
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical bonding effects in Sc compounds studied using X-ray absorption and X-ray photoelectron spectroscopies†
Anna
Zimina
 *ab,
Aline
Léon
c and
Ralph
Steininger
d
*ab,
Aline
Léon
c and
Ralph
Steininger
d
aInstitute of Catalysis Research and Technology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: anna.zimina@kit.edu
bInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology, Engesserstrasse 18-20, 76128 Karlsruhe, Germany
cEuropean Institute for Energy Research, Emmy Noether Strasse 11, 76131 Karlsruhe, Germany
dInstitute for Photon Science, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 4th January 2024
Abstract
Advances on understanding the nature of the chemical bonding and electron correlation effects during the X-ray absorption process in ionic–covalent metal complexes has been achieved for most of the transition elements, except for scandium, due to the lack of a systematic series of spectroscopic reference spectra and the shortage of standard crystallographic data on scandium compounds. To close the gap, the chemical bonding effects in eight Sc compounds are studied using X-ray absorption spectroscopy (XAS) at Sc K and L2,3 absorption edges and X-ray photoelectron spectroscopy (XPS). Indeed, the fine structure of the XAS Sc K edge reflects the chemical sp3-like bond formed between scandium and the ligand while the L2,3 edge and the pre-edge features of the K-edge provide a direct insight into the crystal field parameters at the Sc site in the coordination compound. The XPS data provide the information on binding energies of the core electrons involved in the electron transitions caused by the absorption of high energy X-rays. XAS and XPS complement each other by accessing the information on Sc structure on bulk and the surface. Herein, comprehensive information on the electronic structure of well-known crystalline materials based on Sc is given with spectroscopic fingerprints X-ray data. This will help to predict the formation of chemical bonds in the unknown components via the systematic evaluation of the available spectroscopic fingerprints.
1 Introduction
Scandium is the lightest element in the early transition series and often shows extraordinary behavior due to the presence of empty 3d states in a trivalent state in ionic compounds. The chemical properties of scandium differ significantly from those of other transition elements1 and this unique chemistry is currently exploited in high performance alloys and solid oxide fuel cells.2–4 This results in a growing demand of an exact knowledge of the chemical and structural properties of this element that is currently lacking. Therefore, there is a need for a deep understanding of the coordination chemistry of Sc.The electronic configuration in the ground state of scandium is [Ar]3d14s2 while in a simple ionic compound in the 3+ valence state it has nominally no 3d electrons in the initial state. As a consequence, one can expect that the role of d–d electron correlations is negligible for Sc compounds and the spectral changes along the series of different ligands will be mainly attributed to the changes related to the chemical bonding between the scandium and ligand atoms as well as the Sc–ligand coordination and distances. According to the Pauling rules,5 ionic crystals that contain transition metals (TM) are considered as a set of linked polyhedra. The electronic structure of such ionic crystals is in a first approximation defined by the electronic structure inside the polyhedron where anions build a coordinated complex around the cation. The bonding in TM complexes has a different character compared to ionic compounds like sodium chloride or covalent compounds like methane.5,6 The properties of the complex are strongly dependent on the type of the ligand and the coordination geometry.7 Indeed, crystal field theory considers the behavior of electrons at 3d orbitals in a negatively charged anionic environment which is typically simplified as an array of negative point charges; d electrons try to avoid each other and are repelled by the crystal field (CF). Depending on the symmetry of the CF, the d orbitals energetically split into groups according to their angular symmetry and occupation and the total splitting energy of orbitals for most TM decreases drastically with the increase of the metal–ligand inter-nuclear distance.8 The largest crystal field splitting energy is provided by ligands with a high negative charge and a small radius. Ligands in general can be ordered according to the magnitude of the CF into the so called “spectrochemical series”6 and the correlation between the spectroscopic features and the nature of the ligand was for example studied in cobalt complexes with octahedral arrangements.9 Nevertheless, due to the simplification of ligands as point charges, the experimentally observed fact that the negatively charged ligands induce less splitting of d orbitals compared to the neutral ligands cannot be clarified using solely CF theory. To understand the ligand influence on the electronic structure of TM compounds the bonding in the complex via π and σ molecular orbital (MO) creation should be taken into account. In general, the CF splitting parameter should be considered as a parameter of the whole system including the metal configuration, the ligand type and the character of bonding between them. In particular, the hybridization of Sc 3p and ligand p atomic orbitals as well as an increase of the covalent character of the Sc–ligand bond must be considered as the charge redistribution in this case might influence the d–d and 3d-CF interactions. In this indirect way, the spectroscopic characteristics of elements involving 3d states might be considered as reflecting the character of the chemical bond to the ligand.
X-ray absorption spectroscopy (XAS) is a powerful tool to probe the site-specific distribution of the electronic states in a conduction band.10–13 XAS can provide structural details on element coordination sites under almost any experimental conditions, where other techniques, like X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) or transmission electron microscopy (TEM) for instance, reach their limits. The structural information like coordination numbers, interatomic distances and the type of neighbors can be obtained through analysis of the extended X-ray absorption fine structure (EXAFS) region even for materials with limited long-range order.14 Less straightforward is the analysis of the near edge region of the XAS spectrum (XANES) as the observed transitions obey dipole selection rules and the probed site-specific symmetry-depended states may or may not be involved in the formation of the chemical bond. In the case of scandium, the K edge spectra reflect the 1s → 4p transition which contain little information on 3d states. The 1s → 3d transitions are dipole forbidden and are seen only as a weak pre-edge structure. The L2,3 edge of scandium represents the dipole allowed 2p → 3d transitions and the local chemical surrounding of Sc could be directly evaluated for L-edge. The chemically useful information like parameters of the chemical bond between scandium and the ligand formed by valence electrons is also present in the L2,3 XAS spectra. However, the detailed evaluation of those parameters from the spectroscopic data of ionic compounds with strong electron correlation and with significant interaction between the corresponding core hole and the 3d electron in the excited state is challenging. The screening effects by shared valence electrons may play a significant role in complexes with bonds with a high covalent contribution and should be more pronounced at the K edge compared to the L edge. The strength of the screening effects related to the hybridization effects might be influenced by the state of the core electron in different compounds. Here, XPS can provide comprehensive information on the binding energy of the 2p electron of Sc in the initial state.
In the past, efforts were made to accurately measure and to theoretically explain the atomic-like shape of the XAS L2,3 absorption edge15–22 and the fine structure of the XAS K absorption edge23,24 for several TM compounds. The 2p excitation is usually considered as the intra-atomic intermediate states and are mainly described in various ionic multiplet approaches. The TM 3d electrons participate then in covalent bonding while the hybridization between the TM 3d electrons and the ligand valence electrons leads to the delocalization of the 3d states lowering the dominant role of the 3d electron correlations. The direct evaluation is only possible based on comparison with the spectra of well-defined reference materials, measured in reliable matter.
XAS using high-brilliance synchrotron radiation sources is a very efficient tool for studying the subtle interplay between the hybridization and 3d electrons correlations in the electronic structure. This approach has been applied previously to study several TM compounds16,25 but has not been achieved so far for Sc-based compounds. To fill this gap, the XPS Sc 2p and O 1s and XAS K and L2,3 spectra of scandium metal and seven Sc compounds are measured in this study and discussed regarding the hybridization effects in the chemical bond. In the Sc-based complexes analyzed herein, the Sc ion has an octahedral coordination (Oh) except for the Sc metal that has a close packed hexagonal structure. As ligands in Oh coordination the F−, Cl−, Br−, and O2− single ions and NO3−, CO32−, and SO42− molecules were probed.
2 Experiment
Sc K and L2,3 XAS spectra were acquired for Sc metal, Sc2O3, ScF3, ScBr3, ScCl3, Sc(NO3)3, Sc2(CO3)3 and Sc2(SO4)3 compounds. For the measurements at Sc L2,3 absorption edge ScF3 and Sc metal were thermally evaporated in situ in ultrahigh vacuum onto a Cu substrate while for the measurements at the Sc K absorption edge, Sc metal was sputtered directly on a Si wafer. All the other samples were commercial powder, used as received: oxide (Sc2O3, 99.998% trace rare earth metal basis, Alfa Aeasar), fluoride (ScF3, 99.9% trace rare earth metal basis, MaTeck), bromide (ScBr3, 99.99% trace rare earth metal basis, Sigma-Aldrich), chloride (ScCl3, 99.9% Aldrich), nitrate (Sc(NO3)3·xH2O, 99.9% trace rare earth metal basis, MaTeck), carbonate (Sc2(CO3)3·xH2O, 99.999% trace rare earth metal basis, MaTeck) and sulfate (Sc2(SO4)·3.5H2O, 99.99% trace rare earth metal basis, MaTeck).The XAS Sc L2,3 spectra were acquired at the U49/2-PGM1 beamline26 and at the Russian-German beamline (RGBL)27 at the synchrotron facility BESSY II (Germany) in the total electron yield (TEY), in total fluorescence yield (FY) and in transmission modes. The photon energy resolution was estimated to be 60 meV.
The comparison of XAS Sc L2,3 spectra acquired on Sc metal films (see Fig. S1 in the ESI†) indicates that the chemical state of Sc in films is very sensitive not only to vacuum conditions under which films are prepared but also to the photon beam exposure time. Indeed, spectra measured 10 min after the preparation in both TEY and FY modes are similar to the one obtained by the XAS in transmission detection mode under ultrahigh vacuum conditions (which is the representative spectrum of metallic Sc) while after 50 min exposure to the photon beam at a pressure of 2 × 10−8 mbar the sample is clearly oxidized, highlighting the necessity to minimize the beam effect during the acquisition.
The XAS Sc K spectra were recorded at the SULx beamline at the KIT Light Source28 using a Si(111) double crystal monochromator. The beam was focused with the Kirkpatrick–Baez mirror system to the sample position to a size of approximately 150 (v) × 250 (h) μm2. The XAS spectra were recorded in the transmission mode on pellets using the ionization chambers (OKEN, Japan) and in fluorescence mode using a 7-Element Si(Li) fluorescence detector (Gresham, now RaySpec Ltd) on ScBr3 powder and Sc metal. The photon energy was calibrated to the tabulated value of 4492 eV using the K-edge data of the freshly evaporated metallic Sc. Data were processed using the IFEFFIT package29 for background correction and normalization.
The XPS data were recorded with a laboratory spectrometer30 at Mg Kα1,2 (1253.6 eV) excitation energy, using the flood electron gun for neutralization of the surface charge. The energy calibration was made to the energy of the surface carbon (C 1s at 284.8 eV) and the data processing and analysis was performed using the CasaXPS software.31
XRD measurements were performed using a PANalytical X'pert Pro diffractometer with Cu-Kα and a Ni filter. Measurements are made with a step size of 0.017° in the 2Θ range of 5–120° and the spectra were processed using X'pert Highscore software. The structures of used compounds were confirmed as follows: ScF3 – Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m ICSD 194249, Sc2O3 – Ia ICSD 26841, ScCO3 – amorphous phase, ScNO3 – scandium nitride hydride, CCDC 140821, ScSO4 – scandium sulfate hydrate, CCDC 140820, ScCl3 – mixture of scandium chloride and scandium hydroxide, C2/m ICSD 1018 and P21/c ICSD 134669.
m ICSD 194249, Sc2O3 – Ia ICSD 26841, ScCO3 – amorphous phase, ScNO3 – scandium nitride hydride, CCDC 140821, ScSO4 – scandium sulfate hydrate, CCDC 140820, ScCl3 – mixture of scandium chloride and scandium hydroxide, C2/m ICSD 1018 and P21/c ICSD 134669.
3 Results
The normalized XAS Sc L2,3 absorption spectra of all the scandium-based compounds described in the Experimental section are shown in Fig. 1. For a direct comparison, both surface sensitive TEY-XAS and bulk probing FY-XAS spectra are plotted for all samples except for Sc2O3 where the FY-XAS spectrum was not acquired as differences are not expected between the bulk and the surface of the fully oxidized compound. As expected, according to the dipole selection rules the Sc XAS spectrum of the metal resembles the energy distribution of the unoccupied (3d) – 4p density of states.32,33 All the XAS spectra of Sc ionic compounds with scandium in Oh coordination have a general shape consisting of three groups of peaks with weak peaks or a shoulder at low photon energies (400–401 eV) and two groups of similar “double-peaks” structures (401–405 eV and 405–408 eV). The latter represents the electron transitions to the empty 3d states from 2p3/2 and 2p1/2 core levels and the energy shift between the two groups corresponds to the spin–orbit splitting of the Sc 2p levels. Each group consists of two peaks that correspond to the transitions to different subgroups of 3d orbitals defined by the crystal field and by the hybridization strength of the metal–ligand bond (in Fig. 1 marked with I and II; I* and II*). The absorption peak at a lower photon energy in the group (I and I*) corresponds to the 3d orbital which interacts weakly with the ligand orbitals (t2g, directed between the ligand p orbitals, π-type of bonds). The absorption peak at a higher photon energy in the group (II and II*) corresponds to the 3d orbital which interacts strongly with the ligand orbitals (eg, directed towards the ligand p orbitals and building σ-type of bond). One can notice for some samples discrepancies in the spectra acquired in transmission and fluorescence mode; ScBr3, for example, exhibits a shift of the double peak structure to lower energy in FY-XAS compared to the TEY-XAS. This kind of shift can be mainly attributed either to surface oxidation in the air during the sample preparation or to the radiation damage that is easily detected using the surface sensitive TEY-XAS in contrast to the more bulk sensitive FY-XAS. Additionally, FY-XAS can suffer from saturation effects due to non-appropriate thickness of the sample, which can cause mismatch between the intensity ratio of different absorption features. In the following, only two specific spectral characteristics of the FY-XAS L2,3 spectra will be taken into account: the width of the II* peak as representative of Sc3d-Lp hybridization (L for ligand) and the I*–II* energy splitting as a measure of the CF strength. Table 1 summarizes the position of the peaks as well as the full half width maximum and the energy splitting of peaks mentioned above. For the single ion ligands peaks I* and II* shift to the lower energy, the energy splitting between peaks I* and II* decreases and the width of peak II* increases with the decrease of the assumed ionic character of the bond between the Sc and the ligand. Bond parameters are summarized in Table 5 and as can be seen for compounds with complex ligands the trend is not straightforward.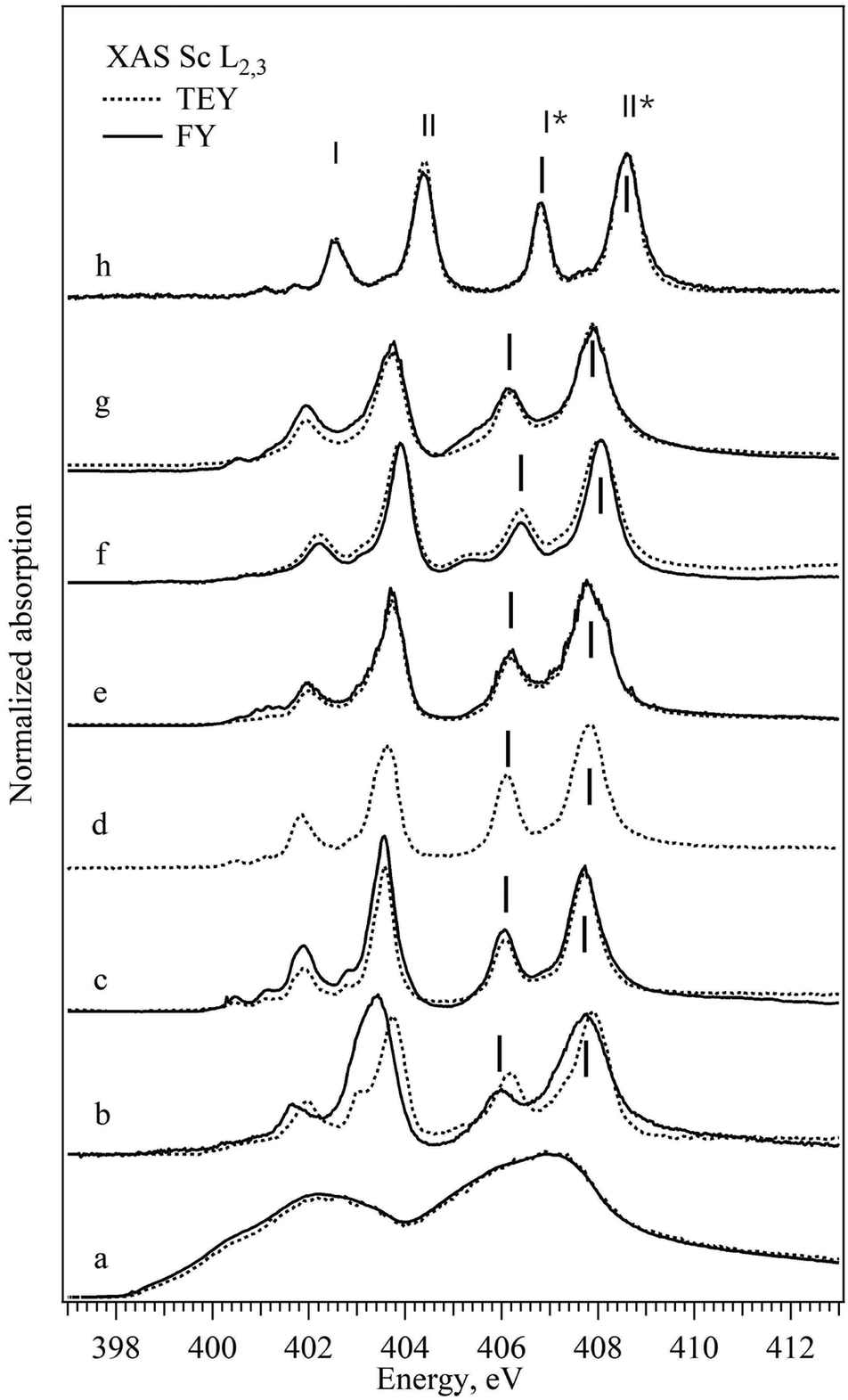 | ||
| Fig. 1 TEY- (dashed lines) and FY-XAS (solid lines) Sc L2,3 spectra of different Sc compounds: (a) Sc metal, (b) ScBr3, (c) ScCl3, (d) Sc2O3, (e) Sc2(CO3)3, (f) Sc2(SO4)3, (g) Sc(NO3)3, and (h) ScF3. Vertical bars mark energy positions of the spectroscopic features of interest. | ||
| Sample | Pos. I* | FWHM I* | Pos. II* | FWHM II* | I* − II* |
|---|---|---|---|---|---|
| ScBr3 | 405.07 | 0.51 | 407.20 | 1.35 | 1.50 |
| ScCl3 | 406.04 | 0.64 | 407.72 | 0.89 | 1.68 |
| Sc2O3 | 406.10 | 0.54 | 407.80 | 0.85 | 1.70 |
| Sc2(CO3)3 | 406.17 | 0.63 | 407.82 | 0.88 | 1.65 |
| Sc2(SO4)3 | 406.41 | 0.59 | 408.03 | 0.64 | 1.65 |
| Sc(NO3)3 | 406.16 | 0.66 | 407.89 | 0.96 | 1.73 |
| ScF3 | 406.82 | 0.44 | 408.58 | 0.65 | 1.76 |
The near-edge and the pre-edge regions of normalized XAS Sc K absorption spectra are shown in Fig. 2, respectively. The shape of the XAS spectrum of Sc metal resembles the density of empty Sc 3d and 4p electronic states in the vicinity of the Fermi energy.34–36 The observed changes in the spectra of ionic compounds are due to the building of the chemical bond to non-metallic ion via formation of the molecular orbitals with the p atomic orbitals of halogens. All the XAS Sc K spectra of ionic compounds have similar features with a pre-edge structure at 4495 eV, a shoulder at around 4502 eV, the main white line at about 4510 eV and a second pronounced peak at about 4517 eV. The position of the absorption edge (defined as the first and major inflection point of the normalized XAS, see Fig. S2 in the ESI†) cannot be assigned here due to the significant electron correlation effects influencing the energy transition from 1s to 3d states.37 The pre-edge feature is related to the symmetry forbidden transitions to the unoccupied Sc 3d orbitals with t2g and eg symmetry, while the peak at around 4517 eV corresponds to the transition from Sc 1s to the Sc 4p states hybridized with corresponding p states of halogen in the conduction band. The pronounced symmetry forbidden transitions to the Sc 3d states are an indication of non-local processes resulting from orbital mixing via the empty p states of the ligands. The Oh coordination double peak structure, similar to the L2,3 edge, is observed in all the ionic compounds although the energy positions and the width of the t2g and eg peaks (marked with III and IV in Fig. 2b accordingly) differ slightly within the row of the ligands. Table 2 summarizes the position of the peaks as well as the full half width maximum and the energy splitting of pre-edge peaks mentioned above. No clear trend on spectroscopic features at pre-edge peaks at the K-edge can be observed within the studied series of ligands.
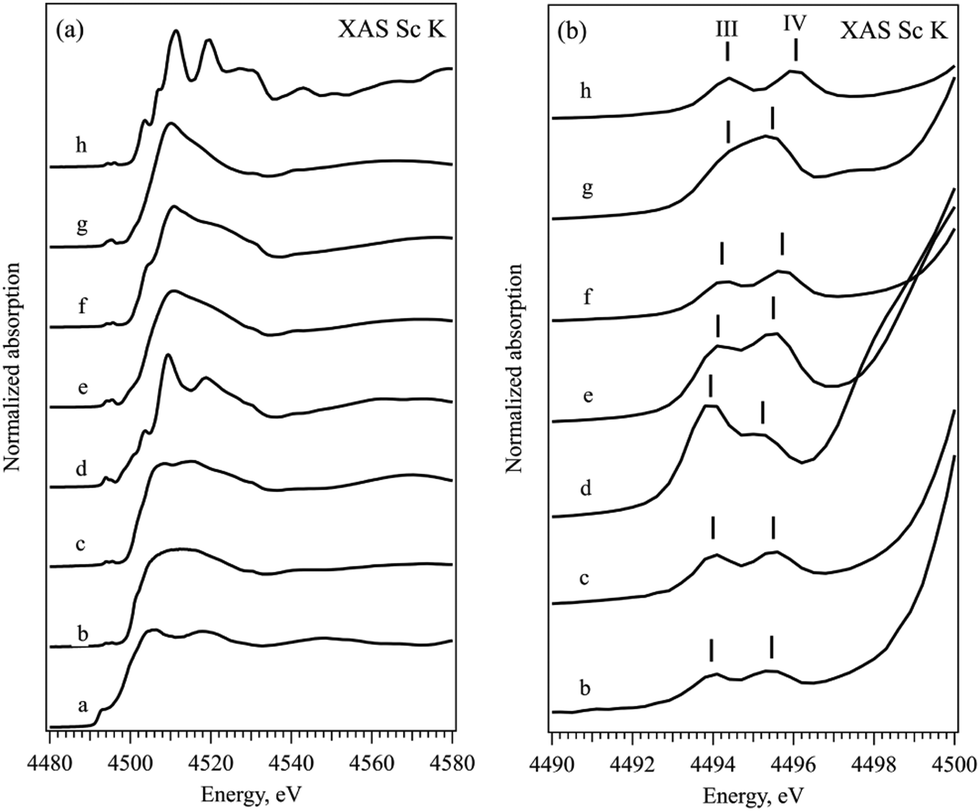 | ||
| Fig. 2 Normalized XAS Sc K spectra (left) and their pre-edge range (right) of different Sc compounds: (a) Sc metal, (b) ScBr3, (c) ScCl3, (d) Sc2O3, (e) Sc2(CO3)3, (f) Sc2(SO4)3, (g) Sc(NO3)3, and (h) ScF3. Vertical bars mark the energy positions of the spectroscopic features of interest. | ||
| Sample | Pos. III | FWHM III | Pos. IV | FWHM IV | III − IV |
|---|---|---|---|---|---|
| ScBr43 | 4493.9 | 1.3 | 4495.4 | 1.3 | 1.4 |
| ScCl3 | 4494.0 | 1.3 | 4495.5 | 1.3 | 1.5 |
| Sc2O3 | 4493.8 | 1.3 | 4495.2 | 1.7 | 1.4 |
| Sc2(CO3)3 | 4494.1 | 1.3 | 4495.5 | 1.4 | 1.4 |
| Sc2(SO4)3 | 4494.2 | 1.4 | 4495.7 | 1.4 | 1.5 |
| Sc(NO3)3 | 4494.3 | 1.4 | 4495.4 | 1.4 | 1.1 |
| ScF3 | 4494.3 | 1.3 | 4496.0 | 1.4 | 1.7 |
Fig. 3 shows the phase corrected Fourier transforms (FT) of k3-weighted EXAFS data in the k range between 3 and 10 Å−1 of all the compounds and the results of the fits selected for Sc metal and ScF3. The fits of all the other compounds studied are shown in the ESI† (Fig. S3–S8). As can be seen, for all the ionic compounds, the first peak between 1.9 and 2.5 Å corresponds to the first coordination shell of the Sc–ligand, and the first peak in the Sc metal at 3.3 Å is related to the Sc–Sc coordination shell in hcp-Sc.32,33 The second coordination shell is only observed in Sc2O3 and ScF3 ionic compounds because of the well-defined structure of ligands and a significant long-range order in these cases.17 In ScBr3, ScCl3 and compounds with complex ligands no other peaks are observed above 4 Å due to the less ordered or even amorphous structures present.38–42 Thereafter, each EXAFS data was fitted to the known structure in the R range between 1.5 and 3.5 Å taking into account the first coordination shell in Oh coordination with the corresponding ligand. For Sc metal, ScF3 and Sc2O3 second coordination shells were included to achieve finer fitting. For scandium bromide the model with a [ScBr6]3− cluster and bond length close to the one of Sc–Cl was used while for calcite, sulfite and nitride [ScO6]9− clusters the bond length of oxide was taken. Table 3 shows the results of the fitting procedures together with the energy of the main absorption edge of XAS K spectra,37 for all the scandium based ionic compounds as well as for scandium metal. For the single ion ligands, the position of the absorption edge (WL E0 value in Table 3) increases with the increase of the ionicity of the Sc–ligand bond and the Sc–ligand distance (R value in Table 3) decreases with the decrease of the size of the ligand ion (see Table 5). The changes in the interatomic distances in the compounds with complex ligands are less pronounced and should be related to the parameters of the bond to oxygen ion inside the ligand complex.
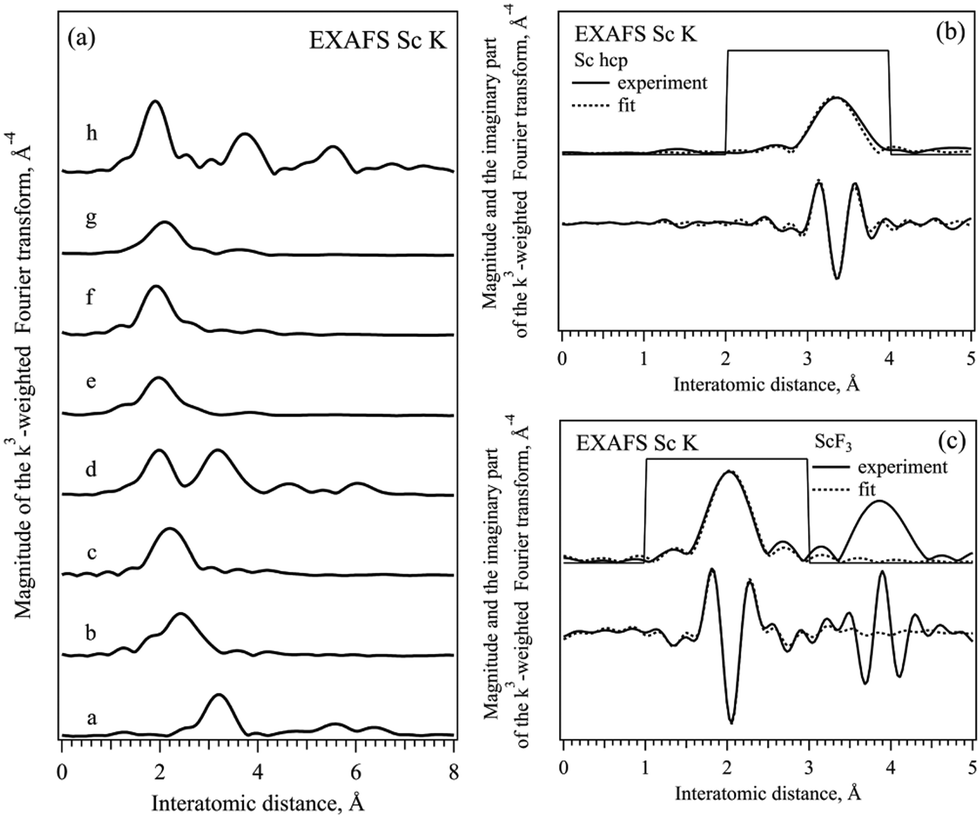 | ||
| Fig. 3 (right) FT of k3-weighted EXAFS Sc K data of different Sc compounds, phase corrected: (a) Sc metal, (b) ScBr3, (c) ScCl3, (d) Sc2O3, (e) Sc2(CO3)3, (f) Sc2(SO4)3, (g) Sc(NO3)3, and (h) ScF3. (left) Experimental (solid line) and fitted (dotted line) magnitude and the imaginary part of the FT of Sc metal and ScF3, the fitting range is marked by a thin solid line. | ||
| Sample | WL E0, eV ±0.05 | R, Å | Sc–ligand CN | σ 2, Å2 × 10−4 |
|---|---|---|---|---|
| a Assumed values. | ||||
| Sc | 4499.0 | 3.32 | 12.0a | 42 ± 20 |
| 3.37 | ||||
| ScBr3 | 4503.1 | 2.61 | 2.9 ± 1.4 | 32 ± 4 |
| ScCl3 | 4504.2 | 2.42 | 4.5 ± 1.0 | 80 ± 3 |
| Sc2O3 | 4507.0 | 2.13 | 4.6 ± 1.8 | 33 ± 3 |
| Sc2(CO3)3 | 4507.0a | 2.17 | 4.1 ± 1.1 | 67 ± 4 |
| Sc2(SO4)3 | 4507.0a | 2.09 | 4.7 ± 0.5 | 30a |
| Sc(NO3)3 | 4507.0a | 2.32 | 4.2 ± 1.3 | 56 ± 5 |
| ScF3 | 4509.1 | 2.04 | 6.0a | 41 ± 6 |
The binding energies of Sc 2p3/2 and O 1s electrons in all measured compounds are listed in Table 4. As expected, the Sc 2p binding energy increases with the increase of the ionicity of the bond between the scandium and a single ligand.43 The situation in the compounds with a complex ion as a ligand is less straightforward. Fig. 4 displays the Sc 2p and O 1s XPS spectra of Sc compounds with the oxygen as a connected ion: the binding energies of Sc 2p and O 1s electrons are both decreasing with the increase of the (assumed) ionicity of the Sc–O bond. The N 1s peak which is the signature for Sc(NO3)3 should appear at around 407 eV and in this case it overlaps with the Sc 2p1/2 peak at around 406 eV.44 Furthermore, the high energy peak is clearly seen in the O 1s XPS spectrum of Sc2O3 most probably due to the surface oxygen45,46 and the low energy peak at around 531 eV in Sc(NO3)3 could be due to the presence of hydroxide47,48 (see the results of the decomposition of the XPS O 1s peaks in Fig. S9 in the ESI†).
| Sample | Sc 2p3/2, eV | O 1s, eV |
|---|---|---|
| ScBr3 | 400.8 | — |
| ScCl3 | 401.2 | — |
| Sc2O3 | 401.6 | 529.6 |
| Sc2(CO3)3 | 402.6 | 531.6 |
| Sc2(SO4)3 | 403.9 | 532.3 |
| Sc(NO3)3 | 403.4 | 533.5/532.7 |
| ScF3 | 405.3 | — |
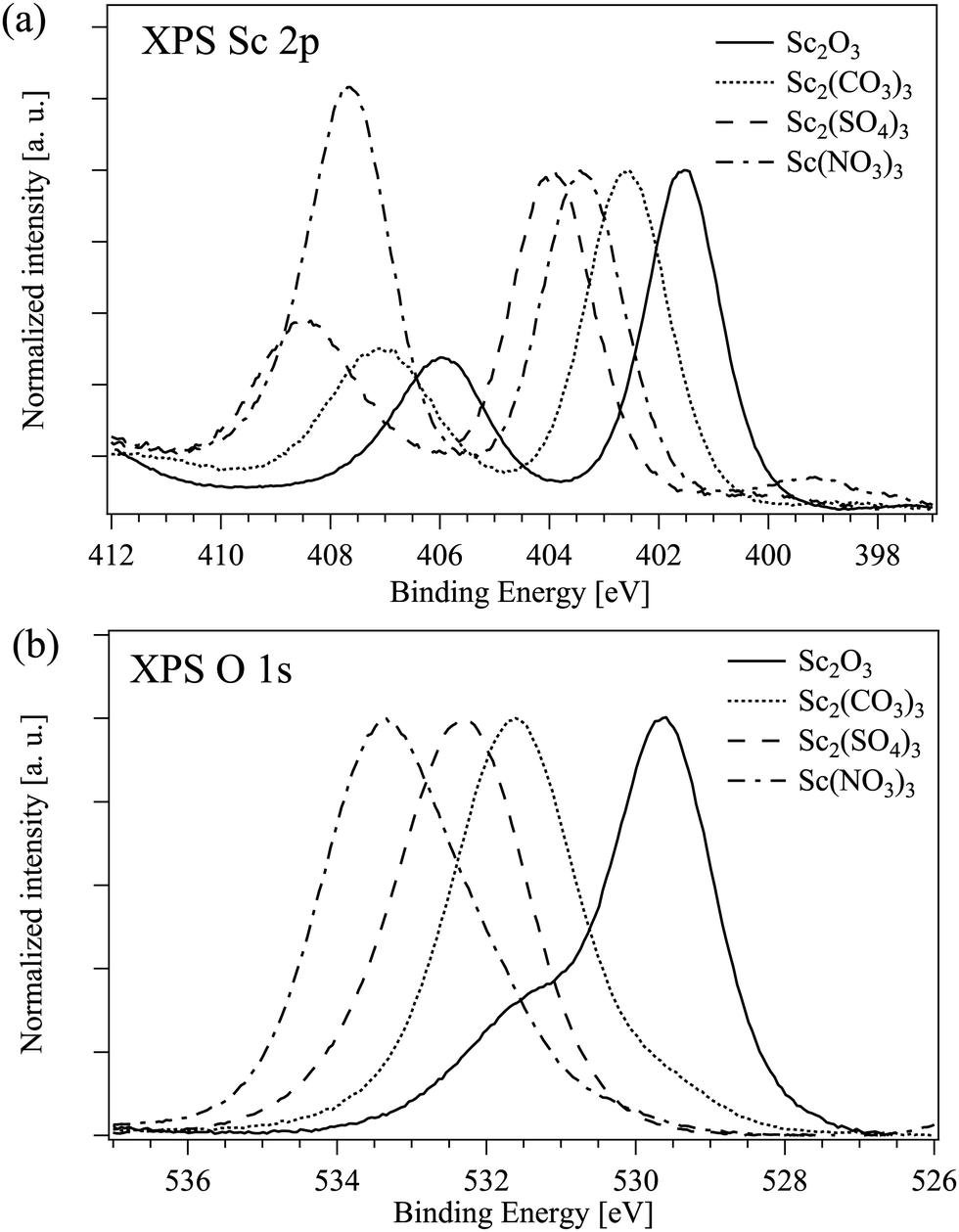 | ||
| Fig. 4 XPS data on selected Sc compounds: (a) Sc 2p and (b) O 1s. | ||
4 Discussion
The core hole effect plays a significant role in strongly correlated materials such as TM compounds. The detailed shape of the L2,3 absorption edge of scandium is currently not fully explained either on the basis of a purely atomic like picture or on the basis of the structure of the modified empty density of states (due to the energy dependence of the matrix elements). It may be recognized from the fact that the intensity ratio of the peaks in theoretical density of electronic states of Sc is closer to the value obtained by Bremsstrahlung isochromate spectroscopy (BIS) (where no core hole is created)49 than to the value acquired by XAS. Nevertheless, there is some uncertainty on how exactly the core hole influences the 3d states. The theoretical calculations including the core hole in the intermediate state often predict a behavior opposite to the experimental observations; as an example, the predicted lower energy shift of the peaks contradict the experimentally observed higher energy shift.15 Other experimental observations which cannot be explained only by the core hole effect are for example a strong deviation of the intensity ratio between spin–orbit counterparts of the absorption band (L3 and L2) compared to the statistical value (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). One can also add the different line shape of L3 and L2 absorption bands and a strong change in the apparent spin–orbit splitting between L3 and L2 edges relative to what is observed in the Sc 2p XPS data.50 Thus, the core hole effect does influence the XAS spectra of transition metals but may interact with other effects like atomic exchange and solid-state band-structure effects.15 The effect of the core hole is less pronounced in XAS K-edge spectra as transition is allowed to the antibonding states with p symmetry that are delocalized.
:1). One can also add the different line shape of L3 and L2 absorption bands and a strong change in the apparent spin–orbit splitting between L3 and L2 edges relative to what is observed in the Sc 2p XPS data.50 Thus, the core hole effect does influence the XAS spectra of transition metals but may interact with other effects like atomic exchange and solid-state band-structure effects.15 The effect of the core hole is less pronounced in XAS K-edge spectra as transition is allowed to the antibonding states with p symmetry that are delocalized.
Early attempts were made to correlate the intensity of the Cu L2,3 peaks with the covalency of the Cu–halogen bond53,54 and a linear relationship was found between the total intensity of the L band and the ligand contribution to the bond. For the Sc3+ the structure of multiplets at the L edge offers the possibility to separate the peaks with different contributions of ligand states, in Sc3+ in Oh surrounding the intensity of L2-t2g with the major Sc d contribution and L2-eg with the expected additional ligand p contribution might be related to the ionicity of the Sc–ligand bonds. The width of the L2-t2g does not change significantly over the row of ligands in contrast to the width of the L2-eg peak (FWHM I* and FWMW II* in Table 1). The ratio between the areas of those peaks plotted against the bond ionicity of single ligands (Table 5) is shown in Fig. 5 (blue crosses). The dependence can be fitted with an exponential function and the area ratio in an ideally ionic Sc compound would be 3.7 according to this fit. The ionicity of the Sc–ligand bond in complex ligands with oxygen as the connecting ion can be adjusted to the fitting curve by taking into account the ionicity of oxygen–heteroatom bonds, assuming linear reduction of the ionicity of the Sc–O bond with the increase of the charge transfer from heteroatom to oxygen (Fig. 5, red triangles). Additionally to the decrease of the width of eg peaks in the XAS L2 spectra, the increase of the CF strength in the row of single ion ligands is reflected as an increase of the t2g–eg energy splitting (I* − II* value in Table 1). The changes in the CF along the series of ligands induce less modification of the pre-edge structure of the Sc XAS K spectra (Table 2), probably due to the limited energy resolution at these energies compared to the soft X-ray energy range. The increase of the Sc–ligand bond length is clearly reflected in the EXAFS Sc K data in compounds with single ion ligands (R values in Table 3). These changes can be associated with the increases of the ionic radii of the ligand reducing the CF strength.21
| Sample | Ionic radii of L, Å | Ionicity Sc–L, % | Bond length Sc–L, Å |
|---|---|---|---|
| a Assumed value. | |||
| ScBr3 | 1.82 (Br−) | 49 | 2.68 |
| ScCl3 | 1.67 (Cl−) | 57 | 2.59/2.61 |
| Sc2O3 | 1.25 (O2−) | 68 | 2.14 |
| Sc2(CO3)3 | 1.25 (O2−) | 68a | 2.08 |
| 0.30 (C4+) | 25 (C–O) | 1.24 (C–O) | |
| Sc2(SO4)3 | 1.25 (O2−) | 68a | 2.06 |
| 0.43 (S6+) | 24 (S–O) | 1.47 (S–O) | |
| Sc(NO3)3 | 1.25 (O2−) | 68a | 3.04 |
| 0.27 (N5+) | 10 (N–O) | 3.06 (–OH) | |
| 1.29 (N–O) | |||
| ScF3 | 1.19 (F−) | 92 | 2.07 |
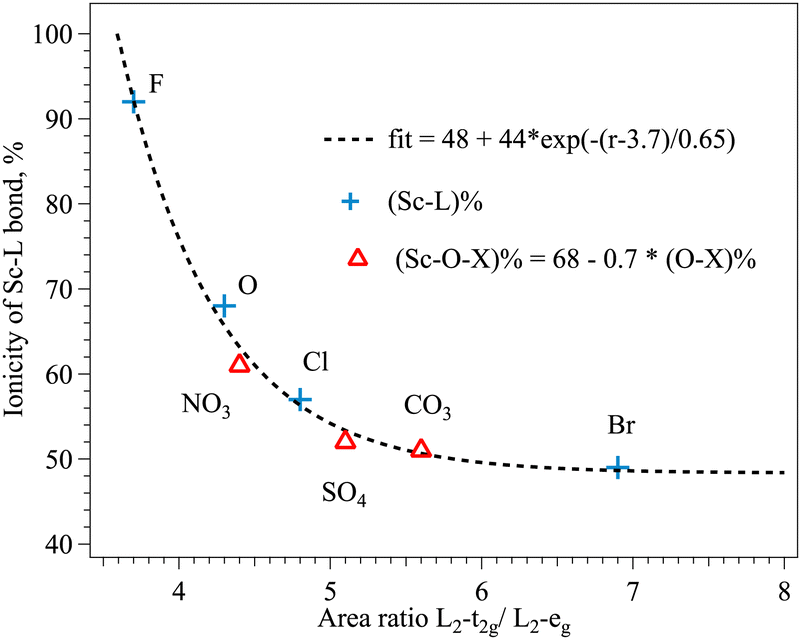 | ||
| Fig. 5 Dependence between the ionicity of the Sc–ligand bond ((Sc–L)%) and the area ratio of L2-t2g/L2-eg peaks in Sc L XANES of different compounds (r). | ||
In the series of complex ligands the t2g–eg energy splitting of XAS L2 lines (I* − II* values in Table 1) in carbonate and sulfate are smaller than in nitrate within the experimental errors. The energy splitting in pre-edge peaks in K XAS (III − IV values in Table 2) is the smallest in nitrate compound. The Sc–O bond length in carbonate and sulfate is close to the value of oxide, and the longest distance measured by EXAFS is in nitrate (R values in Table 3). In general, the usual expectation is that with a more electronegative heteroatom the Sc–O bond is more ionic. The t2g–eg energy splitting increases with the increase of the electronegativity of the central atom in the ligand complex (N5+ = 3.04, S6+ = 2.58, C4+ = 2.55, in Pauling units). The Sc–O distance does not change significantly except for the NO3− complex (Fig. 3 and Table 3), where the weakest splitting of the pre-edge peaks is observed (Fig. 2 and Table 2), hinting at the weakest CF and the highest degree of atomic orbital mixing between N and O in the ligand complex.
The ligands in scandium-based compounds studied in this work can be organized into a spectrochemical series9 based on the ionic radii of the ligand and on the Sc–ligand bond length (measured experimentally and assumed values). The resulting ionicity of the bond to scandium reduces and the CF strength decreases along the series:21
| F− → O2− → Cl− → Br− |
The electronegativity of the heteroatom in complex ligands decreases causing further changes of the ionic character of the Sc–O bond.51 The bond length of the heteroatom to oxygen in (SO4)2− is longer compared to (CO3)2− due to the larger ionic radii of S6+ in the tetrahedral complex than the one of C4+ in the planar structure (Table 5). Therefore, the resulting row of complex ligands with oxygen as a connected ion with reducing ionicity based on experimental and theoretical data can be organized as follows:52
| (NO3)− → (SO4)2− → (CO3)2− |
The electronegativity of the connecting atom directly influences the binding energy of the Sc 2p core electrons in ionic complexes (Fig. 6, black crosses): the more ionic is the Sc–L bond, the higher is the binding energy of the Sc 2p electron.43 Based on the experimental XPS data on compounds with single ligands, it is shown that the dependence is not quite linear. For instance, the linear fit to all the available data points strongly underestimates the ionicity of the Sc–F bond. The electronegativity of the heteroatom in complexes with the oxygen as a connecting ion influences the Sc–O bond in a complex way, the binding energy of the O 2p measured by XPS does not show the direct correlation to the expected ionicity of the Sc–O bond, which further hints to the complex character of the bond formation and its reflection in XAS and XPS spectra.55 The binding energy of the O 1s is increasing according to the electronegativity of the heteroatom in the complex, hinting at the increase of the ionicity of the O–heteroatom bond in this row (Fig. 6, blue circles). The middle value of the Sc 2p3/2 binding energy on nitrate together with the longest Sc–O distance might indicate the redistribution of the charge in this complex. More detailed simulations is needed in the future to explain the experimental findings.
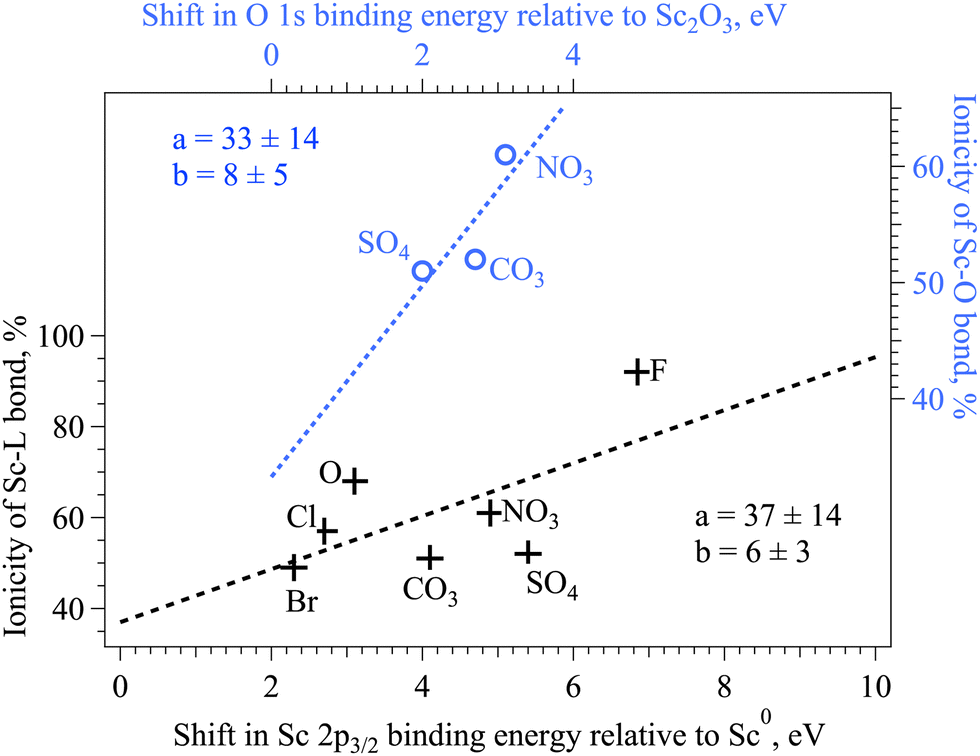 | ||
| Fig. 6 Dependence between the ionicity of the Sc–ligand bond ((Sc–L)%) derived from the analysis of XAS L2 data and the chemical shift of Sc 2p3/2 and O 1s binding energies of different compounds. The resulting fitting parameters for linear functions are marked. | ||
5 Conclusions
The influence of the local environment of the Sc cation on the spectral features observed in XAS Sc K and L2,3 spectra has been studied through the experimental characterization of eight scandium based compounds with known structures. Both edges contain information enabling the chemical specification of the Sc ion through the analysis of the chemical shift and the detailed line shape identification based on comparison with the reference data. The EXAFS analysis and the Sc 2p XPS data complement these findings. This study provides a further set of Sc XAS spectra, which might be of interest to help determining the chemical state and the local arrangement of Sc in other natural or synthetic materials.4,56 Moreover, correlations between the features in highly reliable experimental data and theoretical parameters of chemical bonds in scandium compounds were evaluated and an attempt was made to describe the derived dependencies numerically. This study could give a new impulse to the extended theoretical investigations which will allow us to determine and to predict the electronic structure of the transition metal compound with high precision in the future.Author contributions
A. Z. conceptualized the study and designed the experimental plan. A. L. and A. Z. carried out the XAS experiments. A. Z. conducted the data analysis. All authors discussed the experimental data and equally contributed to the final preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Helmholtz programs From Matter to Materials and Life (MML) and Materials and Technologies for the Energy Transition (MTET) for financial support. We would like to thank the Institute for Beam Physics and Technology (IBPT) for the operation of the storage ring, the Karlsruhe Research Accelerator (KARA). We would like to thank the beamline scientist at RDBL at BESSY Dr. M Brzhezinskay for the support during the experiments. D. Neukum (IKFT/KIT) is acknowledged for performing the XRD measurements.References
- Nomenclature of Inorganic Chemistry: IUPAC Recommendations 2005, ed. N. Connelly, T. Damhus, R. M. Hartshorn and A. T. Hutton, The Royal Society of Chemistry, Cambridge, UK, 2005 Search PubMed.
- J. Emsley, Nat. Chem., 2014, 6, 1025 CrossRef CAS PubMed.
- C. Benel, A. Fischer, A. Zimina, R. Steininger, R. Kruk, H. Hahn and A. Léon, Mater. Horiz., 2019, 6, 727–732 RSC.
- V. Migliorati and P. D'Angelo, Inorg. Chem., 2016, 55, 6703–6711 CrossRef CAS PubMed.
- Krystallochimija (in Russian), ed. G. Bokij, Izdat. Nauka, 1971 Search PubMed.
- Transition metal chemistry: the valence shell in d-block chemistry, ed. M. Gerloch and E. C. Constable, VCH, Germany, 1994 Search PubMed.
- H. B. Gray, J. Chem. Educ., 1964, 41, 2–12 CrossRef CAS.
- Multiplets of transition-metal ions in crystals, ed. S. Sugano, Y. Tanabe and H. Kamimura, Academic, New York, 1970 Search PubMed.
- R. Tsuchida, Bull. Chem. Soc. Jpn., 1938, 13, 388–400 CrossRef CAS.
- Fundamentals of XAFS, ed. M. Newvill, Consortium for Advanced Radiation Sources, University of Chicago, Chicago, IL, 2004 Search PubMed.
- Introduction to XAFS, ed. G. Bunker, Cambridge University Press, 2010 Search PubMed.
- Core Level Spectroscopy of Solids, ed. A. Kotani and F. M. F. de Groot, Taylor and Francis Inc., 2008 Search PubMed.
- S. J. George, O. B. Drury, J. Fu, S. Friedrich, C. J. Doonan, G. N. George, J. M. White, C. G. Young and S. P. Cramer, J. Inorg. Biochem., 2009, 103, 157–167 CrossRef CAS PubMed.
- X-Ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS and XANES (Chemical Analysis), ed. D. C. Koningsber and R. Prins, John and Wiley Sons, 1987 Search PubMed.
- J. Fink, T. Mullerheinzerling, B. Scheerer, W. Speier, F. U. Hillebrecht, J. C. Fuggle, J. Zaanen and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 4899–4904 CrossRef CAS PubMed.
- J. Zaanen, G. A. Sawatzky, J. Fink, W. Speier and J. C. Fuggle, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 4905–4913 CrossRef CAS PubMed.
- G. van der Laan and I. W. Kirkman, J. Phys.: Condens. Matter, 1992, 4, 4189–4204 CrossRef CAS.
- L. Ley, O. B. Dabbousi, S. P. Kowalczyk, F. R. Mcfeely and D. A. Shirley, Phys. Rev. B: Solid State, 1977, 16, 5372–5380 CrossRef CAS.
- D. M. Pease, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 6708–6714 CrossRef CAS PubMed.
- F. M. F. de Groot, J. C. Fuggle, B. T. Thole and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 928–937 CrossRef CAS PubMed.
- F. M. F. de Groot, J. C. Fuggle, B. T. Thole and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 5459–5468 CrossRef CAS PubMed.
- M. A. Arrio, S. Rossano, C. Brouder, L. Galoisy and G. Calas, Europhys. Lett., 2000, 51, 454–460 CrossRef CAS.
- D. Bocharov, M. Krack, A. Kalinko, J. Purans, F. Rocca, E. A. Shehab and A. Kuzmin, J. Phys.: Conf. Ser., 2016, 712, 012009 CrossRef.
- D. Bocharov, P. Zguns, S. Piskunov, A. Kuzmin and J. Purans, Low Temp. Phys., 2016, 42, 556–560 CrossRef CAS.
- T. Yamamoto, X-Ray Spectrom., 2008, 37, 572–584 CrossRef CAS.
- HZB, accessed 25.05.2023, U49-2 PGM-1 beamline https://www.helmholtz-berlin.de/pubbin/igama_output?modus=einzelsprache=engid=1655typoid=75136.
- HZB, accessed 25.05.2023, RGBL Dipole beamline, https://www.helmholtz-berlin.de/pubbin/igama_output?modus=einzelsprache=engid=1623typoid=75136.
- T. Spangenberg, J. Göttlicher and R. Steininger, AIP Conf. Proc., 2009, 1092, 93–97 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- IPS-KIT, accessed 25.05.2023, UHV laboratory, https://www.ips.kit.edu/5881.php.
- N. Fairley, 2020, Casa Software Ltd, https://www.casaxps.com/.
- K. P. Jensen, B. O. Roos and U. Ryde, J. Chem. Phys., 2007, 126, 014103 CrossRef PubMed.
- P. Blaha, K. Schwarz and P. H. Dederichs, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 9368–9374 CrossRef CAS PubMed.
- L. D. Finkel'steyn and S. A. Nemnonov, Fiz. Met. Metalloved., 1966, 2, 49 Search PubMed.
- M. Chassé, A. Juhin, D. Cabaret, S. Delhommaye, D. Vantelon and G. Calas, Phys. Chem. Chem. Phys., 2018, 20, 23903–23912 RSC.
- R. Oberti, S. Quartieri, M. C. Dalconi, F. Boscherini, G. Iezzi, M. Boiocchi and S. G. Eeckhout, Am. Mineral., 2006, 91, 1230–1239 CrossRef CAS.
- N. Finck, M. L. Schlegel and A. Bauer, Phys. Chem. Miner., 2015, 42, 847–859 CrossRef CAS.
- M. Matsubara, Y. Harada, S. Shin, T. Uozumi and A. Kotani, J. Phys. Soc. Jpn., 2004, 73, 711–718 CrossRef CAS.
- A. Léon, N. Finck, J. Rothe, M. Felderhoff and M. Fichtner, J. Phys. Chem. C, 2015, 119, 15810–15815 CrossRef.
- D. J. Millen and J. R. Morton, J. Chem. Soc., 1960, 307, 1523–1528 RSC.
- C. D. Garner and S. C. Wallwork, J. Chem. Soc. A, 1966, 1496–1500 RSC.
- S. P. Petrosyants, A. B. Ilyukhin and A. Y. Sukhorukov, Russ. J. Coord. Chem., 2005, 31, 545–551 CrossRef CAS.
- F. Mercier, C. Alliot, L. Bion, N. Thromat and P. Toulhoat, J. Electron Spectrosc. Relat. Phenom., 2006, 150, 21–26 CrossRef CAS.
- J. Baltrusaitis, P. M. Jayaweera and V. H. Grassian, Phys. Chem. Chem. Phys., 2009, 11, 8295–8305 RSC.
- B. W. Veal and A. P. Paulikas, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 5399–5416 CrossRef CAS PubMed.
- B. Johansson and N. Martensson, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 21, 4427–4457 CrossRef CAS.
- J.-C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC.
- H. W. Nesbitt, D. Legrand and G. M. Bancroft, Phys. Chem. Miner., 2000, 27, 357–366 CrossRef CAS.
- W. Speier, J. C. Fuggle, R. Zeller, B. Ackermann, K. Szot, F. U. Hillebrecht and M. Campagna, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 6921–6930 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- The Nature of the Chemical Bond, ed. L. Pauling, Cornell University Press, Ithaca, NY, 3rd edn, 1960 Search PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- S. J. George, M. D. Lowery, E. I. Solomon and S. P. Cramer, J. Am. Chem. Soc., 1993, 115, 2968–2969 CrossRef CAS.
- I. M. DiMucci, J. T. Lukens, S. Chatterjee, K. M. Carsch, C. J. Titus, S. J. Lee, D. Nordlund, T. A. Betley, S. N. MacMillan and K. M. Lancaster, J. Am. Chem. Soc., 2019, 141, 18508–18520 CrossRef CAS PubMed.
- J. Kawai, K. Maeda, K. Nakajima and Y. Gohshi, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 8560–8566 CrossRef CAS PubMed.
- T. Takahashi, A. Ito, M. Inakuma and H. Shinohara, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 13812–13814 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Sc L2,3 XAS spectra of fresh and oxidized Sc metal, first derivative of normalized XAS Sc K spectra of different Sc compounds, results of decomposition analysis of the O 1s XPS data, and results of the fitting of Sc K EXAFS data (phase corrected). See DOI: https://doi.org/10.1039/d3cp04108e |
| This journal is © the Owner Societies 2024 |