural and dynamical properties of sodium chloride solutions in water-methanol mixtures, J. Mol. Liq., 2016, 221, 842–850 CrossRef CAS.
- M. Meshhal and O. Kühn, Diffusion of Water Confined between Graphene Oxide Layers: Implications for Membrane Filtration, ACS Appl. Nano Mater. , 2022, 5, 11119–11128 CrossRef CAS.
- R. Mills, Self-Diffusion in Normal and Heavy Water 685 Self-Diffusion in Normal and Heavy Water in the Range 1-45°, 1972.
- D. R. Lide and F. H. P. R. Lide, CRC Handbook of Chemistry and Physics, CRC, Boca Raton, FL, 89th edn, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Views of structures, relative abundances of species, and analysis of reactions. See DOI: https://doi.org/10.1039/d3cp04735k |
| This journal is © the Owner Societies 2024 |