
A magnetically reusable Ce-MOF/GO/Fe3O4 composite for effective photocatalytic degradation of chlortetracycline†
Yuting
Bai
 ac,
Derek
Hao
e,
Sisi
Feng
*ab,
Liping
Lu
*a and
Qi
Wang
*d
ac,
Derek
Hao
e,
Sisi
Feng
*ab,
Liping
Lu
*a and
Qi
Wang
*d
aInstitute of Molecular Science, Key Laboratory of Chemical Biology and Molecular Engineering of the Education Ministry, Shanxi University, Taiyuan, Shanxi 030006, P. R. China. E-mail: luliping@sxu.edu.cn
bKey Laboratory of Materials for Energy Conversion and Storage of Shanxi Province, Shanxi University, Taiyuan, Shanxi 030006, P. R. China. E-mail: ssfeng@sxu.edu.cn
cDepartment of Energy Chemistry and Materials Engineering, Shanxi Institute of Energy, Jinzhong, Shanxi 030600, China
dSchool of Environmental Science and Engineering, Zhejiang Gongshang University, Hangzhou 310018, China. E-mail: wangqi8327@zjgsu.edu.cn
eCentre for Technology in Water and Wastewater (CTWW), School of Civil and Environmental Engineering, University of Technology Sydney (UTS), Sydney, NSW 2007, Australia
First published on 26th December 2023
Abstract
Herein, we report a novel 1/GO/Fe3O4 photocatalyst, comprising Ce(BTB)(H2O) (MOF-1, H3BTB = 1,3,5-benzenetrisbenzoic acid), graphene oxide (GO), and iron oxide (Fe3O4) for photocatalytic degradation of chlortetracycline (CTC). This design enables the effective transfer of electrons from the MOF to GO, thereby reducing the photoelectron–hole recombination rate. Therefore, the optimized 1/GO/Fe3O4 photocatalyst with H2O2 shows the highest photocatalytic activity toward CTC. The kinetic constant is 5.4 times that in the system of MOF-1 and hydrogen peroxide, which usually acted as efficient electron acceptors to improve the photocatalytic performance of MOFs. More importantly, light absorption is extended from the ultraviolet to the visible region. Furthermore, 1/GO/Fe3O4 can be quickly recycled under an applied magnetic field and displays outstanding stability and reusability. According to the radical trapping experiments and electron paramagnetic resonance results, hydroxyl radicals, superoxide radicals, and holes all contribute to excellent photocatalytic activity. The possible catalytic mechanism of 1/GO/Fe3O4 is tentatively proposed. This work aims to explore the synergistic effect between metal–organic frameworks (MOFs) and GO, and provide a theoretical basis for MOF-based composites to remove antibiotic contaminants in the environment.
Introduction
Over the past few decades, antibiotics have received increasing attention from the public and scientists as emerging contaminants in the natural water environment.1 Long-term or excessive use of antibiotics may bring them into the environment through direct or indirect means, causing environmental pollution and endangering the health of organisms.2,3 Chlortetracycline (CTC), as a kind of tetracycline antibiotic, is widely used for the disease treatment of animals and humans because of its easy preparation, low price, convenient use, and broad-spectrum antibacterial advantages.4,5 However, only part of the CTC can be metabolized by animals and humans, and the unabsorbed residues will be released into the environment.6 It is difficult to eliminate antibiotics via traditional wastewater treatment technology because of their stable chemical structure and resistance to biodegradation. Consequently, new techniques are urgently needed to effectively remove residual CTC from water.In the past few decades, photocatalysis has been approved as a promising green approach for the removal of antibiotics due to its simple operation, low energy consumption, and no secondary contamination. However, it takes a long time (>200 min) to achieve complete removal of CTC as per some studies.7,8 Therefore, new better photocatalysts are needed to be developed to overcome this barrier. Metal–organic frameworks (MOFs) are chosen as target photocatalytic materials due to their semiconductor-like behavior under light irradiation. Compared with some traditional photocatalysts, MOFs have risen rapidly owing to their three-dimensional porous structure and adjustable electron transmission channels under illumination, which has set off a research frontier in the use of MOFs as photocatalysts.9 The porous structures of MOFs can provide abundant catalytic active sites and have more tight interfacial contact with pollutants, which is beneficial for realizing short-distance to trigger the a catalytic reaction. In addition, the two basic structural units (metal centers and organic ligands) of MOFs have diversified combinations and can be modified, indicating the well-regulated electronic structures. Furthermore, H2O2-involved photocatalysis has been involved in MOF-mediated pollutant degradation, which produces highly oxygen-reactive species through the reaction between MOFs and H2O2, exhibiting improved photocatalytic properties compared to pure MOFs.10
Despite the effectiveness of photocatalysis, it is still not satisfactory due to inefficient visible-light utilization and limited conductivity, so it requires further improvement. Graphene oxide (GO), as one of the ideal electron accepter candidates, has attracted scientific interest due to its superior conductivity and unique optical properties. The combination of MOFs with GO has been demonstrated to be one of the most effective ways to improve the catalytic efficiency of MOFs.11 Benefiting from the synergistic effects between MOFs and GO, MOFs/GO composites can increase the separation efficiency of photogenerated carriers and significantly improve the utilization rate of visible light, which can lead to significant improvement in photocatalytic performance.12,13 Recent studies have indicated that the MOFs/GO composites can degrade antibiotics and display outstanding photocatalytic performance. For example, UiO-67/CdS/rGO was prepared for the degradation of ofloxacin (OFL) under simulated sunlight.14 The GO/NH2-MIL-125(Ti) hybrid showed improved photocatalytic performance towards the oxidation of gaseous pollutants (NOx and acetaldehyde) compared with unmodified NH2-MIL-125(Ti).15 A novel γ-Fe2O3/MIL-53(Fe)/GO composite was also fabricated, which exhibited outstanding photocatalytic performance for the degradation of norfloxacin. The kinetic constant was 4.5 times greater than that of pristine MIL-53 (Fe) and 1.4-fold higher than that in the system with H2O2 and MIL-53 (Fe).16 However, to date, there are a few reports about the elimination of CTC under visible light by MOF-based photocatalysts.
In addition, recycling powder catalysts is another problem for future practical applications. The loss of photocatalysts may happen and bring new secondary pollution. Therefore, developing magnetic catalysts is expected to solve this problem. In this field, some composite photocatalysts, such as ZnIn2S4/CoFe2O4,17 S-scheme heterojunction 3D/2D ZnIn2S4/BiFeO3,18 AgBr/BiOBr/NiFe2O4,19 and Z-scheme heterojunction BiFeO3/ZnFe2O4,20 have been successfully synthesized and exhibited superior photocatalytic activity and excellent magnetic recovery properties.
Herein, a novel 1/GO/Fe3O4 composite was successfully prepared by combining MOF, GO, and Fe3O4 through reflux and ultrasonic procedures. The MOF, Ce(BTB)(H2O) (MOF-1) (H3BTB = 1,3,5-benzenetrisbenzoic acid), is a three-dimensional framework extended by BTB3− ligands with rigid phenyl rings, showing two types of channels with cross-sections of ca. 4.6 Å and 5.6 Å, respectively.21MOF-1 has drawn much attention owing to its high surface area and unique ultramicroporous channels. The obtained novel 1/GO/Fe3O4 composite in this work was applied for the photocatalytic degradation of CTC. Additionally, the effects of several parameters, including the content of GO, catalyst dosage, and H2O2 concentration, on the photocatalytic performance were intensively evaluated. The recyclability of 1/GO/Fe3O4 was also examined. Moreover, the photocatalytic mechanism for CTC degradation was also proposed. To the best of our knowledge, this is the first time that an ultramicroporous MOF-based composite was prepared to degrade CTC. This work opens a new route to prepare novel high-performance MOF-based photocatalysts for water purification and environmental remediation.
Experimental section
The detailed preparation of MOF-1, GO, Fe3O4, and 1/GO/Fe3O4, materials characterization, and evaluation of photocatalytic activity are provided in the ESI† (see the ESI†).Results and discussion
Components and structure
The PXRD patterns of the as-prepared GO, Fe3O4, MOF-1, and 1/GO/Fe3O4 are presented in Fig. 1a and Fig. S1 (ESI†). In the PXRD pattern of GO, there is a robust characteristic peak at 11.1° with a corresponding layer spacing of 0.8 nm.22 It is worth noting that no characteristic diffraction peaks of GO can be detected in the 1/GO/Fe3O4 composite owing to the small amount of GO (9%). Additionally, the PXRD pattern of the 1/GO/Fe3O4 composite shows almost the same characteristic peaks as those of MOF-1, which demonstrates that the incorporation of GO will not change the crystalline phase of MOF-1 and its framework structure is maintained. The diffraction peaks of Fe3O4 can also be found in the 1/GO/Fe3O4 composite, which preliminarily proves the successful preparation of the 1/GO/Fe3O4 composite.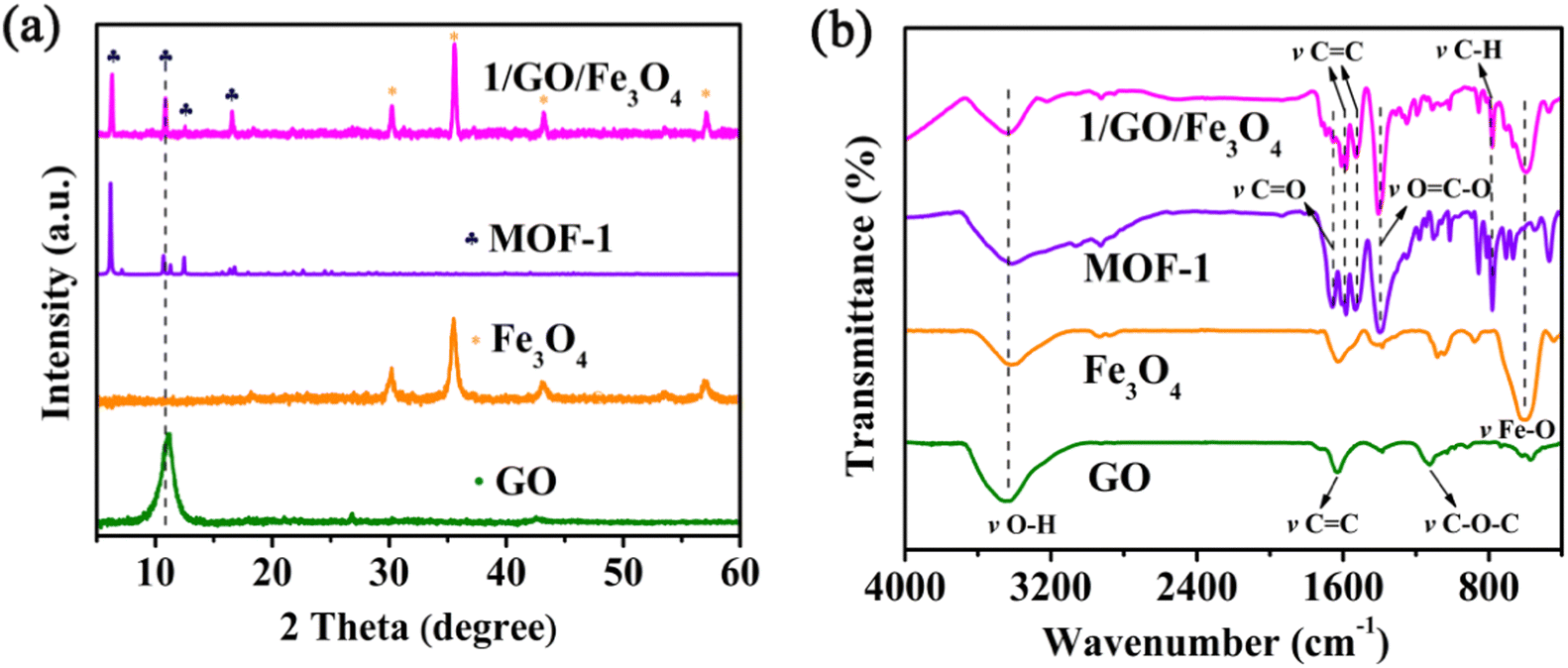 | ||
| Fig. 1 PXRD patterns (a) and FTIR (b) spectra of GO, Fe3O4, MOF-1, and 1/GO/Fe3O4. | ||
To further confirm the existence of GO, Fe3O4, and MOF-1 in the 1/GO/Fe3O4 composite, FTIR spectra were recorded (Fig. 1b). For pure GO, the broadband at 3447 cm−1 corresponds to the O–H stretching vibration, and the bands at 1132, 1382, 1626, and 1734 cm−1 are due to the C–O–C, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and CO stretching vibrations, respectively.23 A significant peak for Fe3O4 can be observed at 597 cm−1, which is associated with Fe–O stretching vibrations. Furthermore, the FTIR spectrum of the 1/GO/Fe3O4 composite displays no change in the characteristic peaks of pure MOF-1, which manifests the incorporation of GO and Fe3O4 has no influence on the original structure of MOF-1 and agrees well with the PXRD patterns. A new peak appeared at about 590 cm−1 in the 1/GO/Fe3O4 owing to the Fe–O stretching vibrations in the Fe3O4. This verifies that MOF-1, GO, and Fe3O4 have been successfully combined.
C, and CO stretching vibrations, respectively.23 A significant peak for Fe3O4 can be observed at 597 cm−1, which is associated with Fe–O stretching vibrations. Furthermore, the FTIR spectrum of the 1/GO/Fe3O4 composite displays no change in the characteristic peaks of pure MOF-1, which manifests the incorporation of GO and Fe3O4 has no influence on the original structure of MOF-1 and agrees well with the PXRD patterns. A new peak appeared at about 590 cm−1 in the 1/GO/Fe3O4 owing to the Fe–O stretching vibrations in the Fe3O4. This verifies that MOF-1, GO, and Fe3O4 have been successfully combined.
Morphological studies of MOF-1, GO, Fe3O4, 1/GO, and 1/GO/Fe3O4 were also conducted through SEM analysis. In Fig. S2a (ESI†), MOF-1 shows a prominent block structure. The image presented in Fig. S2b (ESI†) displays the GO layers with wrinkles.24 Fig. S2c (ESI†) is the morphology of pristine Fe3O4, which consists of sphere-like particles with a diameter of 400 nm. After combining MOF-1 with GO, the irregular MOF-1 deposition on the surface of GO can be observed in Fig. S2d (ESI†), demonstrating the formation of a 1/GO composite. As presented in Fig. S2e and f (ESI†), the Fe3O4 particles are uniformly distributed over the surface of GO, which enables the catalyst to be quickly and conveniently recovered by applying a magnetic field. Also, the energy dispersive X-ray (EDX) elemental mapping images (Fig. S3a–e, ESI†) show Ce, C, O, and Fe elements in the selected area of the composite, and the distribution is uniform.
To further ascertain the elemental composition and chemical state of the 1/GO/Fe3O4 composite, X-ray photoelectron spectroscopy (XPS) was performed. The XPS survey spectra verify the existence of Ce, Fe, O, and C elements in the 1/GO/Fe3O4 composite (Fig. 2a), which matches well with the results of EDX. The high-resolution XPS spectra of Ce in MOF-1 and 1/GO/Fe3O4 are given in Fig. 2b. The Ce 3d XPS spectrum of MOF-1 is composed of four components, which can be attributed to the existence of 904.3/900.5 eV of Ce(III) 3d3/2 and 885.7/881.9 eV of Ce(III) 3d5/2, whereas the XPS spectrum of the 1/GO/Fe3O4 sample contains peaks of 904.5/900.6 eV of Ce(III) 3d3/2 and 886.1/882.2 eV of Ce(III) 3d5/2.25 Peaks corresponding to Ce(III) 3d3/2 and Ce(III) 3d5/2 shift to higher binding energies compared to that of MOF-1 due to the incorporation of GO. The shifts of these binding energies, thanks to the change of electron density when MOF-1, GO, and Fe3O4 are assembled into the 1/GO/Fe3O4 composite, demonstrate that the interface between them is formed by internal interaction. As is well known, a positive shift of the binding energy represents a decrease in the electron density.26 So, it can be inferred that the photogenerated electrons transfer from MOF-1 to GO in the 1/GO/Fe3O4 composite, which is beneficial for constructing the 1/GO/Fe3O4 composite, facilitating the separation of photogenerated carriers and the improvement of photocatalytic performance. Fig. 2c presents the high-resolution XPS spectra corresponding to the Fe 2p of both Fe3O4 and 1/GO/Fe3O4 samples. The Fe 2p XPS spectrum of Fe3O4 can be divided into six peaks at 731.0 eV of satellite Fe(II) 2p1/2, 726.5 eV of Fe(III) 2p1/2, 723.0 eV of Fe(II) 2p1/2, 718.2 eV of satellite Fe(II) 2p3/2, 713.7 eV of Fe(III) 2p3/2, and 709.9 eV of Fe(II) 2p3/2, respectively. Another six peaks of Fe 2p are found in 1/GO/Fe3O4 at 732.9 eV of satellite Fe(II) 2p1/2, 727.5 eV of Fe(III) 2p1/2, 724.0 eV of Fe(II) 2p1/2, 719.1 eV of satellite Fe(II) 2p3/2, 714.5 eV of Fe(III) 2p3/2, and 710.9 eV of Fe(II) 2p3/2, respectively.27 There is a positive shift in the binding energy of the 1/GO/Fe3O4 composite compared to pure Fe3O4, confirming the successful incorporation of Fe3O4 into the composite. The O 1s XPS spectrum of GO is presented in Fig. S4 (ESI†). The O 1s XPS spectrum of MOF-1 can be divided into three bonds with the binding energies of 532.4, 531.8, and 531.1 eV, originating from C–O, CO, and Ce–O, respectively.28 The O 1s spectrum of GO is composed of four components corresponding to C–O–C (533.4 eV), C–OH (532.8 eV), CO (532.2 eV), and O–CO (531.3 eV) groups, which further confirms that oxygen-containing functional groups are present in GO. The oxygen-containing functional groups can serve as reaction sites and interaction sites with MOF-1 and Fe3O4 nanostructures. The peaks due to O–H and Fe–O bonds are observed at 530.2 and 529.0 eV, respectively, in the O 1s XPS spectrum of Fe3O4. In the XPS spectrum of 1/GO/Fe3O4, the peaks due to C–O–C, C–OH, CO, O–CO, Ce–O, and Fe–O appeared at binding energies of 533.3, 532.7, 532.1, 531.5, 530.9, and 530.1 eV, which further confirms the presence of MOF-1, GO, and Fe3O4 in the 1/GO/Fe3O4 composite. So, the XPS analysis of all samples proved the successful formation of the 1/GO/Fe3O4 composite.
 | ||
| Fig. 2 XPS survey spectra of MOF-1, GO, Fe3O4, and 1/GO/Fe3O4 (a). The high-resolution XPS spectra of Ce 3d (b), and Fe 2p (c). | ||
The UV-vis diffuse reflectance spectra (UV-Vis-DRS) of synthesized MOF-1 and 1/GO/Fe3O4 composite were investigated to evaluate the optical properties (Fig. 3). The optical absorption intensity of the 1/GO/Fe3O4 composite in the region of 230–850 nm is much stronger than the pristine MOF-1, indicating that the incorporation of GO can improve the optical absorption of MOF-1.29,30 Also, 1/GO/Fe3O4 can be excited by visible light owing to the formation of Ce–O–C bonds between MOF-1 and GO. The reason is that the oxygenated functional groups are distributed on the surface of GO, which acts as anchoring sites for MOF-1.31 After incorporating black GO, the 1/GO/Fe3O4 composites become darker with more visible light absorption than pure MOF-1, which correspond to a previous report.32 This result combined with XPS characterization results further manifests the successful preparation of the 1/GO/Fe3O4 composite.
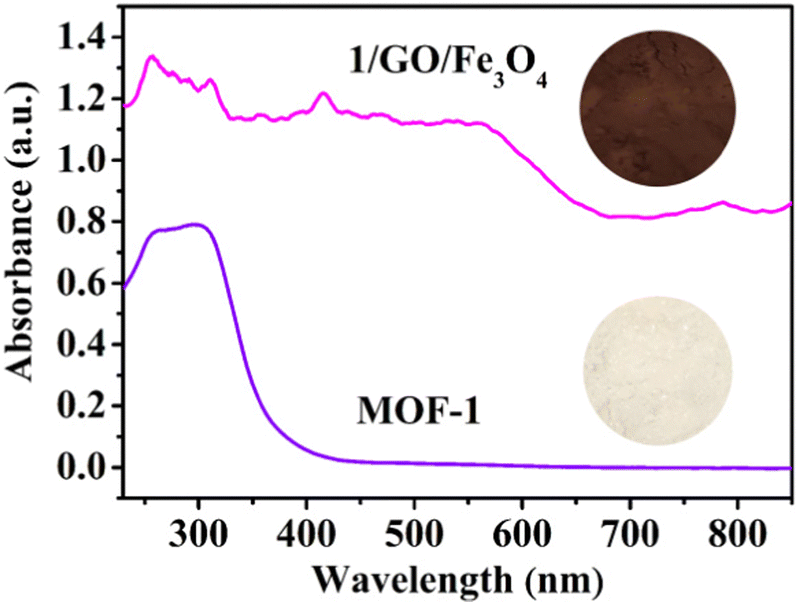 | ||
| Fig. 3 UV-Vis DRS spectra of MOF-1 and 1/GO/Fe3O4. | ||
Photocatalytic performance
The photocatalytic performance of different catalysts towards the degradation of CTC under visible light irradiation was assessed and is presented in Fig. 4a. Without the addition of the catalyst, the photocatalytic reaction did not proceed in 180 min. However, a small quantity of CTC was degraded when H2O2 was present, which may be attributed to the photolysis of H2O2 promoting the formation of reactive ˙OH. Interestingly, the degradation efficiency of CTC in the presence of MOF-1 and H2O2 was significantly higher than that of pristine MOF-1. Since the Ce cation with variable chemical valences (III and IV) can boost the decomposition of H2O2 to produce ˙OH via electron transfer, the photocatalytic performance was improved.33 We also investigated the photocatalytic performance over GO before and after the addition of H2O2, which showed weak photocatalytic activity for CTC. However, the photocatalytic performance could be boosted obviously by the incorporation of GO. And 88.0% degradation of CTC was achieved within 180 min in the 1/GO/Fe3O4/H2O2/Vis system, which was much better than that in MOF-1. This may be attributed to that incorporating GO results in a better response to visible light irradiation and a lower recombination rate of the photogenerated carriers. The kinetic curve of CTC degradation with different photocatalysts followed the pseudo-first-order process by linear transformation ln(C0/C) = kt (Fig. S7a, ESI†). Under identical conditions, the pseudo-first-order reaction kinetics (k) of 1/GO/Fe3O4 was 5.4 times higher than that of pristine MOF-1 in the H2O2/Vis system. By contrast, much boosted CTC degradation efficiency was achieved in photocatalytic reactions with H2O2, indicating accelerated catalytic reactions in these systems. Finally, to confirm the role of Fe3O4 in 1/GO/Fe3O4 in the photocatalytic reaction, the control experiment using only Fe3O4 was also assessed. As shown in Fig. S5 (ESI†), Fe3O4 can degrade CTC with the help of H2O2, but in the 1/GO/Fe3O4 complex, Fe3O4 was only responsible for endowing superparamagnetism to the composite. Extra experiments have also been conducted to figure out the role of the Fe3O4 in the 1/GO/Fe3O4 on the photocatalytic reaction. The photocatalytic properties of 1/GO and 1/GO/Fe3O4 were very close, indicating that the addition of Fe3O4 endowed the catalyst with magnetic properties without affecting the catalytic performance (Fig. S6, ESI†). Using GO as a substrate can increase aggregation and reduce dispersive forces within Fe3O4, forming an electrostatic interaction or chemical bond between Fe3O4 and GO, which was conducive to the protection of Fe3O4. Besides, the tight interfacial contact between Fe3O4 and GO further strengthened the protection of Fe3O4. Thus, the incorporation of Fe3O4 was only responsible for endowing superparamagnetism to the composite, enabling the composite to be recovered by an external magnetic field.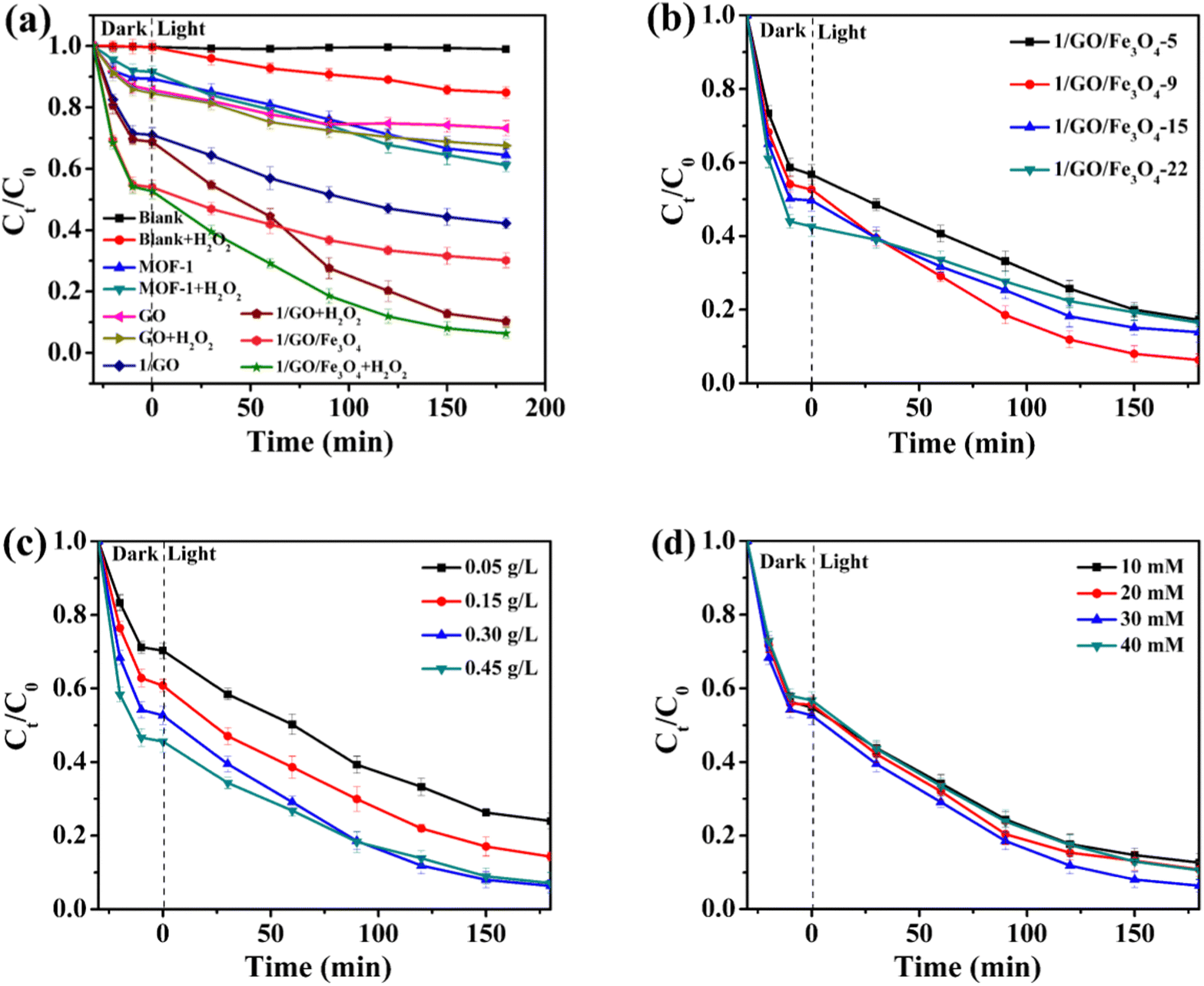 | ||
| Fig. 4 Effect of various parameters: catalyst species (a), the content of GO (b), catalyst dosage (c), and H2O2 concentration (d) on the degradation of CTC in the Fenton-like system. Except for the investigated parameter, other parameters were set as follows: CTC concentration = 20 mg L−1, catalyst dosage = 0.3 g L−1, and H2O2 concentration = 30 mM. | ||
A comparison of the degradation efficiency of CTC in this work with those of other reported catalysts is listed in Table S1 (ESI†). As presented, our designed 1/GO/Fe3O4 catalyst displays a better comprehensive evaluation concerning the degradation efficiency and recyclability than the reported catalysts. This result demonstrates that 1/GO/Fe3O4 appears to be an effective photocatalyst for the degradation of antibiotics in the aquatic environment.
Since the reaction parameters (GO content, catalyst dosage, and H2O2 concentration) have a significant influence on photocatalytic performance, further studies were conducted to study the influence of these reaction parameters on the degradation of CTC by 1/GO/Fe3O4. The degradation of CTC follows two-stage pseudo-first-order kinetics with different reaction parameters (i.e., the initial rapid decline stage and the subsequent slow decay stage). In the first stage, Ce(III) cations can quickly react with H2O2 to generate many ˙OH radicals to attack the target pollutants, thereby helping to degrade CTC rapidly. Owing to the consumption of a large amount of Ce cations in the first stage, the reaction rate in the second stage will be decided by the regeneration rate from Ce(IV) to Ce(III), which caused slower degradation of CTC. Similar results were obtained by another study.34,35
The effects of the GO content in 1/GO/Fe3O4 on the photocatalytic performance toward CTC degradation are presented in Fig. 4b. The photocatalytic performance of the prepared composites based on pseudo-first-order reaction kinetic constants (k) followed the order (Fig. S7b, ESI†): 1/GO/Fe3O4-9 (0.0124 min−1) > 1/GO/Fe3O4-15 (0.0075 min−1) > 1/GO/Fe3O4-5 (0.0069 min−1) > 1/GO/Fe3O4-22 (0.0058 min−1). The 9 wt% 1/GO/Fe3O4 composite was the optimal photocatalyst since a nearly 88.0% degradation of CTC was achieved in 180 min of treatment. This improved performance is chiefly ascribed to the incorporation of GO inhibits the photogenerated charge recombination in MOF-1. The prolonged carrier lifetime produces higher active free radicals than MOF-1, which can help the photocatalytic degradation of CTC. However, a further increase in the content of GO will cause excess GO to compete for active sites, block available optical energy, and lead to a decrease in the photocatalytic degradation efficiency of CTC. Similar phenomena were also reported by previous studies.22,36,37
Fig. 4c shows the effect of catalyst dosages on the CTC degradation. As the photocatalyst dosage increased from 0.05 to 0.30 g L−1, the CTC degradation efficiency increased from 65.9 to 88.0%, and the corresponding k value increased from 0.0062 to 0.0124 min−1 (Fig. S7c, ESI†). One possible reason is that the higher the dosage of the catalyst, the more the number of active sites available for the generation of ˙OH, thereby increasing the degradation efficiency. However, a further increase of catalyst dosages finally led to the decline of degradation efficiency down to 84.4%, since excess photocatalyst will cause an increase in turbidity and light-scattering because of catalyst aggregation. This will inhibit photon penetration and reduce the number of photogenerated oxidants, and thereby the degradation rate of pollutants considerably declined.38 These results prove that the optimal amount of catalyst is about 0.30 g L−1.
As revealed in Fig. 4d, the effect of H2O2 concentration on CTC degradation was studied. The increase of CTC degradation efficiency and the value of k (76.8 to 88.0%, 0.0088 to 0.0124 min−1, respectively) is achieved by increasing H2O2 concentration from 10 to 30 mM (Fig. S7d, ESI†). This may be because the increase in the H2O2 concentration led to the generation of more ˙OH for CTC degradation. However, the decrease in the degradation efficiency and k value (81.3% and 0.0097 min−1) was observed with the H2O2 concentration further increasing to 40 mM. This may result from the self-scavenging effect of excess H2O2 on ˙OH, which corresponds to the previous reports.39,40
Reusability and stability of the composite
The circulating experiments were also performed to confirm the stability of the prepared photocatalyst. As presented in Fig. 5a, 88.0% degradation of CTC under visible light using the 1/GO/Fe3O4 composite was observed for the first run. After ten consecutive runs, the degradation rate of CTC decreased slightly. The minor decrease in the CTC degradation with the increase in the number of cycles could be ascribed to the adsorption of unwashed CTC and partial degradation intermediates on 1/GO/Fe3O4, which impeded further degradation of CTC through the photocatalytic process. Similar phenomena were also observed in previous research.41 Moreover, the PXRD patterns and FTIR spectra of 1/GO/Fe3O4 before and after the degradation (Fig. 5b and c) remained almost identical characteristic peaks, further confirming the excellent stability of 1/GO/Fe3O4. As displayed in Fig. 5d, the CTC degradation efficiency decreased as the pH value increased from 3 to 9, which may be due to the lower oxidation potential of ˙OH radicals (E0 = +2.8 V at pH 0; E0 = +2.0 V at pH 14) and the self-decomposition of H2O2.34 Also, 1/GO/Fe3O4 exhibited relatively outstanding catalytic activity at near-neutral pH, proving that 1/GO/Fe3O4 can be used in a wide pH range. These results indicate that the 1/GO/Fe3O4 composite was the stable, reusable, and efficient photocatalyst with superior photocatalytic performance.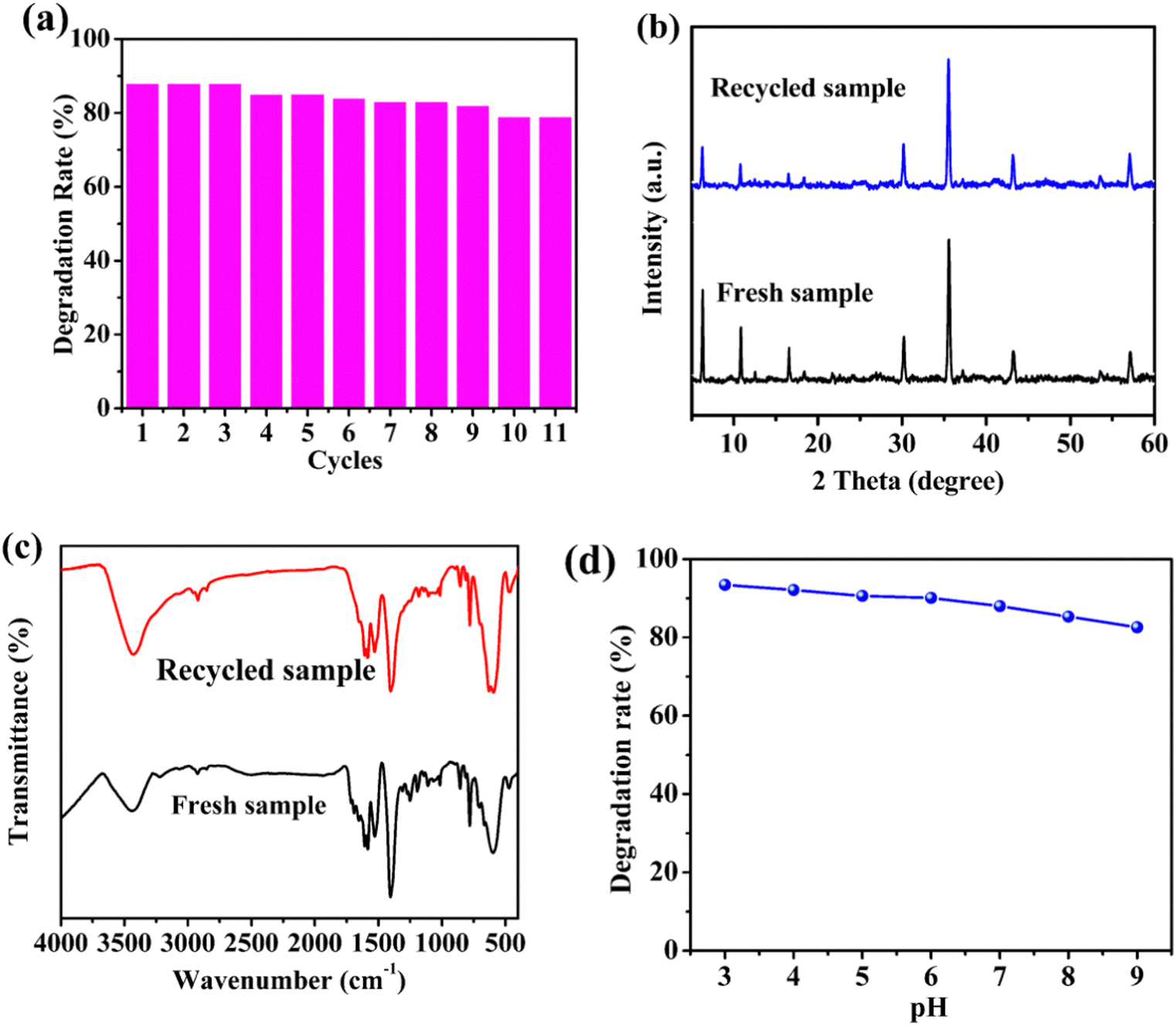 | ||
| Fig. 5 Recycling test of 1/GO/Fe3O4 (a). The PXRD patterns (b) and FTIR spectra (c) of 1/GO/Fe3O4 before and after photocatalytic reactions. Effects of initial pH on degradation of CTC (d). | ||
Possible photocatalytic mechanism
To verify the possible generation of radicals, two possible methods can be considered: (i) electron spin resonance (ESR) and (ii) radical trapping experiments. The ESR was firstly used to verify the existence of ˙OH and ˙O2− during the photocatalytic process in the presence of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) (Fig. 6a and b).42,43 No characteristic signals of ˙OH and ˙O2− can be detected in the dark, while they appeared after 1 min irradiation, and the peak intensity increased with the increase of the irradiation time. Therefore, the production of ˙OH and ˙O2− during CTC degradation can be confirmed. For radical trapping experiments, silver nitrate (Ag(NO)3), p-benzoquinone (BQ), disodium ethylenediaminetetraacetic acid (EDTA-2Na), and isopropanol (IPA) were used to trap e−, ˙O2−, h+, and ˙OH.44–46 As shown in Fig. 6c, the scavenging effects of Ag(NO)3, BQ, EDTA-2Na, and IPA prevented the degradation efficiency by 49%, 40%, 56%, and 48%, respectively, after 180 min of irradiation. EDTA-2Na displayed the most significant decline in the degradation of CTC, demonstrating that the h+ had the greatest influence on CTC degradation, followed by the existence of e− and ˙OH. The presence of ˙O2− displayed the lowest impact on degradation, which may be because the production of ˙O2− is lower than that of e−, h+ and ˙OH. | ||
| Fig. 6 ˙OH (a) and ˙O2− (b) test in methanol with DMPO as radical trapper under dark and visible light conditions. Effects of different radical scavengers on the degradation of CTC in the 1/GO/Fe3O4 system (c). | ||
To explore the charge-separation efficiency, photocurrent measurements (I–t) were performed, and the results displayed that the photocurrent for 1/GO/Fe3O4 enhanced in comparison with that for pure MOF-1 (Fig. 7a), demonstrating that the formation of the 1/GO/Fe3O4 composite contributed to separating the photogenerated electron–hole pairs.47–49 This argument was also supported by the electrochemical impedance spectroscopy (EIS) results (Fig. 7b), where 1/GO/Fe3O4 showed a smaller diameter, indicative of smaller electron-transfer resistance.50–53 This was further proven by cyclic voltammetry curves (CV), which provided valuable hints for the photoexcited electron transfer and recombination. In comparison, the anodic peak current and cathodic one at 1/GO/Fe3O4 significantly increased, which can be attributed to the outstanding conductivity of GO, accelerating electron transfer (Fig. 7c). Such different photoelectrochemical properties in 1/GO/Fe3O4 unequivocally prove that the doping of GO is of great importance.
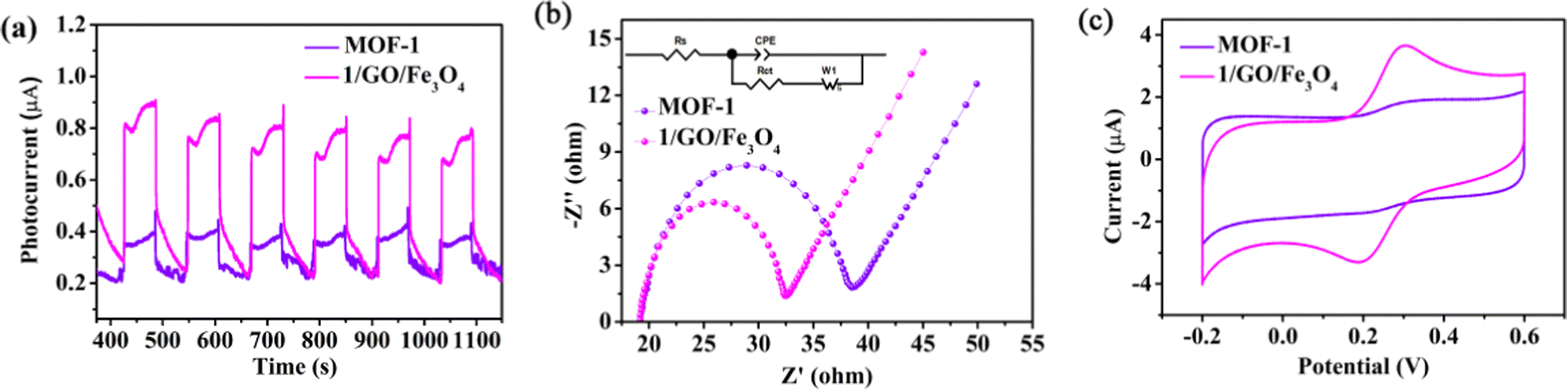 | ||
| Fig. 7 Transient photocurrent responses (I–t) (a), EIS Nyquist impedance (b), and CV plots (c) of MOF-1 and 1/GO/Fe3O4. | ||
Mott–Schottky experiments were implemented to evaluate the conduction band (CB) potential of MOF-1. It was found that MOF-1 was an n-type semiconductor because of the positive slope of the Mott–Schottky plots. The flat band (FB) potential of the semiconductor is calculated by the intercept of the X-axis extended by the Mott curve, which was 0.1 V positive than the CB potential.54,55 As shown in Fig. S8a (ESI†), the CB potential of MOF-1 was extrapolated to be about −1.04 eV vs. Ag/AgCl (−0.84 eV vs. NHE), which was more negative than the reduction potential for O2 to ˙O2− (−0.33 eV vs. NHE), thus indicating that MOF-1 can combine with dissolved O2 to generate ˙O2− (Fig. 8). Combined with the band gap value obtained by UV-Vis DRS spectra (Fig. S8b, ESI†), the corresponding valence band potential (VB) of the composite was calculated to be 2.56 eV vs. Ag/AgCl (2.76 eV vs. NHE).56 Since the VB potential of MOF-1 was more positive than ˙OH/OH− (1.99 eV vs. NHE),57–59 so the photogenerated holes might oxidize OH− to yield ˙OH. In our work, a heterojunction between MOF-1 and GO was formed by reflux, and thus created an inner electric field between them. When it was activated under visible light irradiation, the generated electrons by MOF-1 can be easily delocalized from its π* orbital and captured by GO, which further led to efficient carrier separation and thus an enhanced photocatalytic activity.
 | ||
| Fig. 8 Schematic diagram of the enhanced photocatalytic mechanism over the 1/GO/Fe3O4 composite. | ||
To further investigate the possible photocatalytic degradation mechanism, CTC aqueous solutions before and after degradation were studied by liquid chromatography–mass spectrometry (LC–MS),60–62 and the possible intermediate products (m/z: [M + H]+/z) were determined. Fig. S9a and b (ESI†) exhibit the extraction ion chromatography (EIC) of [CTC + H]+ ions at m/z = 479 ([M + H]+) before irradiation, which was the mass spectrum of CTC. After photocatalytic degradation, the initial concentration of CTC decreased at a retention time of 8.26 minutes, indicating the degradation of CTC (Fig. S9a and c, ESI†). Combined with the previous reports, Fig. S9d–g and Table S2 (ESI†) show the corresponding MS spectra and structure information of the intermediate products. It can be seen that different m/z peaks, including 501, 419, 385, 279, 261, 249, 227, and 185 were detected. The possible degradation pathways are illustrated in Scheme S2 (ESI†) through the comprehensive analysis of the above intermediates and other related references. In the black pathway, ions containing m/z = 501 ([M + Na]+) had a molecular weight difference of 23 from CTC, indicating that it was possible to gain weight from Na+.63 For the red pathway, the intermediate product of m/z = 385 ([M + H]+) was obtained by dechlorination and double bond breaking.63 In the green pathway, CTC was attacked by radicals and converted into products m/z = 419 ([M + H]+)64 and m/z = 249 ([M + H]+)65 by removing an acetyl group and conducting a ring-opening reaction, respectively.66 For the orange pathway, the intermediate products of m/z = 227 ([M + H]+) and m/z = 185 ([M + H]+) were obtained by the degradation.67 In the blue pathway, the intermediate product of m/z = 279 ([M + H]+) was obtained by dechlorination, which was further decomposed after the removal of the aromatic ring, and the intermediate product m/z = 261 ([M + H]+) was obtained due to the breaking of the double bond.68 Ultimately, CO2, H2O, and other inorganic substances could be produced through the mineralization of organic intermediates.
Through the above analysis and the corresponding results, the possible mechanism was proposed, in which the e− in the CB of MOF-1 rapidly transfers to GO due to both the shorter electron-migration distance and the tight solid–solid contact interface, while h+ will remain in the VB of MOF-1. Besides, the transfer pathway was further verified by XPS. The binding energies of Ce 3d in 1/GO/Fe3O4 shifted to higher binding energies compared with those of pure MOF-1 (Fig. 2b), proving that the photoexcited e− in the CB of MOF-1 can flow to the GO. Meanwhile, the accumulated e− in the GO can react with O2 to generate ˙O2− to further decompose CTC, while h+ on the VB of MOF-1 might directly oxidize OH− to yield ˙OH to degrade CTC. Besides, the photogenerated electrons in photocatalytic reactions can also facilitate the Ce(III)/Ce(IV) conversion, and consequently, Ce(III) will sustainably react with H2O2 to produce ˙OH. Accordingly, synergistic effects can be achieved in the H2O2-containing catalytic system for the degradation of CTC. Ultimately, the produced reactive species, including ˙O2−, ˙OH and h+, together oxidize CTC to the smaller molecules or ions (CO2, H2O, Cl−, etc.).
Conclusions
This work reports a facile strategy to synthesize a highly efficient and reusable 1/GO/Fe3O4 catalyst, which displays significantly enhanced photocatalytic activity compared with pure MOF-1. This increase is achieved by utilizing GO, which inhibits the recombination of charge carriers, improves the utilization rate of visible light, and helps to improve the photocatalytic activity of MOF-1. Under the optimized conditions, 88.0% degradation of 20 mg L−1 CTC has been achieved in 180 min under visible light irradiation. In addition, the degradation efficiency of CTC does not change significantly after ten consecutive catalytic degradations, demonstrating the satisfactory stability and reusability of the catalyst. More importantly, the catalyst can be quickly and conveniently separated from the aqueous solution after water treatment benefiting from magnetism endowed by Fe3O4. This research not only provides more insights for expanding the attractive application fields of MOF-based materials but also provides new photocatalysts for the removal of CTC or other antibiotics from wastewater in the future.Author contributions
Yuting Bai: methodology, formal analysis, and writing – original draft; Derek Hao: writing – review & editing; Sisi Feng: conceptualization, writing – review & editing, and supervision; Liping Lu: resources and project administration; and Qi Wang: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (Grant No. 21671124) and the Fund for Shanxi “1331 Project” Key Innovative Research Team (1331KIRT). A portion of this work was performed at the Scientific Instrument Center of the Shanxi University of China.References
- L. Joseph, B.-M. Jun, M. Jang, C. M. Park, J. C. Muñoz-Senmache, A. J. Hernández-Maldonado, A. Heyden, M. Yu and Y. Yoon, Chem. Eng. J., 2019, 369, 928–946 CrossRef.
- K. Changanaqui, E. Brillas, H. Alarcón and I. Sirés, Electrochim. Acta, 2020, 331, 135194 CrossRef CAS.
- X. Deng, S. Zhang, N. Ye, L. Zhang and Y. Xiang, Dyes Pigm., 2022, 206, 110660 CrossRef CAS.
- L. Yi, B. Li, Y. Sun, S. Li, Q. Qi, J. Qin, H. Sun, D. Fang and J. Wang, Sep. Purif. Technol., 2020, 250, 117257 CrossRef CAS.
- J. Zhao, Q. Song, Q. He, D. D. Dionysiou, F. Wu, Y. Feng and X. Zhang, Chemosphere, 2021, 268, 129269 CrossRef CAS PubMed.
- H. Lv, Y. Duan, X. Zhou, G. Liu, X. Wang, Y. Wang, M. Yuan, Q. Meng and C. Wang, Catal. Sci. Technol., 2020, 10, 8230–8239 RSC.
- J. Liu, X. Yu, L. Wang, M. Guo, W. Zhu and S. Tian, Water Sci. Technol., 2019, 80, 1249–1256 CrossRef CAS PubMed.
- J. Fu, X. Yu, Z. Li, Y. Zhang, W. Zhu and J. Liu, Water, Air, Soil Pollut., 2021, 232, 12 CrossRef.
- Y. Wang, L. Feng, J. Pang, J. Li, N. Huang, G. S. Day, L. Cheng, H. F. Drake, Y. Wang, C. Lollar, J. Qin, Z. Gu, T. Lu, S. Yuan and H. Zhou, Adv. Sci., 2019, 6, 1802059 CrossRef PubMed.
- Q. Xia, X. Yu, H. Zhao, S. Wang, H. Wang, Z. Guo and H. Xing, Cryst. Growth Des., 2017, 17, 4189–4195 CrossRef.
- L. Nirumand, S. Farhadi, A. Zabardasti and A. Khataee, Ultrason. Sonochem., 2018, 42, 647–658 CrossRef PubMed.
- B. Bouider, S. Haffad, B. S. Bouakaz, M. Berd, S. Ouhnia and A. Habi, J. Inorg. Organomet. Polym. Mater., 2023, 33, 4001–4011 CrossRef.
- Z. Wu, Z. Chen, J. Chen, X. Ning, P. Chen, H. Jiang and H. Qiu, Environ. Sci.: Nano, 2022, 9, 4609–4618 RSC.
- S. Zhang, Y. Wang, Z. Cao, J. Xu, J. Hu, Y. Huang, C. Cui, H. Liu and H. Wang, Chem. Eng. J., 2020, 381, 122771 CrossRef.
- X. Li, Z. Le, X. Chen, Z. Li, W. Wang, X. Liu, A. Wu, P. Xu and D. Zhang, Appl. Catal., B, 2018, 236, 501–508 CrossRef.
- Q. Wu, Y. Liu, H. Jing, H. Yu, Y. Lu, M. Huo and H. Huo, Chem. Eng. J., 2020, 390, 124615 CrossRef.
- X. Jiang, D. Fan, X. Yao, Z. Dong, X. Li, S. Ma, J. Liu, D. Zhang, H. Li, X. Pu and P. Cai, J. Colloid Interface Sci., 2023, 641, 26–35 CrossRef PubMed.
- D. Zhang, R. Zhang, J. Liu, X. Pu and P. Cai, J. Am. Ceram. Soc., 2023, 106, 4785–4793 CrossRef.
- X. Jiang, D. Kong, B. Luo, M. Wang, D. Zhang and X. Pu, Colloids Surf., A, 2022, 633, 127880 CrossRef.
- X. Jiang, Z. Wang, M. Zhang, M. Wang, R. Wu, X. Shi, B. Luo, D. Zhang, X. Pu and H. Li, J. Alloys Compd., 2022, 912, 165185 CrossRef.
- Z. Lin, R. Zou, J. Liang, W. Xia, D. Xia, Y. Wang, J. Lin, T. Hu, Q. Chen, X. Wang, Y. Zhao and A. K. Burrell, J. Mater. Chem., 2012, 22, 7813–7818 RSC.
- J. Lin, H. Hu, N. Gao, J. Ye, Y. Chen and H. Ou, J. Water Process Eng., 2020, 33, 101010 CrossRef.
- Y. Chen, B. Zhai, Y. Liang and Y. Li, Mater. Sci. Semicond. Process., 2020, 107, 104838 CrossRef.
- R. Al Gaashani, A. Najjar, Y. Zakaria, S. Mansour and M. A. Atieh, Ceram. Int., 2019, 45, 14439–14448 CrossRef.
- N. Zhou, Y. Ma, B. Hu, L. He, S. Wang, Z. Zhang and S. Lu, Biosens. Bioelectron., 2019, 127, 92–100 CrossRef PubMed.
- D. Zhang, Z. Yang, J. Hao, T. Zhang, Q. Sun and Y. Wang, Chemosphere, 2021, 276, 130226 CrossRef PubMed.
- N. Esfandiari, M. Kashefi, M. Mirjalili and S. Afsharnezhad, Mater. Sci. Eng., B, 2020, 262, 114690 CrossRef.
- W. Yi, C. Han, Z. Li, Y. Guo, M. Liu and C. Dong, Environ. Sci.: Nano, 2021, 8, 258–268 RSC.
- C. H. Nguyen, M. L. Tran, T. T. V. Tran and R. S. Juang, Sep. Purif. Technol., 2020, 232, 115962 CrossRef.
- Y. Wang, Z. Qiang, W. Zhu, W. Yao, S. Tang, Z. Yang, J. Wang, J. Duan, C. Ma and R. Tan, ACS Appl. Nano Mater., 2021, 4, 8680–8689 CrossRef.
- Y. M. Hunge, A. A. Yadav, A. G. Dhodamani, N. Suzuki, C. Terashima, A. Fujishima and V. L. Mathe, Ultrason. Sonochem., 2020, 61, 104849 CrossRef PubMed.
- T. Wang, D. Yue, X. Li and Y. Zhao, Appl. Catal., B, 2020, 268, 118399 CrossRef.
- X. Chen, X. Liu, H. Wang, K. Cui, R. Weerasooriya, S. He, G. Li, J. Pan and K. Zhou, Environ. Eng. Res., 2022, 27, 200519 CrossRef.
- J. Tang and J. Wang, Environ. Sci. Technol., 2018, 52, 5367–5377 CrossRef PubMed.
- W. Sharmoukh and H. N. Abdelhamid, J. Cluster Sci., 2023, 34, 2509–2519 CrossRef.
- N. Liu, W. Huang, X. Zhang, L. Tang, L. Wang, Y. Wang and M. Wu, Appl. Catal., B, 2018, 221, 119–128 CrossRef.
- X. Zhang, Y. Yang, W. Huang, Y. Yang, Y. Wang, C. He, N. Liu, M. Wu and L. Tang, Mater. Res. Bull., 2018, 99, 349–358 CrossRef.
- A. R. B. Bayantong, Y. J. Shih, C. D. Dong, S. Garcia Segura and M. D. G. de Luna, Environ. Sci. Pollut. Res., 2021, 28, 5472–5481 CrossRef PubMed.
- M. Liu, D. Zou, T. Ma, Z. Liu and Y. Li, Inorg. Chem. Front., 2019, 6, 1388–1397 RSC.
- J. Tang and J. Wang, Chemosphere, 2020, 241, 125002 CrossRef PubMed.
- Z. Zhu, C. Zhu, C. Hu and B. Liu, J. Colloid Interface Sci., 2022, 607, 595–606 CrossRef PubMed.
- J. Liang, X. Xu, W. Qamar Zaman, X. Hu, L. Zhao, H. Qiu and X. Cao, Chem. Eng. J., 2019, 375, 121908 CrossRef.
- J. Liang, X. Xu, Q. Zhong, Z. Xu, L. Zhao, H. Qiu and X. Cao, J. Hazard. Mater., 2020, 398, 122861 CrossRef PubMed.
- X. Zhao, X. Yi, X. Wang, J. Zhang, B. Liu, X. Liu, S. Guo and W. Chu, Nanotechnology, 2020, 31, 235707 CrossRef PubMed.
- J. Zhou, J. Ding, H. Wan and G. Guan, J. Colloid Interface Sci., 2021, 582, 961–968 CrossRef PubMed.
- X. Bai, X. Wang, X. Lu, Y. Liang, J. Li, L. Wu, H. Li, Q. Hao, B.-J. Ni and C. Wang, J. Hazard. Mater., 2020, 398, 122897 CrossRef PubMed.
- S. Bao, H. Liang, C. Li and J. Bai, J. Photochem. Photobiol., A, 2020, 397, 112590 CrossRef.
- Y. Lv, Y. Liu, J. Wei, M. Li, D. Xu and B. Lai, Chem. Eng. J., 2021, 417, 129188 CrossRef.
- Q. Li, J. Zhao, H. Shang, Z. Ma, H. Cao, Y. Zhou, G. Li, D. Zhang and H. Li, Environ. Sci. Technol., 2022, 56, 5830–5839 CrossRef PubMed.
- F. Wang, H. Fu, F. Wang, X. Zhang, P. Wang, C. Zhao and C. Wang, J. Hazard. Mater., 2022, 423, 126998 CrossRef PubMed.
- N. M. Latiff, X. Fu, D. K. Mohamed, A. Veksha, M. Handayani and G. Lisak, Carbon, 2020, 168, 245–253 CrossRef.
- X. Chen, S. Xiao, H. Wang, W. Wang, Y. Cai, G. Li, M. Qiao, J. Zhu, H. Li, D. Zhang and Y. Lu, Angew. Chem., Int. Ed., 2020, 59, 17182–17186 CrossRef PubMed.
- S. Li, H. Shang, Y. Tao, P. Li, H. Pan, Q. Wang, S. Zhang, H. Jia, H. Zhang, J. Cao, B. Zhang, R. Zhang, G. Li, Y. Zhang, D. Zhang and H. Li, Angew. Chem., Int. Ed., 2023, 62, e202305538 CrossRef PubMed.
- H. Derikvand, N. Tahmasebi and S. Barzegar, J. Phys. Chem. Solids, 2023, 181, 111528 CrossRef.
- C. Wan, L. Zhou, L. Sun, L. Xu, D. Cheng, F. Chen, X. Zhan and Y. Yang, Chem. Eng. J., 2020, 396, 125229 CrossRef.
- Y. Fu, K. Zhang, Y. Zhang, Y. Cong and Q. Wang, Chem. Eng. J., 2021, 412, 128722 CrossRef.
- A. Gordanshekan, S. Arabian, A. R. Solaimany Nazar, M. Farhadian and S. Tangestaninejad, Chem. Eng. J., 2023, 451, 139067 CrossRef.
- S. Meena, M. Sethi, S. Meena, P. Saini, K. Kumar, S. Saini, S. Shekhawat, M. L. Meena, A. Dandia, S. D. Lin and V. Parewa, Environ. Res., 2023, 231, 116181 CrossRef CAS PubMed.
- X. Chen, Z. Li, J. Zhou, S. Chen, Y. Huang, W. Wang, W. Wang, Q. Xu and X. Xi, J. Alloys Compd., 2023, 960, 170892 CrossRef.
- R. Tang, D. Gong, Y. Deng, S. Xiong, J. Zheng, L. Li, Z. Zhou, L. Su and J. Zhao, J. Hazard. Mater., 2022, 423, 126944 CrossRef PubMed.
- Y.-X. Li, W.-L. Duan, B.-Y. Ren, J. Luan and F. Guo, Sep. Purif. Technol., 2023, 311, 123337 CrossRef.
- K. Yan, R. Li, Z. Yang, X. Li, Y. Wang and G. Wu, iScience, 2021, 24, 102421 CrossRef PubMed.
- Y. Liu, Y. Gao, B. Yao and D. Zou, Chemosphere, 2020, 238, 124543 CrossRef PubMed.
- Y. Zhang, F. Ma, M. Ling, H. Zheng, Y. Wu and L. Li, Chem. Eng. J., 2023, 464, 142762 CrossRef.
- R. Zhang and K. Zeng, Diamond Relat. Mater., 2021, 115, 108343 CrossRef.
- F. Guo, X. Huang, Z. Chen, L. Cao, X. Cheng, L. Chen and W. Shi, Sep. Purif. Technol., 2021, 265, 118477 CrossRef.
- R. Pulicharla, R. Drouinaud, S. K. Brar, P. Drogui, F. Proulx, M. Verma and R. Y. Surampalli, Chemosphere, 2018, 207, 543–551 CrossRef PubMed.
- N. Liu, W. Huang, M. Tang, C. Yin, B. Gao, Z. Li, L. Tang, J. Lei, L. Cui and X. Zhang, Chem. Eng. J., 2019, 359, 254–264 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04499h |
| This journal is © the Owner Societies 2024 |