
UV-spectrum and photodecomposition of peroxynitrous acid in the troposphere†
Wiem
Chebbi
 *ab,
Najoua
Derbel
a,
Alexander
Alijah
b and
Thibaud
Cours
b
*ab,
Najoua
Derbel
a,
Alexander
Alijah
b and
Thibaud
Cours
b
aLSAMA, Laboratoire de Spectroscopie Atomique, Moléculaire et Applications, Department of Physics, University Tunis - El Manar, 1060 Tunis, Tunisia
bGSMA, Groupe de Spectrométrie Moléculaire et Atmosphérique, UMR CNRS 7331, University of Reims Champagne-Ardenne, 51100 Reims, France. E-mail: wiem.chebbi@univ-reims.fr
First published on 27th November 2023
Abstract
The UV spectrum of peroxynitrous acid, HOONO, was computed at the B3LYP/AVTZ and MCSCF/AVTZ levels using the fewest switches surface hopping algorithm. Due to large-amplitude vibrational motions of this molecule, the maxima in the simulated spectra are displaced from the positions of vertical excitations. The three lowest excited electronic singlet states, which are all repulsive, can be reached by UV absorption. The photolysis products are determined, and the photolysis rate constant is provided for the first time. We found that near the tropopause the photolysis rate constant J ≈ 6 × 10−4 s−1, exceeds that for thermal decomposition by two orders of magnitude. The photolysis lifetime is about 30 minutes. Thus, photolysis is an important process and should be included in atmospheric models.
1 Introduction
Radicals containing hydrogen, OH and OOH, and nitrogen, NO and NO2, are present in the earth's atmosphere where they participate in a large number of radical chain reactions.1 More than fifty years ago, Crutzen2 suggested that nitrogen oxides arriving in the stratosphere due to anthropogenic activities contribute to the catalytic conversion of ozone into dioxygen, thus damaging the stratospheric ozone layer. The nitrogen oxides can be removed partially from this cycle by recombination with the hydroxyl radical in the presence of a third body, such as N2, O23,4 or water.5| HO + NO2 + M → HONO2 + M | (1) |
| ⇌ HOONO + M | (2) |
Peroxynitrous acid, known for a long time to exist in aqueous solution,13 was first isolated in an argon matrix by Y. P. Lee's group14,15 and identified by IR spectroscopy. In their experiment, the acid was formed by recombination of the photolysis products of nitric acid. They also recorded the UV spectrum16 of peroxynitrous acid. Identification of this molecule was guided by theoretical predictions provided by McGrath et al.17 More recently, Zhang and coworkers18,19 reported the experimental vibrational overtone spectrum of the matrix-isolated cis–cis conformer, supported by ab initio computations. First experimental evidence for the existence of peroxynitrous acid in the gas phase was provided by Donahue et al.20 and by Hippler et al.,8 when they studied the kinetics of the OH + NO2 reaction. The molecule was then observed directly in the gas phase by Nizkorodov and Wennberg21 by vibrational photodissociation spectroscopy, following excitation of the OH stretching mode. In another overtone action-spectroscopic experiment, Pollack et al.22 identified the less stable trans-perp conformer. Fry et al.23 also detected the trans-perp conformer and noticed rapid isomerization to the more stable cis–cis conformer. Bean and coworkers,6 in a kinetics study carried out in a low-pressure discharge flow reactor, observed both peroxynitrous acid in the cis–cis conformation and nitric acid by infrared cavity ringdown spectroscopy. Experimental and computational evidence for the existence of a third conformer, called cis-perp, was presented by Li et al.24 Drouin, Fry and Miller25,26 investigated the cis–cis conformer by submillimeter spectroscopy and derived rotational constants.
Substantial theoretical work was also published. The first thorough investigation of the potential energy surface was reported by McGrath and coworkers,17,27 who also proposed a nomenclature for the structures of the stationary points on the potential energy surface. The most stable geometrical configuration of HOONO is the one classified as cis–cis, where both the OONO and the HOON entities are in cis-positions, see Fig. 1. This structure is stabilized by a hydrogen bond. There are two other local minima on the potential energy surface, denoted as trans-perp and cis-perp, as shown in Fig. 1. The cis-perp conformer is separated from the cis–cis conformer by a very weak rotational barrier.28 The potential energy surface and geometrical structures of these conformers and transition states were studied by a number of authors, see for example., ref. 23, and 28–33 Accurate thermochemical data were reported by Szakács and coworkers.34 In thermal equilibrium at room temperature, only the cis–cis conformer is populated. A global potential energy surface in full dimensionality was constructed by Chen et al.33 and used for trajectory calculations.
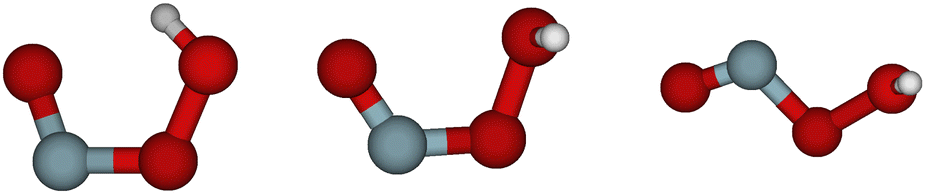 | ||
| Fig. 1 Structures of the cis–cis, cis-perp and trans-perp configurations, from left to right. | ||
The success of overtone action spectroscopy has stimulated theoretical investigations of excited rovibrational states and vibrational transition dipole moments.24,35–38 Such calculations are non-trivial due to the presence of three low-frequency vibrational modes, the HO–ONO and HOO–NO torsional modes and the ONO bending vibration, and multidimensional models were devised for their description.24,37,39
In contrast to the impressive work on the characterisation of the ground state potential energy surface, vibrational spectroscopy and kinetic modelling, the electronic spectroscopy of peroxynitrous acid has received very little attention. The UV spectrum of peroxynitrous acid in an argon matrix was recorded almost forty years ago by W.-J. Lo and Y. P. Lee.16 We are aware of a single theoretical publication, by Li and Francisco,40 who investigated the cis–cis conformer and computed the vertical transition energies to the three lowest excited singlet states and two triplet states. Tentative assignment of the experimental bands was made on the basis of these transition energies. An important conclusion was that peroxynitrous acid, once formed in the atmosphere, will likely be photolysed. The aim of the present work is to re-analyse the experimental UV spectrum and to compute the photolysis rate constant under conditions found in the atmosphere near and above the tropopause. Such data are required for atmospheric modelling.7,10,41
2 UV spectrum
W.-J. Lo and Y. P. Lee,15 studying the photolysis of nitric acid, HONO2, in a solid argon matrix, observed the subsequent formation of peroxynitrous acid in the cis–cis and trans-perp configurations by recombination of the fragments according to reaction (2). They also recorded the IR15 and UV spectra16 of peroxynitrous acid. The latter spectra were reported in the range of 200 nm < λ < 450 nm and show broad absorption signals that were attributed to the cis–cis and the trans-perp conformers. No further experiments have been conducted to the best of our knowledge.To gain insight into the observed spectra, we have computed the oscillator strengths for electronic transitions from the ground state to the lowest three excited singlet state at the MCSCF/AVTZ level using the Molpro package.42 Two active spaces were tested: a large active space CAS(24e,17o), i.e. 24 electrons of the valence shell in 17 active orbitals. Only the 1s orbitals of the heavy atoms were kept inactive. The other active space was CAS(16e,13o), in which also the 2s orbitals were kept inactive‡. A second set of computations were performed with Gaussian G1643 at the density functional level with the same AVTZ basis set for comparison. The B3LYP functional was chosen after a systematic test with a number of alternative density functionals, M05-2X and M06-2X, as it produces reasonably well the relative intensities found in the experiment for the two isomers. The B3LYP functional has been widely used by previous workers. We note here that Berski and coworkers,44 examining the electronic structure and bonding properties of this molecule with the same functionals, found that only B3LYP produces the geometrical structures correctly. The global potential energy surface presented by Chen et al.33 is also based on the B3LYP functional.
The results of the two sets of computations are compared in Table 1. They are quite close, except for the transition to the third electronic state in the case of the trans-perp conformer. The reason is that the excitation energies of the trans-perp conformer are very sensible to the position of the hydrogen atom. The transitions are analysed in detail in Table 2. The most intense transition is towards the second excited electronic state, where the principal electronic excitation is between two π orbitals and parallel to the molecular plane. Transitions to the first and third electronic states are σ → π and π → σ excitations, respectively, perpendicular to the molecular plane and thus less intense. As can be inferred from Table 2, the σ or π character of the HOMO, orbital 16, is changed when passing from the cis–cis configuration to the trans-perp configuration. This can be rationalised in the following way: in the planar cis–cis configuration, the hydrogen 1s-orbital cannot participate at the molecular 3π orbital by symmetry. As the hydrogen atom is rotated out of plane, its 1s orbital interacts strongly with the π orbital at the oxygen atom so that the molecular 3π orbital becomes stabilized and passes below the 13σ orbital.
| Transition | B3LYP | MCSCF, CAS(16e,13o) | MCSCF, CAS(24e,17o) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| E (eV) | λ (nm) | f | E (eV) | λ (nm) | f | E (eV) | λ (nm) | f | |
| cis–cis | |||||||||
| 1 → 2 | 4.097 | 302.6 | 0.0020 | 4.181 | 296.5 | 0.0035 | 4.142 | 299.3 | 0.0027 |
| 1 → 3 | 5.114 | 242.5 | 0.0393 | 5.577 | 222.3 | 0.0292 | 5.505 | 225.2 | 0.0265 |
| 1 → 4 | 5.932 | 209.0 | 0.0004 | 5.896 | 210.3 | 0.0013 | 5.958 | 208.1 | 0.0010 |
| trans-perp | |||||||||
| 1 → 2 | 3.215 | 385.6 | 0.0010 | 3.478 | 356.4 | 0.0022 | 3.351 | 370.0 | 0.0016 |
| 1 → 3 | 5.212 | 237.9 | 0.0252 | 6.066 | 206.4 | 0.0340 | 5.637 | 219.9 | 0.0349 |
| 1 → 4 | 5.623 | 220.5 | 0.0017 | 7.381 | 156.5 | 0.0016 | 7.116 | 174.2 | 0.0010 |
| Transition | Cis–cis | Trans-perp | ||
|---|---|---|---|---|
| Orbitals | Contribution [%] | Orbitals | Contribution [%] | |
| 1 → 2 | 15 (13σ) → 17 (4π) | 71 | 16 (13 σ) → 17 (4π) | 71 |
| 1 → 3 | 16 (3π) → 17 (4π) | 68 | 15 (3π) → 17 (4π) | 66 |
| 15 (13 σ) → 18 (14σ) | 15 | 16 (13 σ) → 20 (16σ) | 15 | |
| 1 → 4 | 16 (3π) → 18 (14σ) | 70 | 14 (12 σ) → 17 (4π) | 66 |
However, the vertical transition energies and oscillator strengths can only give a crude approximation to the UV spectrum of the title molecule, because the molecule is not static but performs large-amplitude torsional motions. For a more realistic simulation of the UV spectrum, displacement of the nuclei from their equilibrium positions needs to be taken into account. Such a simulation was performed using the Newton-X package45 coupled to Gaussian G16.43 Within Newton-X, the photoabsorption cross-section is computed as
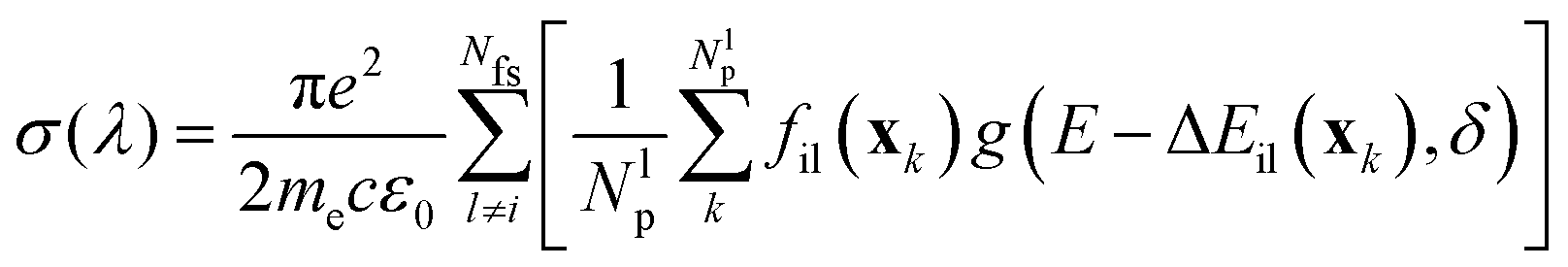 | (3) |
The computed UV spectra of the two isomers are presented in Fig. 2. Though the maxima of the signals are displaced from the experimental positions, which are 275 ± 35 nm for the cis–cis conformer and 220 nm for the trans-perp conformer, the spectra allow a useful analysis of the experimental results.
 | ||
| Fig. 2 UV spectrum and its decomposition in terms of electric states, computed at the B3LYP/AVTZ level. Upper: cis–cis and lower: trans-perp. Different axis scales were noted. As the experimental data were given in arbitrary units, they were scaled to match the maximum heights for each conformer, with scaling factors of 0.042 and 0.022 respectively. | ||
After irradiation of the argon matrix with a 308 nm source during one hour, while observing the IR spectrum, the signals of the cis–cis conformer became nearly unobservable, whereas those of the trans-perp conformer were still present. Our computed UV spectrum shows that radiation with a 308 nm laser reaches the first excited electronic state. This wavelength is close to the maximum of the signal for the cis–cis conformer, but is in the tail region of the signal for the trans-perp conformer, which therefore decomposes slower than the cis–cis conformer.
Looking now at the intense signals, we note that they are very broad, which is likely caused by torsional motion. To analyse this finding, we have computed, at the MCSCF/AVTZ level, the vertical excitation energies as a function of the two torsional angles, and they are shown in Fig. 3. The cis–cis conformer is located at the centre of the figure. When either of the two torsional modes, NOOH or ONOO, is excited, the molecule samples nuclear configurations at which the vertical transition energies towards the first excited state are red-shifted. In contrast, excitation energies towards the second and third electronic states appear blue-shifted by the NOOH mode and red-shifted by the ONOO mode. As a consequence, the maximum for excitation to the first excited state is found around 330 nm, whereas the vertical excitation value is 300 nm. The signals for excitations to the second and third excited states, are broadened, but not displaced strongly from the vertical excitation value due to the counteracting effects of the two torsional motions. In the case of the trans-perp conformer, located at τONOO ≈ ±180° and τNOOH ≈ ±90°, excitation to the first excited state is close to the vertical excitation energy, as the two torsional motions counteract each other. However, these motions largely broaden the signal. Concerning excitation to the second or third excited states, both torsional motions shift the maximum of the signals towards larger wavelengths.
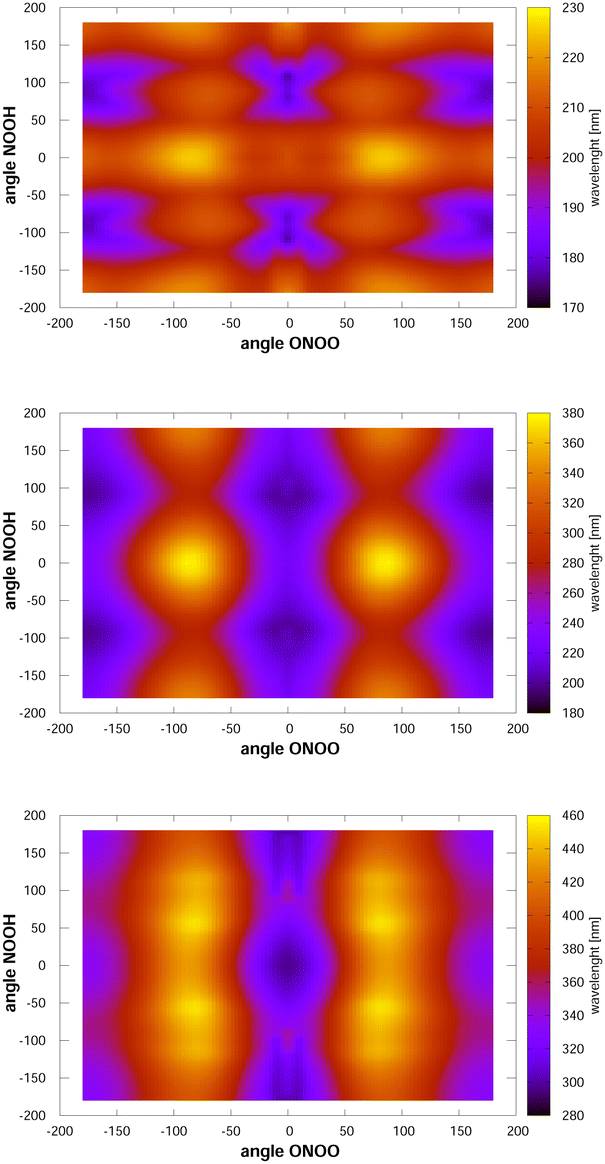 | ||
| Fig. 3 Vertical excitation energies between the ground state and the first (lower), second (middle) and third (upper) excited singlet states. Note that the configuration space is covered twice such that the triangles τNOOH ≥ τONOO and τNOOH ≤ τONOO represent duplicated configurations. The cis–cis conformer is located at the centre. Of the trans-perp conformer, which has symmetry C1, two enantiomeric forms exist, see the ESI.† They are at τNOOH ≈ 90° and τNOOH ≈ −90° with τONOO ≈ −180° using the first triangle. | ||
3 Photodecomposition
The thermal decomposition of HOONO on the electronic ground state surface was investigated by Dixon et al.11 HOONO can break up mainly in two ways, HOONO → HO + NO2 and HOONO → HO2 + NO with energies of ΔE = 6925 cm−1 and 9548 cm−1 with respect to the cis–cis conformer, though a third channel, HOONO → HNO + O2(a1Δg), may become accessible at much higher energies ΔE = 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 768 cm−1.
768 cm−1.
The aim of the present work is to understand the photodecomposition pathways of HOONO in the cis–cis and trans-perp configurations in the lowest three excited singlet states that were reached in W.-J. Lo and Y. P. Lee's15 experiment. The asymptotic energies can be obtained from combinations of the product energies according to their electronic states, which are reported in Table 3.
![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) and à states, are from Melnikov et al.48 All other data are from NIST49
and à states, are from Melnikov et al.48 All other data are from NIST49
| NO2 + OH channel | HO2 + NO channel | ||||||
|---|---|---|---|---|---|---|---|
| NO2 | OH | HO2 | NO | ||||
| 2A1 |
0 | X2Π | 0 | 2A′′ |
0 | X2Π | 0 |
| Ã2B2 | 10393 |
A2Σ+ | 32684.1 |
Ã2A′ | 7101 | A2Σ+ | 43965.7 |
![[B with combining tilde]](https://www.rsc.org/images/entities/char_0042_0303.gif) 2B1 2B1 |
14615 |
B2Σ+ | 69774 |
2A′′ |
48800 |
B2Π | 45910 |
The lowest dissociation channels including those from excited electronic states are thus:
 | (4) |
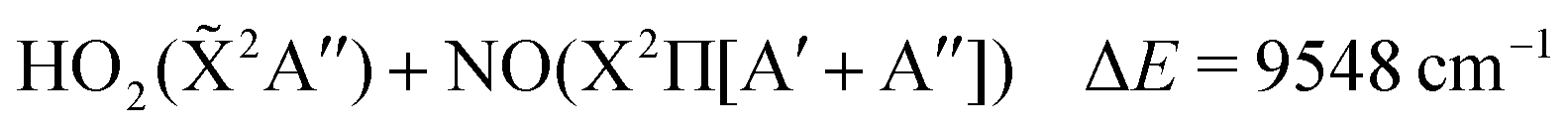 | (5) |
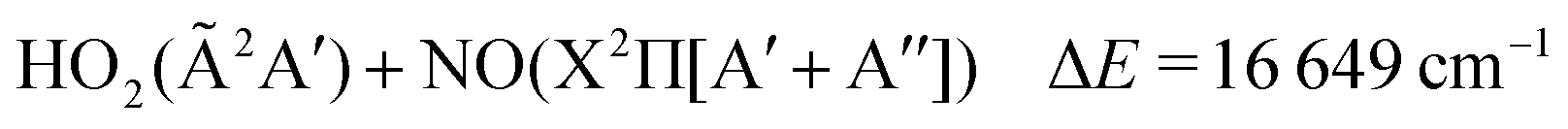 | (6) |
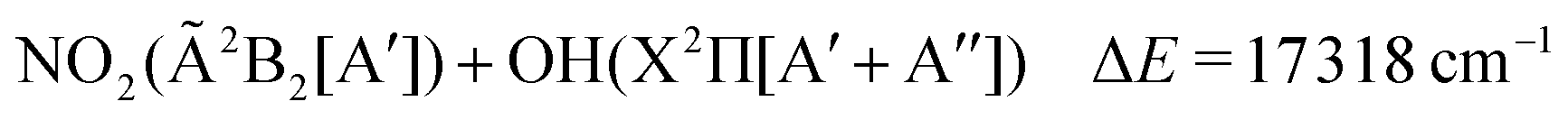 | (7) |
In the above expressions, the symmetry labels in the Cs point group are added in parentheses. By correlation, each combination of two fragments gives rise to one state of A′ symmetry and one state of A′′ symmetry of HOONO in the cis–cis configuration. The trans-perp conformer has symmetry C1 and thus all symmetry labels become A. Both singlet and triplet combinations are allowed. As a result, the electronic ground state and the first excited singlet state have common asymptotes, NO2(2A1) + OH(X2Π) and HO2(2A′′) + NO(X2Π), and the second and third excited electronic singlet states, NO2(Ã2B2) + OH(X2Π) and HO2(Ã2A′′) + NO(X2Π). Dissociation to OH or NO in their first excited states and the triatomics in their electronic ground states only occurs at much higher energy.
To investigate the photodecomposition process, we have run semiclassical trajectories with the Newton-X package45 starting at an excited singlet state. The initial configurations for a set of 100 trajectories were obtained from a Wigner distribution in phase space. For each of these trajectories, the potential energy surfaces and non-adiabatic coupling terms were computed on the fly. Finally, the hopping probability for passing from the current potential energy surface to any of the other surfaces is computed according to Tully's50 fewest switches surface hopping (FSSH) procedure. Decoherence correction with simplified decay of mixing51 was applied. Potential energy surfaces and coupling terms were obtained with the time-dependent density functional theory (TD-DFT) formalism as implemented in Gaussian G1643 and also at the ab initio CASSCF level using Molpro.42 For computational reasons, two different basis sets had to be used, both of them were of triple-zeta quality. In the Molpro calculations,§ a large CAS(16e,13o) was used together with the diffuse 6-311++G(3df,3pd) segmented basis set, which is approximately equivalent to the aug-cc-VTZ basis. However, the latter basis set cannot be handled by the Cadpac gradient programme within Molpro to compute state-averaged MCSCF gradients. In the TD-DFT calculations, the minimally augmented maug-cc-pVTZ basis set was chosen. This basis set is recommended for DFT calculations53 as it yields results of almost aug-cc-pVTZ quality at a significantly lower computational cost.
Dissociation of peroxynitrous acid in two fragments of doublet spin multiplicity is intrinsically a multi-reference problem, and thus the MCSCF approach is well suited. However, trajectory calculations at the MCSCF level become rapidly prohibitive in terms of CPU time, in particular if four electronic states are considered simultaneously. Fortunately, it is not necessary to follow the trajectories until “complete” separation of the fragments in order to identify the final products. In the present work, we have found that up to intermediate distances the open-shell singlet system is also described well by density functional theory. This can be seen from the Mulliken charges and spin densities presented in Table 4 for one exemplary product configuration, Fig. 4, of each of the two dissociation channels.
| Channel | Property | H(1) | O(2) | O(3) | N(4) | O(5) |
|---|---|---|---|---|---|---|
| H(1)O(2)O(3) + N(4)O(5) | SD | −0.01 | 0.23 | 0.78 | −0.71 | −0.29 |
| Q | 0.25 | −0.09 | −0.16 | 0.08 | −0.08 | |
| H(1)O(2) + O(3)N(4)O(5) | SD | 0.02 | −1.02 | 0.27 | 0.45 | 0.28 |
| Q | 0.27 | −0.27 | −0.24 | 0.55 | −0.31 |
 | ||
| Fig. 4 Typical product configurations at the end of the trajectories. | ||
After some experimentation, we chose the CAM-B3LYP functional, developed by Tanai et al.,54 for better description of charge-transfer processes. For our problem, CAM-B3LYP improves the stability of the trajectory propagation compared to B3LYP.
The trajectories were run over tmax = 100 fs with a propagation step of tmax = 0.25 fs. Visual analysis of the molecular structures along the trajectories did not show a single case in which the fragments would rejoin. On this basis, we attributed the fragments to one of the channels, NO2 + OH or HO2 + NO, depending whether the O–O or O–N distances were increased by at least 25% of their initial values. Trajectories that did not satisfy these conditions were analysed manually. The product distributions, or quantum yields, are collected in Table 5. Error bounds were obtained by a statistical analysis, and the results are reported as 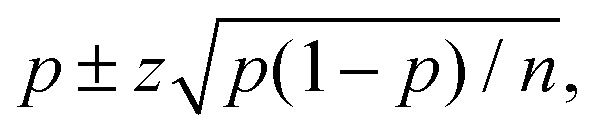 where p denotes the fraction of trajectories leading to a particular channel and n is the total number of trajectories. z = 1.96 for the 95% confidence interval. A small difference was found in the product distributions for the cis–cis conformer following excitation to the first excited state. With the larger CAS(16e,13o), a larger yield of NO2 + OH is found. The two additional molecular orbitals in the active space, when compared with the smaller CAS(16e,11o), orbitals 20 and 21, both have antibonding O–O characters. Apparently, their inclusion facilitates the O–O bond rupture. The TD-DFT calculation gives the same product distribution as the small CAS. For other cases, the results are practically the same. With respect to the third excited state, only the TDDFT method was computationally feasible.
where p denotes the fraction of trajectories leading to a particular channel and n is the total number of trajectories. z = 1.96 for the 95% confidence interval. A small difference was found in the product distributions for the cis–cis conformer following excitation to the first excited state. With the larger CAS(16e,13o), a larger yield of NO2 + OH is found. The two additional molecular orbitals in the active space, when compared with the smaller CAS(16e,11o), orbitals 20 and 21, both have antibonding O–O characters. Apparently, their inclusion facilitates the O–O bond rupture. The TD-DFT calculation gives the same product distribution as the small CAS. For other cases, the results are practically the same. With respect to the third excited state, only the TDDFT method was computationally feasible.
| Exc. state | CAM-B3LYP | MCSCF | ||
|---|---|---|---|---|
| HO2 + NO | NO2 + OH | HO2 + NO | NO2 + OH | |
| Cis–cis | ||||
| 1 | 0.95 ± 0.04 | 0.05 ± 0.04 | 0.71 ± 0.09 | 0.29 ± 0.09 |
| 0.92 ± 0.05 | 0.08 ± 0.05 | |||
| 2 | 0.66 ± 0.09 | 0.34 ± 0.09 | 0.57 ± 0.10 | 0.43 ± 0.10 |
| 3 | 0.32 ± 0.09 | 0.68 ± 0.09 | — | — |
| Trans-perp | ||||
| 1 | 1.00 | 0.00 | 1.00 | 0.00 |
| 1.00 | 0.00 | |||
| 2 | 0.84 ± 0.07 | 0.16 ± 0.07 | 0.87 ± 0.06 | 0.13 ± 0.06 |
| 3 | 0.56 ± 0.10 | 0.44 ± 0.10 | — | — |
4 Rate constant for actinic photodecomposition
Photo-decomposition of peroxynitrous acid may occur in the troposphere induced by absorption of sunlight. The unimolecular decay constant with the formation of a product A, JA, can be computed as | (8) |
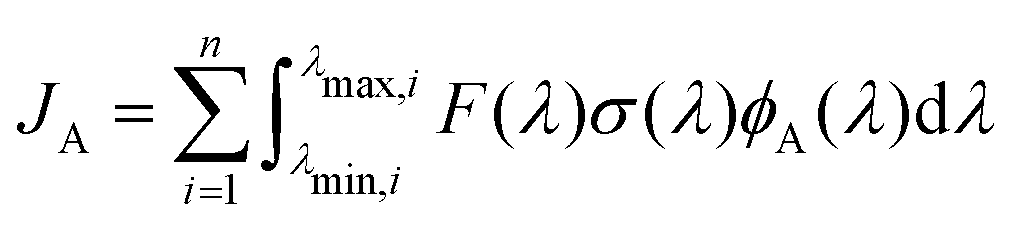 | (9) |
| Exc. state | CAM-B3LYP | MCSCF | ||
|---|---|---|---|---|
| HO2 + NO | NO2 + OH | HO2 + NO | NO2 + OH | |
| Cis–cis | ||||
| 1 | 3.89 × 10−4 | 8.13 × 10−7 | 4.93 × 10−4 | 1.26 × 10−4 |
| Trans-perp | ||||
| 1 | 1.26 × 10−3 | — | 1.14 × 10−3 | — |
In thermal equilibrium at room temperature, 99.9% of peroxynitrous acid is found in the cis–cis form. The trans-perp configuration can therefore be neglected in atmospheric modelling. The photolysis rate constant J, adding up contributions from the two decay channels, i.e. J = JHO2+NO + JNO2+OH, is about J ≈ 6 × 10−4 s−1 at the MCSCF level. This rate constant may be compared with the unimolecular thermal decay rate constant on the electronic ground state into OH + NO2, for which Golden, Barker and Lohr7 report kuni ≈ 10−6 s−1 in the region around the tropopause at about 10 km of altitude, with T ≈ 220 K and p ≈ 0.26 bar. Photolysis is thus the principal atmospheric decomposition mechanism of peroxynitrous acid and must be included in atmospheric modelling.
5 Conclusions
In the present work, we have reanalysed the experimental UV spectrum of peroxynitrous acid recorded by W.-J. Lo and Y. P. Lee15 forty years ago. Their broad signals result from excitations to the second excited singlet state, with smaller, overlapping contributions from the third electronic state. Excitation to the first excited state appears in the long-wavelength tail region and was not resolved in the experiment. Since the excited electronic states are repulsive, the molecule will disintegrate upon UV absorption, and the decomposition mechanism was investigated in the present work. In contrast to thermal decomposition, which mainly yields NO2 + OH, the principal photolysis products are HO2 + NO. In the earth's atmosphere, only excitation to the first excited singlet state occurs, as higher-energy solar radiation is absorbed in the ozone layer. The rate constant for atmospheric photodecomposition, which is urgently needed for use in atmospherical models,7,10,41 was computed here for the first time. Our results show that under conditions near the tropopause and above, photodecomposition is significantly more rapid than thermal decomposition, J ≈ 6 × 10−4 s−1, whereas k ≈ 10−6 s−1 near the tropopause.7Author contributions
All authors contributed equally to this work. W. C. is a PhD student supervised by the other three authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Alex Brown and Zhibo Wang for kindly providing their Molpro–Newton-X interface, and Basile Curchod and Mario Barbatti for enlightening discussions. We are grateful for financial support obtained from the “PHC Utique” programme of the French Ministry of Foreign Affairs and Ministry of Higher Education and Research and the Tunisian Ministry of Higher Education and Scientific Research, project number 18G1302, and from the CNRS, IEA project number 317871. W. C. acknowledges a PhD studentship from the Tunisian Ministry of Higher Education and Scientific Research. N. D. acknowledges financial support from the French Consulate in Tunis and from the CNRS. Supercomputer time was provided by the ROMEO HPC Center at the University of Reims Champagne-Ardenne and by CRIANN (Centre des Ressources Informatiques et Applications Numériques de Normandie).Notes and references
- B. Finlayson-Pitts and J. James Pitts, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, 2000 Search PubMed.
- P. J. Crutzen, Q. J. R. Meteorol. Soc., 1970, 96, 320–325 CrossRef.
- C. F. Williams, S. K. Pogrebnya and D. C. Clary, J. Chem. Phys., 2007, 126, 154321 CrossRef PubMed.
- J. Troe, J. Phys. Chem. A, 2012, 116, 6387–6393 CrossRef CAS PubMed.
- D. Amedro, M. Berasategui, A. J. C. Bunkan, A. Pozzer, J. Lelieveld and J. N. Crowley, Atmos. Chem. Phys., 2020, 20, 3091–3105 CrossRef CAS.
- B. D. Bean, A. K. Mollner, S. A. Nizkorodov, G. Nair, M. Okumura, S. P. Sander, K. A. Peterson and J. S. Francisco, J. Phys. Chem. A, 2003, 107, 6974–6985 CrossRef CAS.
- D. M. Golden, J. R. Barker and L. L. Lohr, J. Phys. Chem. A, 2003, 107, 11057–11071 CrossRef CAS.
- H. Hippler, S. Nasterlack and F. Striebel, Phys. Chem. Chem. Phys., 2002, 4, 2959–2964 RSC.
- A. K. Mollner, S. Valluvadasan, L. Feng, M. K. Sprague, M. Okumura, D. B. Milligan, W. J. Bloss, S. P. Sander, P. T. Martien, R. A. Harley, A. B. McCoy and W. P. L. Carter, Science, 2010, 330, 646–649 CrossRef CAS PubMed.
- F. A. F. Winiberg, K. Zuraski, Y. Liu, S. P. Sander and C. J. Percival, J. Phys. Chem. A, 2020, 124, 10121–10131 CrossRef CAS PubMed.
- D. A. Dixon, D. Feller, C.-G. Zhan and J. S. Francisco, J. Phys. Chem. A, 2002, 106, 3191–3196 CrossRef CAS.
- Y. Zhao, K. N. Houk and L. P. Olson, J. Phys. Chem. A, 2004, 108, 5864–5871 CrossRef CAS.
- D. J. Benton and P. Moore, J. Chem. Soc. A, 1970, 3179–3182 RSC.
- B. M. Cheng, J. W. Lee and Y. P. Lee, J. Phys. Chem., 1991, 95, 2814–2817 CrossRef CAS.
- W. Lo and Y. P. Lee, J. Chem. Phys., 1994, 101, 5494–5499 CrossRef.
- W.-J. Lo and Y.-P. Lee, Chem. Phys. Lett., 1994, 229, 357–361 CrossRef CAS.
- M. P. McGrath, M. M. Francl, F. S. Rowland and W. J. Hehre, J. Phys. Chem., 1988, 92, 5352–5357 CrossRef CAS.
- X. Zhang, M. R. Nimlos, G. B. Ellison, M. E. Varner and J. F. Stanton, J. Chem. Phys., 2006, 124, 084305 CrossRef PubMed.
- X. Zhang, M. R. Nimlos, G. B. Ellison, M. E. Varner and J. F. Stanton, J. Chem. Phys., 2007, 126, 174308 CrossRef PubMed.
- N. M. Donahue, R. Mohrschladt, T. J. Dransfield, J. G. Anderson and M. K. Dubey, J. Phys. Chem. A, 2001, 105, 1515–1520 CrossRef CAS.
- S. A. Nizkorodov and P. O. Wennberg, J. Phys. Chem. A, 2002, 106, 855–859 CrossRef CAS.
- I. B. Pollack, I. M. Konen, E. X. J. Li and M. I. Lester, J. Chem. Phys., 2003, 119, 9981–9984 CrossRef CAS.
- J. L. Fry, S. A. Nizkorodov, M. Okumura, C. M. Roehl, J. S. Francisco and P. O. Wennberg, J. Chem. Phys., 2004, 121, 1432–1448 CrossRef CAS PubMed.
- E. X. J. Li, I. M. Konen, M. I. Lester and A. B. McCoy, J. Phys. Chem. A, 2006, 110, 5607–5612 CrossRef CAS PubMed.
- B. J. Drouin, J. L. Fry and C. E. Miller, J. Chem. Phys., 2004, 120, 5505–5508 CrossRef CAS PubMed.
- J. L. Fry, B. J. Drouin and C. E. Miller, J. Chem. Phys., 2006, 124, 084304 CrossRef PubMed.
- M. P. McGrath and F. S. Rowland, J. Phys. Chem., 1994, 98, 1061–1067 CrossRef CAS.
- M. P. McGrath and F. S. Rowland, J. Chem. Phys., 2005, 122, 134312 CrossRef.
- R. Sumathi and S. D. Peyerimhoff, J. Chem. Phys., 1997, 107, 1872–1880 CrossRef CAS.
- R. S. Zhu and M. C. Lin, J. Chem. Phys., 2003, 119, 10667–10677 CrossRef CAS.
- R. D. Bach, O. Dmitrenko and C. M. Estévez, J. Am. Chem. Soc., 2003, 125, 16204–16205 CrossRef CAS PubMed.
- H. W. Jin, Z. Z. Wang, Q. S. Li and X. R. Huang, J. Mol. Struct.: THEOCHEM, 2003, 624, 115–121 CrossRef CAS.
- C. Chen, B. C. Shepler, B. J. Braams and J. M. Bowman, J. Chem. Phys., 2007, 127, 104310 CrossRef PubMed.
- P. Szakács, J. Csontos, S. Das and M. Kállay, J. Phys. Chem. A, 2011, 115, 3144–3153 CrossRef PubMed.
- J. Matthews, A. Sinha and J. S. Francisco, J. Chem. Phys., 2004, 120, 10543–10553 CrossRef CAS PubMed.
- I. M. Konen, I. B. Pollack, E. X. J. Li, M. I. Lester, M. E. Varner and J. F. Stanton, J. Chem. Phys., 2005, 122, 094320 CrossRef PubMed.
- D. P. Schofield, H. G. Kjaergaard, J. Matthews and A. Sinha, J. Chem. Phys., 2005, 123, 134318 CrossRef PubMed.
- M. A. Boyer and A. B. McCoy, J. Chem. Phys., 2022, 157, 164113 CrossRef CAS PubMed.
- A. B. McCoy, M. K. Sprague and M. Okumura, J. Phys. Chem. A, 2010, 114, 1324–1333 CrossRef CAS PubMed.
- Y. Li and J. S. Francisco, J. Chem. Phys., 2000, 113, 7976–7981 CrossRef CAS.
- D. M. Golden and G. P. Smith, J. Phys. Chem. A, 2000, 104, 3991–3997 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, et al., MOLPRO, version 2021.1, a package of ab initio programs, 2021, see https://www.molpro.net Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, et al., Gaussian-16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- S. Berski, Z. Latajka and A. J. Gordon, J. Comput. Chem., 2011, 32, 1528–1540 CrossRef CAS PubMed.
- M. Barbatti, M. Ruckenbauer, F. Plasser, J. Pittner, G. Granucci, M. Persico and H. Lischka, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 26–33 CAS.
- M. Barbatti, A. J. A. Aquino and H. Lischka, Phys. Chem. Chem. Phys., 2010, 12, 4959–4967 RSC.
- S. Ndengué, E. Quintas-Sánchez, R. Dawes and D. Osborn, J. Phys. Chem. A, 2021, 125, 5519–5533 CrossRef.
- V. V. Melnikov, P. Jensen and T. Hirano, J. Chem. Phys., 2009, 130, 224105 CrossRef PubMed.
- P. Linstrom and W. Mallard, Proceedings of the 10th International Chemical Information Conference and Exhibition, Nimes, FR, 1998.
- J. C. Tully, J. Chem. Phys., 1990, 93, 1061–1071 CrossRef CAS.
- G. Granucci and M. Persico, J. Chem. Phys., 2007, 126, 134114 CrossRef PubMed.
- Z. Wang, Master thesis, University of Alberta, Department of Chemistry, Supervisor Alex Brown, 2019 Search PubMed.
- E. Papajak, H. R. Leverentz, J. Zheng and D. G. Truhlar, J. Chem. Theory Comput., 2009, 5, 1197–1202 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Figure of the trans-perp enantiomers, plot of orbitals, and parameters of Newton-X calculations. See DOI: https://doi.org/10.1039/d3cp04580c |
| ‡ Figures of the orbitals are provided as supplementary information. |
| § A Molpro-Newton-X interface was provided by Alex Brown and Zhibo Wang.52 |
| ¶ https://www.acom.ucar.edu/Models/TUV/Interactive_TUV/ |
| This journal is © the Owner Societies 2024 |