
Highly efficient hydrogen production and selective CO2 reduction by the C3N5 photocatalyst using only visible light†
Kosei
Ito
and
Kei
Noda
 *
*
Department of Electronics and Electrical Engineering, Keio University, 3-14-1, Hiyoshi, Kohoku-ku, Yokohama, 223-8522, Japan. E-mail: nodakei@elec.keio.ac.jp
First published on 30th November 2023
Abstract
The production of energy sources by metal-free photocatalysts based on graphitic carbon nitride (g-C3N4) has garnered substantial attention. In this study, nitrogen-rich carbon nitride (C3N5) was successfully synthesized through the thermal polycondensation of 3-amino-1,2,4-triazole. The structural and physical characterization has suggested that a portion of the triazine rings, which constitute the structural framework of g-C3N4, may be substituted with five-membered rings in C3N5. Furthermore, the polymerization of C3N5 proceeded more extensively than that of g-C3N4 from melamine precursors. The increased nitrogen content in C3N5 resulted in a heightened number of π-electrons and a narrowed energy bandgap, with the potential of the valence band maximum being negatively shifted. Additionally, photocatalytic assessments encompassing nitro blue tetrazolium reduction, H2 production from triethanolamine aqueous solution, and CO2 reduction in the liquid phase were performed. All findings demonstrated that C3N5 exhibits significantly superior photocatalytic properties compared to g-C3N4. It is particularly noteworthy that C3N5 selectively generates methanol and H2 from oversaturated CO2 solutions under visible light irradiation, while g-C3N4 selectively generates formaldehyde. These outcomes strongly indicate that C3N5 serves as a metal-free, visible-light-responsive photocatalyst, capable of contributing to both the production of renewable energy sources and the reduction of greenhouse effect gases.
Introduction
Since the first report of photocatalytic hydrogen generation using graphitic carbon nitride (g-C3N4) in 2009, g-C3N4 has attracted attention as an inexpensively, semi-permanently and easily synthesized metal-free photocatalytic material.1–3 Prior research endeavors have reported more efficient H2 production and CO2 reduction by, for example converting g-C3N4 from a two-dimensional material to a three-dimensional one and loading noble metal co-catalysts on it.4,5 At present, the reaction efficiency of g-C3N4 is still notably inferior to that of metal compound photocatalysts. As long as continuing g-C3N4 studies just follow conventional approaches and methodologies that have been applied for metal compound photocatalysts with overwhelmingly high performance, it seems quite difficult to make carbon nitride materials exceed metal compound photocatalysts. However, if carbon nitride can exhibit photocatalytic properties comparable to those of metal compound photocatalysts, it would be an economically-viable choice and highly valuable for use from the viewpoints of sustainability and resource saving. Therefore, a fundamental structural reformulation of g-C3N4 is first required to create a metal-free photocatalyst that has capabilities like metal photocatalysts.In the latest study, a new type of nitrogen-rich carbon nitride (C3N5) has been considered as an emerging photocatalytic material, because of its attractive features such as the smaller BG (ca. 2.1 eV) than that of g-C3N4, easier adsorption of organic compounds, faster charge transfer to metal cocatalysts, and robustness against secondary contamination.6–9 Furthermore, C3N5 was combined with other metal photocatalytic materials, which resulted in efficient H2 production, CO2 reduction, and organic decomposition.10–12
On the other hand, the reported C3N5 studies used metal-containing materials such as KBr during the synthesis process, and its photocatalytic properties are also evaluated in combination with other metal photocatalysts. This cancels out the attractive features of carbon nitrides that are easy-to-synthesize and metal-free. To effectively utilize the functions of carbon nitride, it is important to evaluate and compare the inherent natures of existing materials (g-C3N4) and new ones (C3N5) and find important directions for material development, rather than focusing on immediate property improvements. To date, we have not been able to find any papers that experimentally prove that C3N5 alone has better photocatalytic properties than g-C3N4 alone.
In this study, first, C3N5 was synthesized by only thermal polymerization from a precursor, without the use of metal-containing materials. Next, the detailed crystal structure and energy bands were evaluated. Finally, the photocatalytic properties of C3N5 were evaluated by H2 production and CO2 reduction, which were theoretically indicated as possible photocatalytic reactions in visible light over C3N5 owing to its conduction band (CB) and valence band (VB) edge positions. Similar experiments were also performed for g-C3N4 and the results for both C3N5 and g-C3N4 were compared and discussed. These experimental studies show that C3N5 is a better material than g-C3N4 for solving environmental and energy issues such as H2 production and CO2 reduction, and provide new guidelines for photocatalyst material design based on carbon nitride.
Experimental methods
Materials and reagents
In this work, the following chemicals and reagents were purchased and used without any further purification: 3-amino-1,2,4-triazole (AT, C2H4N4, Tokyo Chemical Industry), melamine monomer (C3H6N6, Tokyo Chemical Industry), triethanolamine (TEOA, N(CH2CH2OH)3, Nacalai Tesque; >98%), nitro blue tetrazolium (NBT) chloride (C40H30Cl2N10O6, Tokyo Chemical Industry), terephthalic acid (C6H4(COOH)2, Nacalai Tesque), and sodium hydroxide solution (NaOH, Nacalai Tesque; 5 M).Synthesis of g-C3N4 and C3N5
C3N5 was synthesized by the following method. First, AT (3 g) was dissolved in 30 ml of pure water and stirred for 30 min. After that, the pure water was evaporated using an oil bath at 100 °C for 3 h and the remaining powder was dried under vacuum at 70 °C overnight. Finally, it was heated at a rate of 5 °C min−1, kept at 550 °C for 2.5 h, and cooled naturally to room temperature. g-C3N4 was synthesized by thermal polycondensation of melamine (3 g) at a heating rate of 5 °C min−1 and at a holding temperature of 550 °C for 2.5 h.Characterization
The morphology of the samples was investigated with a transmission electron microscope (TEM, Tecnai G2,FEI). Specific surface area measurements were performed using a commercially available adsorption analyzer (ASAP2020, Micromeritics). The chemical state information was obtained by an X-ray photoelectron spectroscopy instrument (XPS, JPS-9010TR, JEOL) with Al-Kα radiation and a Fourier transform infrared spectroscopy (FTIR) system (ALPHA, Bruker). The crystal structure was specified by an X-ray diffractometer (XRD, D8 ADVANCE, Bruker) with Cu-Kα radiation. To examine the detailed energy band structure, diffuse-reflectance ultraviolet-visible (UV-Vis) absorption spectra were monitored by a UV-visible-near-infrared (UV-Vis-NIR) spectrophotometer (UV-3600Plus, Shimadzu) with an integration sphere. In addition, the determination of the flat band (FB) potential was performed by Mott–Schottky analysis with an electrochemical impedance analyzer (VersaSTAT3, AMETEK). For electrochemical impedance (EI) measurements, a 0.5 M Na2SO4 aqueous solution was employed as a liquid electrolyte, and an Ag/AgCl and a platinum wire were used as the reference and counter electrode, respectively.Photocatalytic hydrogen production using TEOA solution and photoreducing power assessment with NBT solution
45 ml of pure water, 5 ml of TEOA, and 300 mg of carbon nitride were put into a 50 ml beaker and stirred for 30 min. After the beaker was placed inside a home-made measurement cell with gas circulation, the gas lines were purged with argon and gas was circulated in a closed system. Subsequently, visible light (λ > 385 nm) from a xenon lamp (MAX-303, Asahi Spectra) was irradiated onto the photocatalyst samples through the quartz window of the measurement cell, and the generated gas was analysed every 1 hour after the onset of the light irradiation, by a gas chromatograph (GC-8A, Shimadzu) equipped with a thermal conductivity detector (TCD) and packed columns (ShinCarbonST, Shinwa Kako).30 ml of 5.0 × 10−5 M NBT aqueous solution and 10 mg of the synthesized carbon nitrides were added into a 50 ml beaker and stirred in the dark for 1 h. After visible light (λ > 385 nm) from a xenon lamp (MAX-303, Asahi Spectra) was irradiated for 5 min, 3 ml of the solution was centrifuged to separate the carbon nitride powder from the NBT solution. Then, the absorbance of NBT at 260 nm was measured with a UV-Vis-NIR spectrophotometer. NBT is oxidized to formazan by superoxide anions (˙O2−) formed by photocatalytic reduction of dissolved oxygen in solution.13 NBT has a maximum absorption wavelength at around 260 nm, while the formazan reveals a maximum absorption at around 530 nm.14 Since formazan is prone to be adsorbed on the catalyst surface and cannot be easily removed from the catalyst sample, the change in the absorbance of NBT was employed as an indicator of the photoreducing power of the catalyst in this study.15
CO2 photoreduction in the liquid phase
50 ml of pure water and 300 mg of carbon nitrides were added into a 50 ml beaker and bubbled with pure CO2 (99.9%) for 8 min. After that, a similar procedure as mentioned in the photocatalytic H2 production measurement was employed to detect the products generated during CO2 photoreduction. The solution in the beaker after the experiments was analysed by another gas chromatograph (GC2014, Shimadzu) with a TCD and packed columns (Sunpak-H, Shinwa Kako) using helium carrier gas. Gas analysis was also performed by the same gas chromatograph using packed columns (ShinCarbonST, Shinwa Kako) with argon carrier gas. The OH radicals formed on the photocatalyst surface were measured using the photoluminescence (PL) method. For this experiment, 10 mg of carbon nitrides, 0.04 g of terephthalic acid as a probe molecule, and 0.3 ml of NaOH, which is a reagent dissolving the terephthalic acid and preventing quenching of the fluorescent material, were added to 30 ml of an oversaturated CO2 solution. Subsequently, visible light (λ > 385 nm) was irradiated for 10 minutes, and only 3 ml of the solution was collected after centrifugation. When the OH radicals generated by the photocatalytic reaction interact with terephthalic acid, a highly fluorescent compound, 2-hydroxyterephthalic acid, is formed. The PL emission spectrum of this solution containing 2-hydroxyterephthalic acid was analysed using a fluorescence spectrophotometer (RF-6000, Shimadzu), with excitation and emission wavelengths set at 315 nm and 425 nm, respectively.Results and discussion
Morphology and material characterization
The TEM observations presented in Fig. 1 show that the synthesized C3N5 and g-C3N4 have plate-like layer structures (Fig. 1a and b) and that C3N5 has a much larger plate size (ca. 5 μm) than g-C3N4 (ca. 1 μm) (Fig. 1c and d). This means that the polymerization in C3N5 proceeded more prominently than in g-C3N4. Brunauer–Emmett–Teller (BET) specific surface areas of the C3N5 and g-C3N4 samples were measured to be 2.6 and 5.4 m2 g−1, respectively, which are well-correlated with the plate sizes estimated from Fig. 1. | ||
| Fig. 1 TEM images of (a) C3N5 and (b) g-C3N4 particles. Low magnification TEM images of the same (c) C3N5 and (d) g-C3N4 particles are also presented. | ||
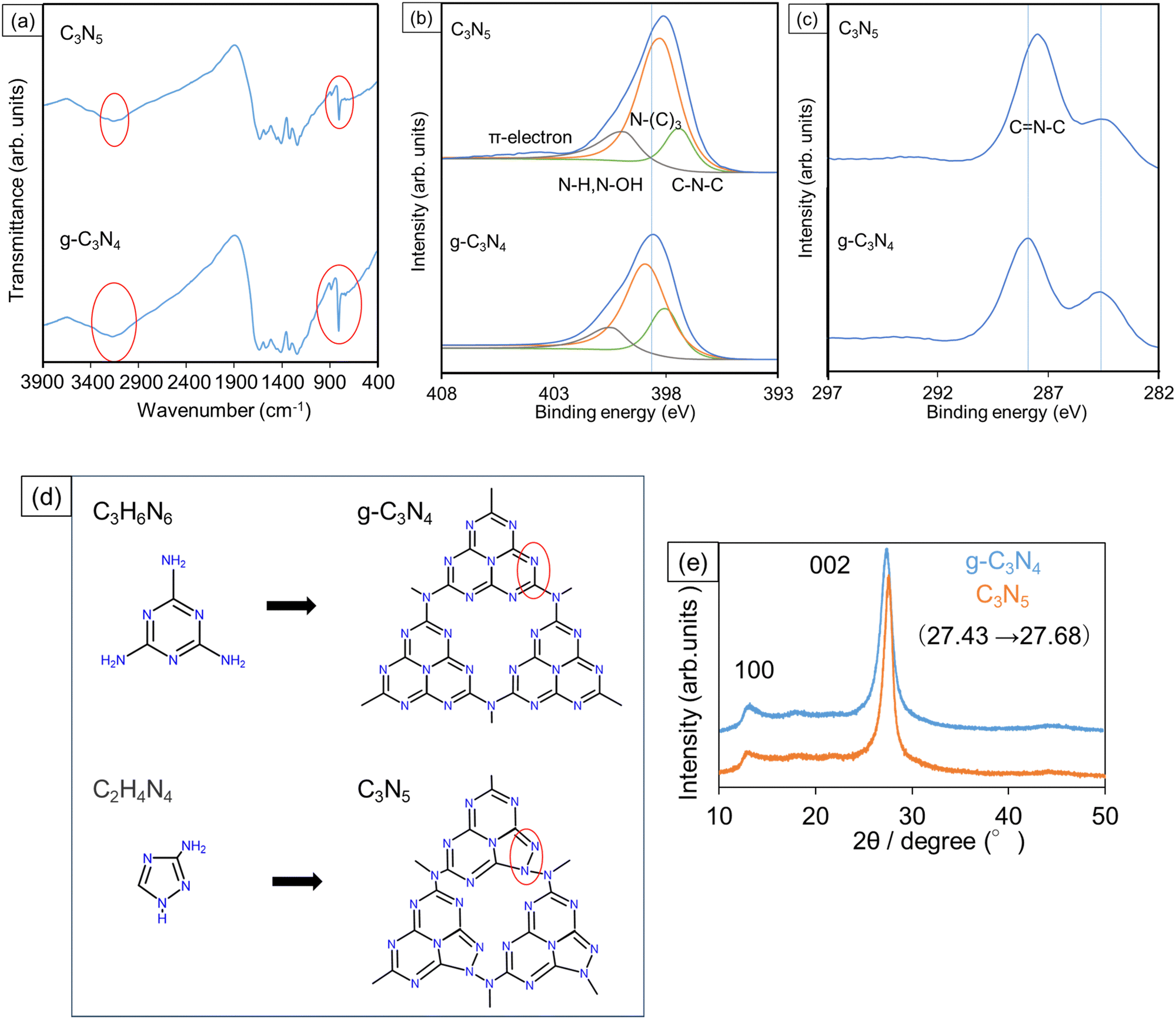
In the FTIR transmission spectra of the synthesized C3N5 and g-C3N4 (Fig. 2a), some characteristic absorption bands for carbon nitride species appeared. An absorption peak at 800–900 cm−1 originates from triazine rings, broad bands in the range from 1100 to 1700 cm−1 derive from C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds, and the bands at 2900–3400 cm−1 are due to the end groups such as C–N–H, N–H, and O–H.16,17 Although the broad absorption bands from 1100 to 1700 cm−1 did not show any significant difference between these two samples, the peak at 800–900 cm−1 in C3N5 was smaller than that in g-C3N4, presumably because a portion of the triazine rings was replaced with five-membered rings derived from the AT precursor in C3N5. Several literatures have reported that some of the precursor backbones exist in the synthesized samples, and the C3N5 precursor (AT) molecule owns a five-membered ring.18–20 Also, the absorbance of the bands from 2900 to 3400 cm−1 in C3N5 seems somewhat smaller than that in g-C3N4. As we discussed with the TEM images (Fig. 1) and the BET specific surface areas, the C3N5 plate size was larger than that of g-C3N4, resulting in the smaller number of end groups in the C3N5 sheets.
N bonds, and the bands at 2900–3400 cm−1 are due to the end groups such as C–N–H, N–H, and O–H.16,17 Although the broad absorption bands from 1100 to 1700 cm−1 did not show any significant difference between these two samples, the peak at 800–900 cm−1 in C3N5 was smaller than that in g-C3N4, presumably because a portion of the triazine rings was replaced with five-membered rings derived from the AT precursor in C3N5. Several literatures have reported that some of the precursor backbones exist in the synthesized samples, and the C3N5 precursor (AT) molecule owns a five-membered ring.18–20 Also, the absorbance of the bands from 2900 to 3400 cm−1 in C3N5 seems somewhat smaller than that in g-C3N4. As we discussed with the TEM images (Fig. 1) and the BET specific surface areas, the C3N5 plate size was larger than that of g-C3N4, resulting in the smaller number of end groups in the C3N5 sheets.
 | ||
| Fig. 2 (a) FTIR spectra and XPS spectra of (b) N1s and (c) C1s signals measured for C3N5 and g-C3N4 samples. (Peaks at 284.6 eV were used for charge correction.) (d) Structural diagrams of polymerization for C3N5 and g-C3N4. (e) XRD patterns for C3N5 and g-C3N4. | ||
The compositional ratio of carbon and nitrogen atoms (C/N) in the synthesized C3N5 and g-C3N4 was calculated from the XPS spectra (Fig. 2b and c), where the N1s and C1s signals were deconvoluted by using a Gaussian–Lorentzian function. The C/N ratio was given as the ratio between the peak area of the C1s signal divided by the sensitivity of 4.079 and that of the N1s signal divided by the sensitivity of 7.041. As a result, the calculated C/N ratio was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.93 for C3N5 and 3:3.96 for g-C3N4, respectively, supporting that the chemical composition is almost stoichiometric for both C3N5 and g-C3N4.
:4.93 for C3N5 and 3:3.96 for g-C3N4, respectively, supporting that the chemical composition is almost stoichiometric for both C3N5 and g-C3N4.
In the narrow-scan N1s spectra (Fig. 2b), the C–N–C peaks are assigned to be triazine frameworks, the N–(C)3 peaks are a heptazine ring nucleus and the bridges between the heptazine rings, and the N–H and N–OH are end groups.21–23 The ratios among the respective signals (C–N–C:N–(C)3:N–H and N–OH) were 14:67:19 for C3N5 and 28:50:22 for g-C3N4, respectively. The larger ratio of N–(C)3 and the smaller ratio of N–H and N–OH for C3N5 indicate the grain size enlargement, which was in line with the TEM observation results (Fig. 1) and measured specific surface areas. Additionally, the relative intensity of C–N–C in C3N5 was smaller than that in g-C3N4, which probably reflects the partial replacement of six-membered rings in the triazine frameworks with five-membered rings, as we discussed in the FTIR spectra (Fig. 2a). Furthermore, a small π-electron peak was observed at around 404 eV in only C3N5, where n–π* transition can be promoted by the increase in the number of unshared electron pairs with an increasing nitrogen content.24 Besides, the main peak position of N1s binding energy in C3N5 (398.2 eV) is shifted negatively from that of g-C3N4 (398.7 eV). Since C3N5 has more π electrons, the electron density in C3N5 becomes larger than that in g-C3N4. As a result, the binding energy is considered to become shifted negatively.25,26 As presented in Fig. S1 (ESI†), the peak area of the O1s signal in C3N5 (4.55 × 104) is smaller than that of g-C3N4 (5.70 × 104). Since the oxygen peaks are derived from adsorbed oxygen species and end OH groups,27,28 this decrease in the O1s peak intensity of C3N5 correlates with the larger C3N5 particle size.
Based on these FTIR and XPS data, the structures of C3N5 and g-C3N4 are schematically drawn in Fig. 2d. XRD profiles in Fig. 2e reveal that both samples displayed 100 and 002 reflections. The 100 peak indicates the in-plane ordering of the two-dimensional direction, and the 002 peak indicates interphase stacking of the carbon nitride sheets.29,30 The 002 peak position of C3N5 (27.68°) was shifted to a higher diffraction angle than that of g-C3N4 (27.43°). Considering that C3N5 has more π-electrons, the C3N5 layers are attracted to each other by π–π interactions more strongly than the g-C3N4 layers. Therefore, the 002 peak of C3N5 was shifted to a higher diffraction angle.
Energy band structure
Tauc plot analysis was carried out for estimating the band gap (BG) energy of the samples. Fig. 3a presents the Tauc plots obtained from the measured diffuse-reflectance UV-Vis absorption spectra under the assumption that C3N5 and g-C3N4 have indirect bandgaps.31,32 In Fig. 3a, α is the light absorbance, h the Planck constant, and v the light frequency, respectively. The estimated BG values of C3N5 and g-C3N4 are 2.15 and 2.75 eV.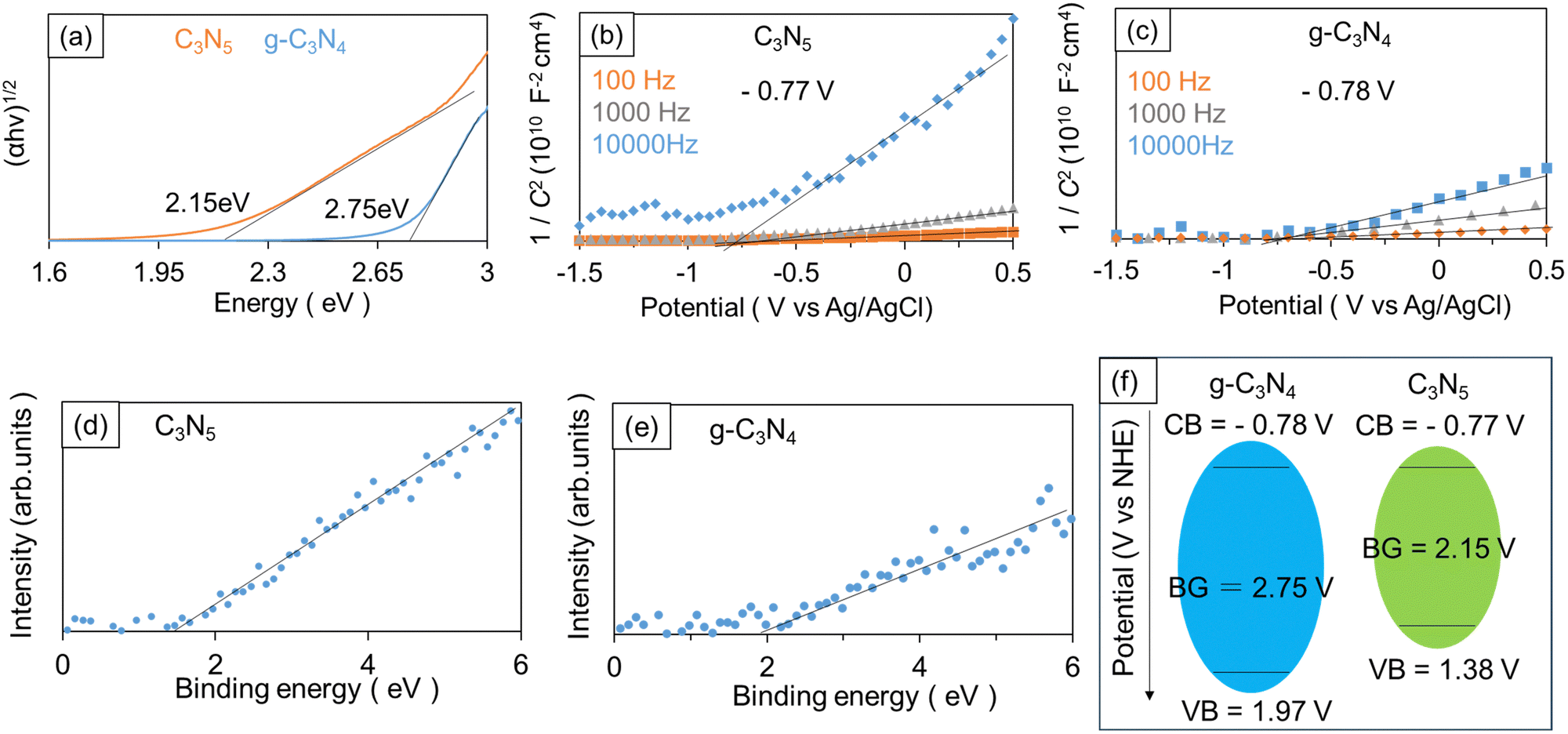 | ||
| Fig. 3 (a) Tauc plots obtained from UV-Vis absorption spectra for C3N5 and g-C3N4 (the obtained BG values are given in the figure). Mott–Schottky plots measured for (b) C3N5 and (c) g-C3N4 (the obtained FB potentials were provided in the figures). XPS valence band spectra of (d) C3N5 and (e) g-C3N4. (f) Schematic drawing of energy band diagrams of C3N5 and g-C3N4. | ||
Mott–Schottky (M–S) analysis was performed to further investigate the energy band structure. The carbon nitrides were deposited onto fluorine-doped tin oxide (FTO) substrates in the same way as for the powder synthesis, except that an FTO substrate was placed on top of the powder sample during the high-temperature treatment. The applied potential (V) and measured space charge capacitance (C) at various modulation frequencies produced M–S plots (1/C2–V curves), as shown in Fig. 3b and c. The positive slopes in the obtained M–S plots indicate that the synthesized C3N5 and g-C3N4 are n-type semiconductors.33,34 If we assume that the conduction band (CB) edge is about 0.2 V more negative than the FB level of n-type semiconductors, the CB edge positions of C3N5 and g-C3N4 films were calculated to be −0.77 V (vs. normal hydrogen electrode (NHE)) and −0.78 V (vs. NHE), respectively.35,36 The valence band (VB) edge positions of C3N5 and g-C3N4 were also determined to be 1.38 V (vs. NHE) and 1.97 V (vs. NHE), respectively, by taking their BG energy values into account. XPS valence band spectra (Fig. 3d and e) revealed that the VB edge of C3N5 was more negative than that of g-C3N4, which was consistent with the above-described results. The energy band diagrams of the synthesized C3N5 and g-C3N4 are schematically depicted in Fig. 3f. The BG of carbon nitride materials is determined by the N2p orbital for VB and the C2p orbital for CB.37,38 The narrower BG of C3N5 is attributed to the shallower VB level because C3N5 is richer in nitrogen than g-C3N4 and has more nitrogen-derived π electrons.
Photocatalytic hydrogen production over C3N5 and g-C3N4
Fig. 4 shows the photocatalytic H2 production from TEOA solution over C3N5 and g-C3N4, respectively. The H2 production rate of C3N5 (ca. 1.2 μmol h−1) was almost twice as high as that of g-C3N4 (ca. 0.6 μmol h−1). Photocatalysis always involves a similar number of holes used for oxidation reactions and electrons used for reduction reactions. Both C3N5 and g-C3N4 meet the oxidation potential (1.23 V (vs. NHE)) and reduction potential (0 V (vs. NHE)) for hydrogen production from water.39,40 But when water contains sacrificial reagents, they are preferentially oxidized and this photooxidation reaction proceeds fast.41 In this case, the rate-determining process must be a two-electron reduction of protons produced by the oxidation reaction. Therefore, the reason why C3N5 could generate hydrogen more efficiently than g-C3N4 can be ascribed to the improved reducing power of C3N5.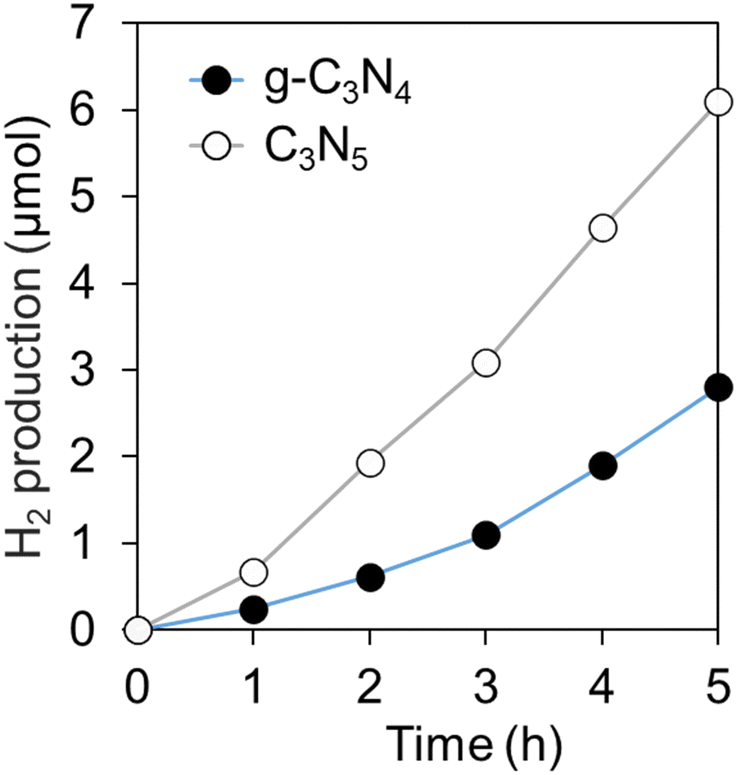 | ||
| Fig. 4 Amount of H2 production from TEOA aqueous solution over C3N5 and g-C3N4 under VIS irradiation. | ||
NBT measurements (Fig. 5) also suggested that C3N5 has better reducing power than g-C3N4. The reducing power of C3N5 is enhanced by the large reduction in BG by the negative shift of the VB edge and by the increase in the number of excited electrons by visible light irradiation, despite the similar structures and almost identical CB edge positions between C3N5 and g-C3N4.
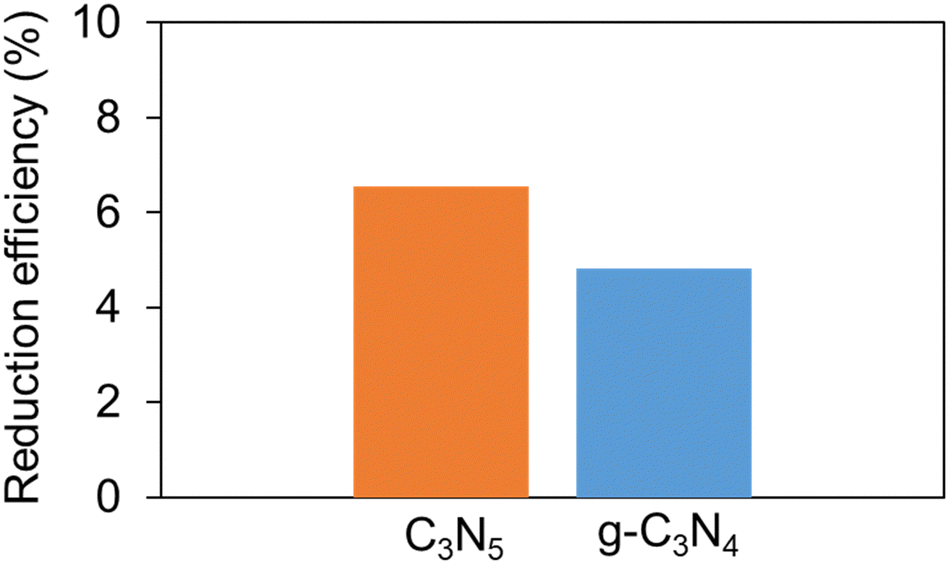 | ||
| Fig. 5 Efficiency of NBT reduction caused by O2 radicals produced over C3N5 and g-C3N4. | ||
Liquid phase CO2 photoreduction over C3N5 and g-C3N4
The experimental results of CO2 photoreduction in water under oversaturated conditions are provided in Fig. 6a and b. C3N5 selectively produced methanol (CH3OH) and H2, while g-C3N4 produced only formaldehyde (CH2O). No other gases and liquids, except CO2, were observed (Fig. 6c). In Fig. 6c, the pH of CO2 aqueous solution was 3.9 immediately after preparing the oversaturation condition and was gradually raised up to 5.6 after 2 h in the dark. In general, the pH of pure water is 6 to 6.5, and when CO2 is dissolved, it tends to be more acidic depending on its concentration.42,43 In other words, the detection of CO2 gas and the change in pH verified that CO2 was present in excess in the water.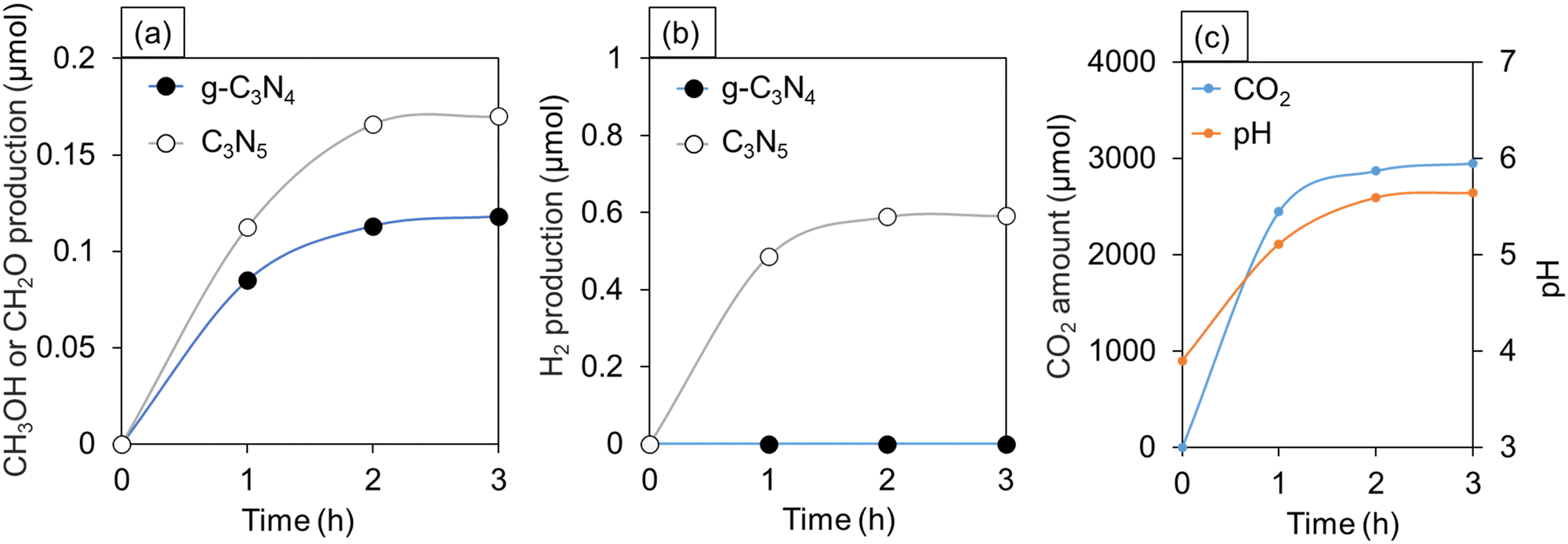 | ||
| Fig. 6 Amounts of (a) CH3OH or CH2O and (b) H2 production from oversaturated CO2 aqueous solution over C3N5 and g-C3N4 under VIS irradiation. (c) Temporal changes in CO2 amount detected by gas chromatograph and pH of the CO2 aqueous solution with no catalyst in the dark. | ||
As shown in Fig. 6, the time variation of the gas production (CH3OH, CH2O, H2) amounts, pH of the solution, and CO2 amount become almost unchanged after 2 h passed from the start of the measurements. The reaction kinetics depends on temperature, activation energy, and concentration of gases.44 In the present experiment, the temperature was constant during 3 h after the start of the measurements. On the contrary, the concentration of CO2 dissolved in the solution decreased significantly over time. Hence, the chemical reaction rate of CO2 reduction was relatively high at the initial stage of the photocatalytic experiment due to the presence of CO2 in an oversaturated state, while the reaction rate became lowered with the rapid decrease in the CO2 concentration in the solution.
In the case of simple CO2 reduction, CO is usually detected as an intermediate.45,46 However, CO was not detected in our measurements (Fig. 6). This indicates that CO2 directly produces CH3OH and CH2O by multi-electron reduction.
| CO2 + 6H+ + 6e− → CH3OH + H2O | (1) |
| CO2 + 4H+ + 4e− → CH2O + H2O | (2) |
Six electrons are required for the generation of CH3OH, and four electrons are required for the generation of CH2O. Nevertheless, the quantity of CH3OH produced by C3N5 (0.17 μmol) is greater than that of CH2O produced by g-C3N4 (0.12 μmol) (Fig. 6a). Some possible reasons why these reactions could occur are provided as follows.
It has been reported that when the CO2 concentration near the catalyst is high, the final substance is formed directly from CO2.47 Additionally, both g-C3N4 and C3N5 have narrow BGs, and they have a larger number of excited electrons. These situations may have induced multi-electron reduction and allowed direct reduction to the final material.48 C3N5 may have produced CH3OH by six-electron reduction because of its narrower BG than g-C3N4.49 It is also worth noting that no byproduct radical species were generated. In general, the generation of radical species could be one of the key factors that promote the formation of intermediates such as CO and CH4 in the reduction of CO2.50 However, OH radicals were not produced under our experimental conditions (Fig. S2, ESI†). This is because the VB edges of C3N5 and g-C3N4 are far from the redox potential to generate OH radicals [E(H2O/OH˙) = 2.72 V (vs. NHE)], and therefore, they would not be able to produce OH radicals from water.51 In addition, under the conditions of the current experiment, O2 radicals are not produced because dissolved oxygen is no longer present due to CO2 bubbling.
CO2 dissolved in water is known to exist in the form of CO2 (aq), carbonic acid (H2CO3), and carbonate ions (HCO3−).52
| CO2 + H2O ⇄ CO2 (aq) + H2CO3 ⇄ H+ + HCO3− | (3) |
It has been reported that the CO2 concentration in CO2 (aq) is approximately 500 times as large as that of H2CO3, and the concentration of H2CO3 in the solution is from 23 to 71 times greater than that of HCO3−.53 Although the concentration of H2CO3 is quite low, the reaction pathway via H2CO3 formation should be regarded. Taking thermodynamics into consideration, as the reduction potential of the H2CO3/CH3OH pair (0.044 V (vs. NHE)) is more positive than that of CO2/CH3OH (−0.38 V (vs. NHE)), H2CO3 reduction will be more favorable than CO2 reduction.54,55 The same is true for the CH2O formation because the reduction potentials of H2CO3/CH2O and CO2/CH2O are −0.05 V (vs. NHE) and −0.52 V (vs. NHE), respectively.55 The reaction pathways of H2CO3 reduction are not routed through carbon monoxide (CO) as an intermediate, which is consistent with our results without CO generation (Fig. 6).53,55 From Fig. 3f, the CB edges of both C3N5 and g-C3N4 meet the reduction potentials to produce CH3OH and CH2O from H2CO3. Accordingly, CO2 reduction via H2CO3 formation is one of the plausible scenarios in this work.
The oxidation reaction route can generate protons from pure water, that are essential for the formation of CH3OH and CH2O.
| 2H2O + 4h+ → 4H+ + O2 | (4) |
Here, we would like to note that protons can be slightly produced by reversible reaction of CO2 in water (eqn (3)). Also, no change in the amount of CH3OH and H2 produced after 2 and 3 h (Fig. 6a and b) reflects that the oversaturated CO2 solution is the source of H2, and that CH3OH does not contribute to the production of H2.
Additional control experiments (Fig. S3, ESI†) clearly show that this photocatalytic CO2 reduction was caused by C3N5 and g-C3N4. As for the photocatalytic cycling test, a CO2 reduction experiment over C3N5 was performed three times, which resulted in no change in photocatalytic activity (Fig. S4, ESI†). Furthermore, XRD and TEM observations of the samples after the photocatalytic measurements revealed no significant changes in the morphology and crystal structure of C3N5 (Fig. S5 and S6, ESI†). The above results indicate that C3N5 is chemically stable while g-C3N4 is reported to be chemically unstable. The increased number of π-electrons may allow orbital interactions and improve the chemical stability of C3N5.56
Conclusions
We have successfully synthesized C3N5, a nitrogen-abundant carbon nitride, by thermal polycondensation using 3-amino-1,2,4-triazole as a precursor. TEM images and specific surface area measurements showed that the synthesized C3N5 has a larger grain size than g-C3N4 prepared from melamine precursors, and that polymerization proceeds more easily in C3N5. XPS and FTIR studies suggested that part of the triazine frameworks in nitrogen-rich C3N5 is composed of five-membered rings. Then, the increase in nitrogen atoms with unshared electron pairs leads to the generation of more π-electrons in C3N5. This increase is reflected in the shorter interlayer distance of C3N5 with stronger π–π interactions. Furthermore, experimentally obtained energy band alignments revealed that C3N5 has a narrower band gap than g-C3N4 owing to a large negative shift of the valence band maximum dominated by N2p orbitals, which may also be attributed to the increased π-electrons in C3N5.In the photocatalytic H2 production from TEOA solution, the rate of H2 production for C3N5 was about twice as high as that for g-C3N4. Considering the results of the NBT experiment, the promotion of the rate-determining proton reduction might lead to more efficient hydrogen production. Finally, in the liquid phase CO2 photoreduction, C3N5 selectively produced CH3OH and H2, while g-C3N4 selectively produced CH2O. The amount of CH3OH produced by C3N5 was greater than that of formaldehyde produced by g-C3N4, indicating that C3N5 is more capable of multi-electron reduction than g-C3N4. To understand the observed product selectivity in the CO2 photoreduction, two possible reaction pathways were considered and discussed based on the reversible changes of CO2 in the water.
Eventually, all photocatalytic experiments in this study supported our idea that C3N5 alone has better photocatalytic properties than g-C3N4 alone. The results of this research will guide the synthesis of novel metal-free photocatalysts and contribute, in part, to triggering new breakthroughs for their practical applications.
Author contributions
Conceptualization: K. I. and K. N., methodology: K. I. and K. N., investigation: K. I., data curation: K. I., supervision: K. N., project administration: K. N., writing – original draft: K. I. and K. N., writing – review & editing: K. I. and K. N.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (KAKENHI No. 19H02174) of the Japan Society for the Promotion of Science (JSPS). K. I. is very grateful for the support from JST SPRING Grant Number JPMJSP2123.Notes and references
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- P. Chen, X. Dong, M. Huang, K. Li, L. Xiao, J. Sheng, S. Chen, Y. Zhou and F. Dong, ACS Catal., 2022, 12, 4560–4570 CrossRef CAS.
- N. Jiang, L. Lyu, G. Yu, L. Zhanga and C. Hu, J. Mater. Chem. A, 2018, 6, 17819–17828 RSC.
- X. Wang, J. Gong, Y. Dong, S. An, X. Zhang and J. Tian, Mater. Today Phys., 2022, 27, 100806 CrossRef CAS.
- S. Tasleem and M. Tahir, Int. J. Hydrogen Energy, 2021, 46, 20995–21012 CrossRef CAS.
- P. Kumar, E. Vahidzadeh, U. K. Thakur, P. Kar, K. M. Alam, A. Goswami, N. Mahdi, K. Cui, G. M. Bernard, V. K. Michaelis and K. Shankar, J. Am. Chem. Soc., 2019, 141, 5415–5436 CrossRef CAS PubMed.
- J. Zhang, B. Jing, Z. Tang, Z. Ao, D. Xia, M. Zhu and S. Wang, Appl. Catal., B, 2021, 13, 24907–24915 Search PubMed.
- H. Che, J. Wang, X. Gao, J. Chen, P. Wang, B. Liu and Y. Ao, J. Colloid Interface Sci., 2022, 627, 739–748 CrossRef CAS PubMed.
- J. Liu, S. Wang, C. Zhao and J. Zheng, Nanomaterials, 2023, 13, 499 CrossRef CAS PubMed.
- L. Wang, R. Chen, Z. Zhang, X. Chen, J. Ding, J. Zhang, H. Wan and G. Guan, J. Environ. Chem. Eng., 2023, 11, 109345 CrossRef CAS.
- S. Vadivel, M. Fujii and S. Rajendran, Chemosphere, 2022, 307, 135716 CrossRef CAS PubMed.
- S. Li, M. Cai, Y. Liu, J. Zhang, C. Wang, S. Zang, Y. Li, P. Zhang and X. Li, Inorg. Chem. Front., 2022, 9, 2479–2497 RSC.
- W. Yang, K. Sun, J. Wan, Y.-A. Ma, J. Liu, B. Zhu, L. Liu and F. Fu, Appl. Catal., B, 2023, 320, 121978 CrossRef CAS.
- C. Zhua, Y. Zhanga, Z. Fana, F. Liua and A. Li, J. Hazard. Mater., 2020, 393, 122395 CrossRef PubMed.
- C.-H. Chang, C.-L. Wang and B.-R. Li, Biosens. Bioelectron., 2023, 236, 115403 CrossRef CAS PubMed.
- J. Xu, L. Zhang, R. Shi and Y. Zhu, J. Mater. Chem. A, 2013, 1, 14766–14772 RSC.
- Z. Xiong, Y. Liang, J. Yang, G. Yang, J. Jia, K. Sa, X. Zhang and Z. Zeng, Sep. Purif. Technol., 2023, 306, 122522 CrossRef CAS.
- A. Krishnan, M. Yoosuf, K. Archana, A. S. Arsha and A. Viswam, J. Energy Chem., 2023, 80, 562–583 CrossRef CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- Y. Zhao, J. Zhang and L. Qu, ChemNanoMat, 2015, 1, 298–318 CrossRef CAS.
- X. Wu, R. Zhong, X. Lv, Z. Hu, D. Xia, C. Li, B. Song and S. Liu, Appl. Catal., B, 2023, 330, 122666 CrossRef CAS.
- J. Ding, Q. Tang, Y. Fu, Y. Zhang, J. Hu, T. Li, Q. Zhong, M. Fan and H. H. Kung, J. Am. Chem. Soc., 2022, 144, 9576–9585 CrossRef CAS PubMed.
- X. Guan, M. Fawaz, R. Sarkar, C.-H. Lin, Z. Li, Z. Lei, P. D. Nithinraj, P. Kumar, X. Zhang, J.-H. Yang, L. Hu, T. Wu, S. Chakraborty, J. Yi and A. Vinu, J. Mater. Chem. A, 2023, 11, 12837–12845 RSC.
- X. Li, D. Shen, C. Liu, J. Li, Y. Zhou, X. Song, P. Huo, H. Wang and Y. Yan, J. Colloid Interface Sci., 2019, 554, 468–478 CrossRef CAS PubMed.
- L. Yang, X. Liu, Z. Liu, C. Wang, G. Liu, Q. Li and X. Feng, Ceram. Int., 2018, 44, 20613–20619 CrossRef CAS.
- L. V. Goncharova, Basic Surfaces and their Analysis, Morgan & Claypool Publishers, USA, 2018, 6, 1–17 Search PubMed.
- Q. Zhang, Z. Zhang, S. Xu, A. Liu, L. Da, D. Lin and C. Jiang, Anal. Chem., 2023, 95, 4536–4542 CrossRef CAS.
- X. Huang, X. Xu, R. Yang and X. Fu, Colloids Surf., A, 2022, 643, 128738 CrossRef CAS.
- X. An, Q. Tang, H. Lan, H. Liu, X. Yu, J. Qu, H. Lin and J. Ye, Angew. Chem., Int. Ed., 2022, 134, e202212706 CrossRef.
- B. Yang, J. Han, Q. Zhang, G. Liao, W. Cheng, G. Ge, J. Liu, X. Yang, R. Wang and X. Jia, Carbon, 2023, 202, 348–357 CrossRef CAS.
- P. V. Viet, T.-D. Nguyen, D.-P. Bui and C. M. Thi, J. Materiomics, 2022, 8, 1–8 CrossRef.
- M. Mohammadikish and N. Mosleh, Appl. Organomet. Chem., 2023, e7177 CrossRef CAS.
- Z. Cai, Y. Huang, H. Ji, W. Liu, J. Fu and X. Sun, Sep. Purif. Technol., 2022, 280, 119772 CrossRef CAS.
- R. Ren, G. Liu, J. Y. Kim, R. E. A. Ardhi, M. X. Tran, W. Yang and J. K. Lee, Appl. Catal., B, 2022, 306, 121096 CrossRef CAS.
- X. Ma, Z. Ma, H. Zhang, D. Lu, J. Duan and B. Hou, J. Photochem. Photobiol., A, 2022, 426, 113772 CrossRef CAS.
- Y. Liang, X. Wu, X. Liu, C. Li and S. Liu, Appl. Catal., B, 2022, 304, 120978 CrossRef CAS.
- W. Hoa, Z. Zhanga, M. Xub, X. Zhang, X. Wang and Y. Huang, Appl. Catal., B, 2015, 179, 106–112 CrossRef.
- J. Chen, Y. Xiao, N. Wang, X. Kang, D. Wang, C. Wang, J. Liu, Y. Jiang and H. Fu, Sci. China Mater., 2023, 66, 3165–3175 CrossRef CAS.
- D. Dai, X. Liang, B. Zhang, Y. Wang, Q. Wu, X. Bao, Z. Wang, Z. Zheng, H. Cheng, Y. Dai, B. Huang and P. Wang, Adv. Sci., 2022, 9, 2105299 CrossRef CAS PubMed.
- Y.-J. Yuan, N. Lu, L. Bao, R. Tang, F.-G. Zhang, J. Guan, H.-D. Wang, Q.-Y. Liu, Q. Cheng, Z.-T. Yu and Z. Zou, ACS Nano, 2022, 16, 12174–12184 CrossRef CAS PubMed.
- V. Kumaravel, M. D. Imam, A. Badreldin, R. K. Chava, J. Y. Do, M. Kang and A. A. Wahab, Catalysts, 2019, 9, 276 CrossRef CAS.
- H. Jun, S. Choi, M. Y. Yang and Y. S. Nam, J. Mater. Chem. A, 2019, 7, 17254–17260 RSC.
- J. Byun, W. Huang, D. Wang, R. Li and K. A. I. Zhang, Angew. Chem., Int. Ed., 2018, 57, 2967–2971 CrossRef CAS PubMed.
- B. Liu, X. Zhao, C. Terashima, A. Fujishima and K. Nakata, Phys. Chem. Chem. Phys., 2014, 16, 8751–8760 RSC.
- N. F. Khusnun, A. A. Jalil, T. A. T. Abdullah, S. S. M. Latip, C. N. C. Hitam, A. A. Fauzi, N. S. Hassan, M. A. H. Aziz, A. F. A. Rahman, F. F. A. Aziz, M. Bahari, R. H. Adnan and R. Saravanan, J. CO2 Util., 2022, 58, 101908 CrossRef.
- X. Wanga, Y. Wanga, M. Gaob, J. Shenb, X. Puc, Z. Zhangb, H. Linb and X. Wang, Appl. Catal., B, 2020, 270, 118876 CrossRef.
- A. B. Anderson and H. A. Asiri, Phys. Chem. Chem. Phys., 2014, 16, 10587–10599 RSC.
- X. Wu, Y. Li, G. Zhang, H. Chen, J. Li, K. Wang, Y. Pan, Y. Zhao, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2019, 141, 5267–5274 CrossRef CAS PubMed.
- A. Li, T. Wang, C. Li, Z. Huang, Z. Luo and J. Gong, Angew. Chem., Int. Ed., 2019, 58, 3804–3808 CrossRef CAS PubMed.
- M. Ma, Z. Huang, R. Wang, R. Zhang, T. Yang, Z. Rao, W. Fa, F. Zhang, Y. Cao, S. Yu and Y. Zhou, Green Chem., 2022, 24, 8791–8799 RSC.
- K. Ito, R. Uchida and K. Noda, J. Photochem. Photobiol., A, 2023, 443, 114824 CrossRef CAS.
- T. Zhao, J. Li, J. Liu, F. Liu, K. Xu, M. Yu, W. Xu and F. Cheng, ACS Catal., 2023, 13, 4444–4453 CrossRef CAS.
- S. Pocoví-Martínez, I. Zumeta-Dube and D. Diaz, J. Nanomater., 2019, 6461493 Search PubMed.
- A. L. Soli and R. H. Byrne, Mar. Chem., 2002, 78, 65–73 CrossRef CAS.
- X. Yang, T. Xiao and P. P. Edwards, Int. J. Hydrogen Energy, 2011, 36, 6546–6552 CrossRef CAS.
- S. Shen, Y. Zeng, X. Li, L. Meng and X. Zhang, Int. J. Quantum Chem., 2018, 118, e25521 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04431a |
| This journal is © the Owner Societies 2024 |