
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidation of protein–ligand interactions by multiple trajectory analysis methods†
Nian
Wu‡
 *a,
Ruotian
Zhang‡
a,
Xingang
Peng
a,
Lincan
Fang
b,
Kai
Chen
c and
Joakim S.
Jestilä
b
*a,
Ruotian
Zhang‡
a,
Xingang
Peng
a,
Lincan
Fang
b,
Kai
Chen
c and
Joakim S.
Jestilä
b
aInstitute for Interdisciplinary Information Sciences, Tsinghua University, Beijing, China. E-mail: wunianwhu@gmail.com
bDepartment of Applied Physics, Aalto University, Espoo, Finland
cInstitute of Catalysis, Zhejiang University, Hanghzhou, China
First published on 5th February 2024
Abstract
The identification of interaction between protein and ligand including binding positions and strength plays a critical role in drug discovery. Molecular docking and molecular dynamics (MD) techniques have been widely applied to predict binding positions and binding affinity. However, there are few works that describe the systematic exploration of the MD trajectory evolution in this context, potentially leaving out important information. To address the problem, we build a framework, Moira (molecular dynamics trajectory analysis), which enables automating the whole process ranging from docking, MD simulations and various analyses as well as visualizations. We utilized Moira to analyze 400 MD simulations in terms of their geometric features (root mean square deviation and protein–ligand interaction profiler) and energetics (molecular mechanics Poisson–Boltzmann surface area) for these trajectories. Finally, we demonstrate the performance of different analysis techniques in distinguishing native poses among four poses.
1 Introduction
Predicting interaction between protein and ligand is an essential issue in drug discovery, which determines the possibility of candidates as effective drugs toward specific targets. To accelerate drug discovery, many computer-aided drug design methodologies1,2 have been developed to estimate the binding affinity as an alternative to experiments to shortlist promising candidates. These methods range from coarse to fine, including machine learning,3–6 molecular docking, end-point calculations like linear interaction energy,7 molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) simulations,8–11 and rigorous methods like thermodynamic integration (TI).12–16Among these methods, various scoring functions have been developed to estimate the binding affinity of given 3D structures.17,18 These scoring functions could be majorly classified into three types. i.e., (1) physics-based: fitting energies with a weighted sum of several energy terms based on force field parameters. (2) Empirical: counting the number of interactions that could be defined based on geometry criteria of bond length or angles. (3) Knowledge-based: analyzing intermolecular interactions between certain types of atoms or functional groups. However, the ability of these models to predict binding poses and affinities is still limited, which could predominantly be ascribed to three factors: molecular flexibility, binding score and computing time.19–22 In terms of the enormous chemical phase space, the geometrical optimization of ligands and proteins during the docking tends not to drastically change the initial structure due to the prohibitive cost. To some extent, molecular dynamics (MD) may be used to address the issue of flexibility via relaxing the initial structure based on Newton's laws.
Based on the MD trajectories, a series of analytical methods have been applied to evaluate the conformations in terms of residence time or binding free energy.23,24 For the former, the stability of a given conformation is normally indicated by the structural deviation over time. Parameters could range from global structure (whole complexes, proteins or ligand) to selected fragments or pairs (like atom pairs or residue pairs with hydrophobic interaction,25–27 hydrogen interaction,28 halogen bonds29). Root-mean square deviation (RMSD) is a typical characteristic capable of estimating the movement of each atom, Liu and coworkers30 found that about 94% of the native poses (experimental crystal structures) remain stable during the simulation, while only 56–62% of decoy poses performs comparable stability. The number and strength of interaction pairs28,31 could also be regarded as critical descriptors of stability. In addition, Sakano and coworkers24 discussed the impact of simulation time which revealed that MD simulations longer than 100 ns can deal with membrane proteins or highly-flexible proteins, whereas MD shorter than 5 ns is insufficient to accurately predict the stable structure of protein–protein. For the latter, MM/PBSA or MM/GBSA is one of the typical methods used to predict binding affinity by treating the solvent effect implicitly without atomistic detail. Numerous studies also proved the ability of MM/PBSA or MM/GBSA in distinguishing between the native structures from decoy poses through ranking the binding affinity of different ligands binding to one protein in specific cases.32,33
These previous papers have provided considerable insights for specific complex systems with few methods of analysis. However, studies performing comprehensive analysis based on relatively numerous trajectories is still lacking, and some corresponding knowledge is not necessarily transferable to new systems. Therefore, many issues associated with the information in trajectories remains elusive. For example, whether combining these analysis techniques outperforms the use of a single technique on the performance of distinguishing poses, whether the decoyed poses deviating from the native pose to various degrees share some similarity or distinctions, whether combining various analysis methods could contribute to the identification of native pose, as well as the impact of the simulation time of MD, whether the longer time means better performance.
To address these questions, we developed a framework named Moira (molecular dynamics trajectory analysis) by integrating molecular docking, molecular dynamics and various analysis methods with more details shown in the Methods. Moira not only enabled the automation of the whole process, but also allowed batch processing for a large number of systems. All complex samples were collected from the refined PDBbind repository due to their high quality in crystal structures. To make a trade off between computational cost and generalization for either proteins or ligands, we randomly selected 100 samples. For each complex, based on the extent of deviation of the ligand structure from those in the corresponding native structures, four conformations (c_native, c_2a, c_5a, c_10a, representing native structures, while ligand conformations are represented by the RMSD values, which are close to 2 Å, 5 Å, 10 Å relative to the native structures) were considered. To summarize, we calculated 400 MD trajectories for these 100 complexes with four initial conformations of each ligand. Subsequently, multiple perspectives analysis methods (geometric and energetic) involving RMSD, protein–ligand interaction profiler (PLIP), MM/PBSA were carried out to analyze these trajectories. Finally, we evaluated these techniques for their performance in distinguishing native conformations from four initial conformations.
1.1 Data analysis
To evaluate the performance of AutoDock Vina on predicting binding positions, we analyzed 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 450 complexes in the PDBbind database. For these complexes, we directly generated ten initial three-dimensional ligand structures using RDKit34 with SMILES as input. AutoDock Vina was then employed to generate the top 10 ranked conformations for the protein–ligand complexes based on the scoring function from the 10 initial ligand structures, thereby generating 100 docked conformations for each sample. The RMSD of the conformation referred to the native structure was used to assess the performance. The various poses with RMSD values ranging from 2 Å to 8 Å, relative to c_native pose (depicted in grey) are displayed in Fig. 1(a). In general, the majority of fragments overlap entirely at 2 Å, except for one or two fragments caused by few torsion angles. Therefore, they can be considered as the true native structure. At 6 Å, there are still few overlapping fragments with more torsion angles, resulting in dramatic changes. Meanwhile, at an RMSD of 8 Å, the orientations of most fragments differ completely, highlighting a significant divergence from the native structure. For the 100 complexes, less than 50% achieved high accuracy where the RMSD would be within 1 Å, indicating almost perfect overlap with corresponding c_native poses using the aforementioned conformation generation method without experimental guidance. When we loosen the criteria to 2 Å and even 8 Å, the ratio of complexes satisfying the criteria increase to 72% and near 100%, respectively.
450 complexes in the PDBbind database. For these complexes, we directly generated ten initial three-dimensional ligand structures using RDKit34 with SMILES as input. AutoDock Vina was then employed to generate the top 10 ranked conformations for the protein–ligand complexes based on the scoring function from the 10 initial ligand structures, thereby generating 100 docked conformations for each sample. The RMSD of the conformation referred to the native structure was used to assess the performance. The various poses with RMSD values ranging from 2 Å to 8 Å, relative to c_native pose (depicted in grey) are displayed in Fig. 1(a). In general, the majority of fragments overlap entirely at 2 Å, except for one or two fragments caused by few torsion angles. Therefore, they can be considered as the true native structure. At 6 Å, there are still few overlapping fragments with more torsion angles, resulting in dramatic changes. Meanwhile, at an RMSD of 8 Å, the orientations of most fragments differ completely, highlighting a significant divergence from the native structure. For the 100 complexes, less than 50% achieved high accuracy where the RMSD would be within 1 Å, indicating almost perfect overlap with corresponding c_native poses using the aforementioned conformation generation method without experimental guidance. When we loosen the criteria to 2 Å and even 8 Å, the ratio of complexes satisfying the criteria increase to 72% and near 100%, respectively.
 | ||
| Fig. 1 (a) Performance of docking (AutoDock Vina) for 13,450 complexes in PDBbind bank. (b) Distribution of experimental binding affinity for selected 100 complexes. (c) Example of four conformations c_native, c_2a, c_5a, c_10a (pdbid: 1kpm). (d) Number of ranked first poses from four pose groups for 100 complexes based on scoring function of Vina. | ||
Further, to compare the performance of various scoring strategies for ranking different poses, including both static and dynamic methods, we tested these on a randomly selected set of 100 complexes. The distribution of experimental binding affinities −logKd for these 100 complexes is illustrated in Fig. 1(b), ranging from 2 M to 11 M, where higher values indicate strong interactions between ligands and proteins. For each complex, we considered four poses (c_native, c_2a, c_5a, c_10a) as shown in Fig. 1(c), with increasing deviation from the native poses with RMSD near 0 Å, 2 Å, 5 Å, 10 Å. When ranking c_native or c_2a poses as the stablest structures out of the aforementioned four poses using the classifical scoring function in AutoDock Vina (Fig. 1(d)), the probability of failure could reach up to 30%, thus demonstrating the limited accuracy of AutoDock Vina on the evaluation of static poses.
1.2 Molecular dynamics
To investigate the structural evolution of the protein–ligand complexes, we performed molecular dynamics for 100 complexes with four poses (c_native, c_2a, c_5a, c_10a) over a time span of 25 ns. The following subsections summarize our findings based on both geometric and energetic perspectives.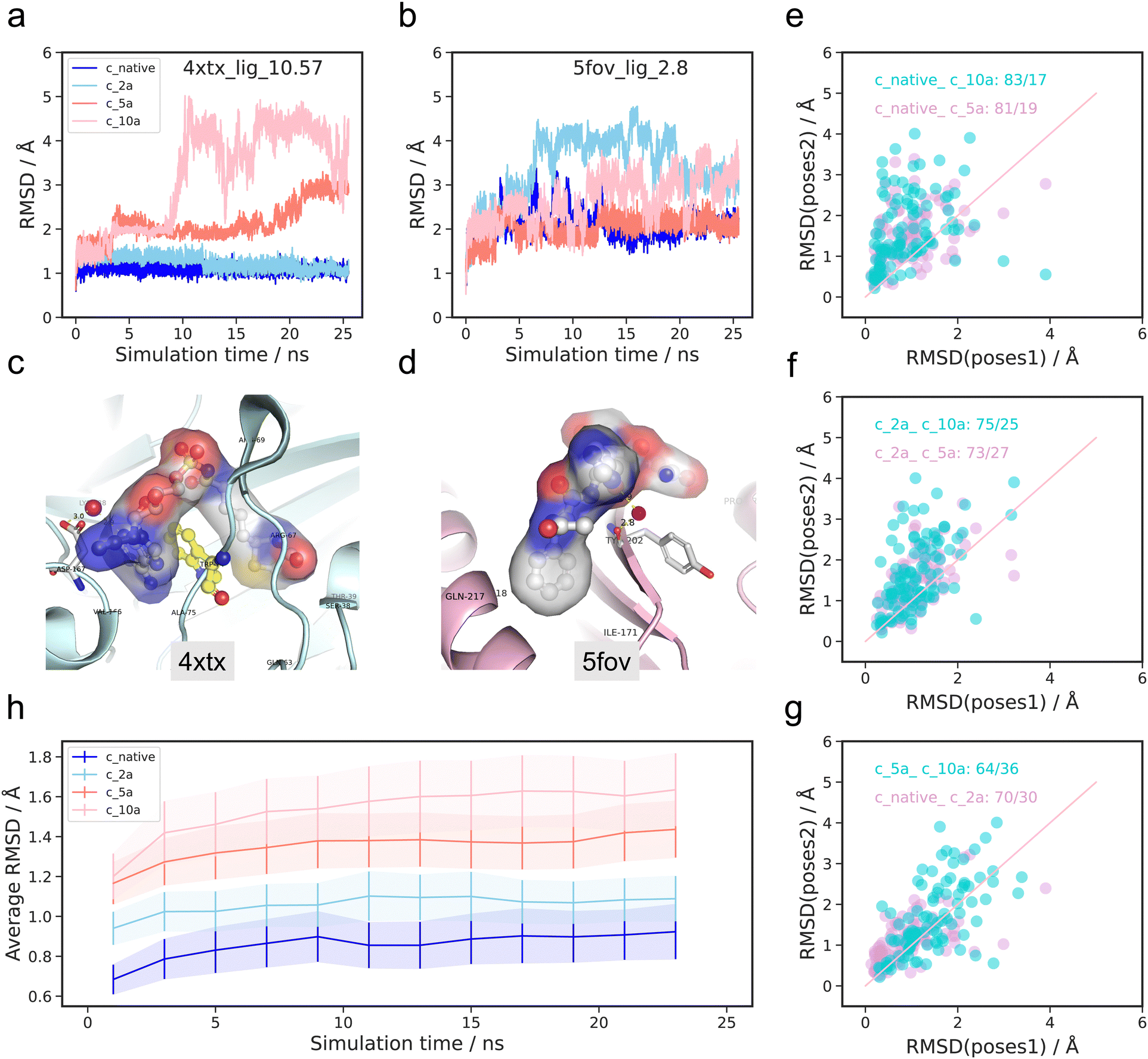 | ||
| Fig. 2 RMSD performance for four initial conformations (c_native, c_2a, c_5a, c_10a) during 25 ns molecular dynamics. (a) RMSD profile of ligand and protein with pdbid 4xtx with largest experimental binding affinity −logKd 10.57 M. (b) RMSD profile of ligand and protein of pdbid 5fov with smallest experimental binding affinity −logKd 3.74 M. (c) 3D crystal structure of complexes with pdbid 4xtx. (d) 3D crystal structure of complexes with pdbid 5fov. (e)–(g) Averaged RMSD during the final 5 ns of the MD simulations between four initial poses. (h) The averaged RMSD relative to the initial structures for all selected 100 complexes at the interval of 2 ns during 25 ns MD simulations. The shaded section represents the standard deviation. | ||
There are significant conformational variations among the ligands with the four initial poses. For the complex with pdbid 4xtx, the RMSD values relative to the initial structures for the c_native and c_2a initial structures remain consistently low at less than 2 Å, with minimal fluctuations throughout the simulation. In contrast, the RMSD values for the c_5a and c_10a pose initial structures are not only noticeably larger, but also exhibit substantial fluctuations, particularly for the c_10a pose after 8 ns. In contrast, the RMSD rankings of four initial poses for the complex of pdbid 5fov display different patterns. The c_2a pose exhibits the highest RMSD value with the largest fluctuation, followed by the c_10a pose. The c_5a pose and the c_native pose consistently remain the lowest RMSD values over the whole simulation period. Overall, while there are no definitive universal trends regarding the timing of sharp changes and rankings during the RMSD evolution of the four poses for both complexes, the results suggest that higher RMSD values are typically associated with larger fluctuations. Conversely, the RMSD evolution of the protein (shown in Fig. S1 and S2, ESI†) demonstrates a similar pattern over time for the four poses, with minimal deviations in conformation. Given the high similarity and minimal changes in protein conformation over time across the four poses, we will focus our discussion on the results pertaining to the ligand in the following section of the RMSD analysis.
Firstly, we extracted 2500 snapshots during the 25 ns simulations at 100 ps intervals for each trajectory. This was done for all 100 complexes with the four initial poses, where the RMSD describes each respective snapshot in relation to the initial pose.
To provide a qualitative interpretation of the overall conformational changes in the 100 complexes, we depict the distributions of the averaged RMSD values from the last 5 ns of the simulations for the four initial poses in Fig. S3 (ESI†). The analysis revealed that the mean RMSD for the complexes was the lowest, less than 1 Å, with the c_native initial pose for the MD simulation. This was closely followed by pose c_2a, outperforming the mean values associated with c_5a and c_10a. Furthermore, the boxplots representing each initial pose show how lower mean values are accompanied by less scattered distributions. These results indicate that the c_native pose exhibits a lower RMSD compared to the other three initial poses across the majority of the complexes.
To further evaluate the structural fluctuations in 100 complexes quantitatively, a comparative analysis was done on the average RMSD values during the last 5 ns of the trajectories, referred to initial structures for four initial poses. In Fig. 2(e)–(g), each point represents a comparison between trajectories from the same ligand but with varying initial structures. For clarity, we grouped two pairs together, distinguished by different colors (dark turquoise and plum). The horizontal axis represents the RMSD values from the pose before underscore, while the vertical axis indicates the RMSD values from the pose after underscore. For example, in Fig. 2(e), the dark turquoise points “native_c_10a: 83/17” represent a comparison between the RMSD of trajectories from c_native poses (horizontal axis) and c_10a poses (vertical axis) as initial poses. The value “83/17” signifies that 83 complexes had a higher RMSD value when c_10a was the initial pose in comparison to the native initial pose. The findings imply that, for the majority of 100 complexes, the c_native poses exhibit greater stability than the c_10a poses. Similarly, the distribution of the plum colored points in Fig. 2(e) reveals that the majority of points lie above the diagonal line, indicating that the c_native pose is generally more stable than the c_5a pose, with approximately 90% of complexes following this trend. However, a few points (10%) show the opposite trend.
In Fig. 2(f), the c_2a poses exhibit a similar pattern as the c_native poses when compared to the c_5a and c_10a poses. This observation is further supported by the relatively even distribution near the diagonal line in Fig. 2(g). In contrast, the distribution of points representing the c_10a versus c_5a poses does not have any apparent bias (dark turquoise points in Fig. 2(g)).
Subsequently, we accounted for temporal factors by dividing the 2500 snapshots into 12 parts with 2 ns increments, and calculating the averaged RMSD for these intervals. Fig. 2(h) displays the RMSD evolution for all 100 complexes across four poses (native: blue, c_2a: sky blue, c_5a: salmon, c_10a: pink), where the full line indicates the average RMSD values, while the shaded section represents the corresponding standard deviation. The figure reveals slightly increasing mean RMSD values for all four poses over time, with the value at the starting point consistently higher than that at the ending points, despite minor fluctuations. Furthermore, similar to patterns observed in the boxplot analysis (Fig. S3, ESI†), the c_native initial poses consistently exhibit higher stability compared to c_2a, c_5a and c_10a during each period across the 100 complexes, having the lowest mean RMSD and standard deviation values. Additionally, the c_native and c_2a poses display more similar trends, while c_5a is seen to be closer to c_10a in terms of trends. The results illustrate that partitioning the MD simulations into given periods leads to similar tendencies in determining the stability of conformations across the four poses in general, despite sample specific deviations.
| Algorithm 1 Counting the occurrence frequency of interaction pairs detected by PLIP |
|---|
| Input: S = snapshots I = interactions P = pairs |
| Output: C = count |
| Do PLIP for all snapshots, find all pairs and all interactions |
| Define count = np.zeros((len(P),len(I)) |
| Forj = 1, 2,…, len(S); j = 1, 2,…,len(P); k = 1, 2,…,len(I) |
| If I[k] exists in P[j]; count[j,k] = 1 Elsecount[j, k] = 0 |
| Return C = count/len(S) |
 | ||
| Fig. 3 Examples of interaction analysis. (a) Schematic of ligand–protein interactions determined by ligplot. (b) 4xtx and (d) 5fov: the occurrence probability of three type interactions (hydrophobic interactions, hydrogen bonds and water bridges) for four trajectories with different initial poses of ligand. (c) Illustration of three interaction types. | ||
To analyze the dynamic evolution of the interaction patterns, PLIP was employed to assess the 2500 snapshots from each trajectory, and subsequently these were compared across the four poses. Unlike interactions in the static simulations, certain interaction patterns may appear or disappear during the process. Consequently, we calculated the occurrence probability of each interaction pattern over the 25 ns of simulation time. In this analysis, we considered the protein residue–ligand pair only. Note that the entire ligand was treated as a single unit, although the software may also be used to gain information on specific atom pairs. In cases where multiple atoms within a residue could interact with the ligand, the occurrence probability may exceed 1.0.
Furthermore, we examined the probability of interaction pair occurrence within the seven interaction groups for 4xtx (Fig. 3(b)) and 5fov (Fig. 3(d)) over the 25 ns of MD simulation time (blue, sky blue, salmon, and pink representing native, c_2a, c_5a, and c_10a, respectively). In the figures, the horizontal axis represents the residue numbers involved in ligand interactions. In Fig. S2 (ESI†), all seven interaction patterns, including hydrophobic interactions, hydrogen bonds, and water bridges (as depicted in the cartoon schematic Fig. 3(c)), exhibit noticeable frequencies compared to the other patterns. In the 4xtx case, the c_native pose demonstrates both a higher number of interactions, as well as their corresponding probabilities, surpassing the other three initial poses. This is particularly prominent for hydrogen bonds and salt bridges. Meanwhile, the trend is less pronounced for the 5fov complex.
To ensure a sufficiently large sample size, we examined the probabilities of all interaction pairs for the four initial poses across the 100 complexes. As the residue–atom pairs involved in interactions may completely be different, we opted to compare the probabilities among all types of interactions, independent of the residue–atom pairs. Furthermore, the interactions giving rise to the largest occurrence probabilities tend to significantly influence the overall interaction dynamics, representing the dominant interactions for the complex in question. Therefore, further analysis is mostly based on the interaction modes with the largest occurrence probability (top one), while the results based on top two and top five are provided in Fig. S7 and S8 (ESI†).
Fig. 4 illustrates the differences between the largest occurrence probabilities of specific interaction types in two poses (pose1 and pose2). A positive value indicates that the most stable interaction pair in pose1 has a higher probability than in pose2, while a negative value suggests the opposite.
 | ||
| Fig. 4 PLIP performance on 100 complexes for four initial conformations (native, c_2a, c_5a, c_10a) during 25 ns of molecular dynamics. The difference of the highest occurrence probability for pairwise comparisons in four poses. (a) Hydrophobic interaction type; (b) hydrogen bonds interaction type; (c) water bridges interaction type. | ||
As was the case for the number of interactions, hydrophobic interactions, hydrogen bonds, and water bridges are also seen to far exceed other types of interactions (pi–stacking, pi–cation interactions, salt bridges, halogen bonds). These three interaction patterns emerge as the most prominent driving forces in ligand–protein binding. However, due to the limited number of cases, the latter four interaction patterns cannot be reliably discussed based on the results depicted in Fig. S9 (ESI†).
As shown in Fig. 5(a), the stablest hydrophobic interaction pairs in the MD trajectory for the c_native case tend to have higher occurrence probabilities in comparison to the other three initial poses for the majority of the 100 complexes. Furthermore, the positive/negative value ratios increase in the following order: c_2a, c_5a, and c_10a. The highest ratio, with a value of 75:20, is observed when comparing the c_native and the c_10a. Meanwhile, the ratio reduces to 59:36 for the c_5a and c_10a case.
 | ||
| Fig. 5 MM/PBSA performance on 100 complexes for four initial conformations (c_native, c_2a, c_5a, c_10a) during the 25 ns of molecular dynamics. (a)–(f) The comparisons of MM/PBSA binding affinities in terms of static poses (initial snapshot) and dynamic poses (last 5 ns) for four poses. (g)–(k) The comparisons of four decomposed MM/PBSA binding affinity for c_native poses and c_5a poses. (l) The Pearson correlation coefficient of MM/PBSA binding affinities among four statics poses and dynamics trajectories of last 5 ns plus experimental values. | ||
Hydrogen bond interactions are dominant contributors to the electrostatic interactions. Fig. 5(b) exhibits similar patterns as hydrophobic interactions, albeit with some differences. The gap between the c_native pose and the c_2a pose narrows down to 48:50, as well as the gap between the c_5a pose and the c_10a pose with a ratio of 54:44. Moreover, the gaps between the c_native pose (or c_2a pose) and the c_5a pose (or c_10a pose) are larger, particularly for the second stablest hydrogen interaction pair, as shown in Fig. S9 (ESI†).
The hydrogen bond network between the ligand and the protein facilitated by water molecules plays a significant role in the overall structural stability. In contrast to the aforementioned interaction patterns, the water bridge interactions (Fig. 5(c)) exhibit different patterns. There is no clear gap observed in any of the comparisons among the four poses, and no explicit trend indicating which initial pose is more likely to possess the water bridge interaction pair with the largest occurrence probability among complexes.
First, we started by calculating the MM/PBSA binding affinities for the four initial structures (Fig. 5(a)–(f), lime green points). Similar to the RMSD values, the MM/PBSA binding affinities for the native structure are significantly lower than those for the other three poses in most complexes, followed by the c_2a pose. However, no clear trend was observed for c_5a and c_10a. Therefore, MM/PBSA can often distinguish the native structure from the other poses in most complexes, despite the positive values indicating an imperfect match between the ligand and protein in static snapshots, which could mainly be ascribed to different force fields being used for the docking method and for MM/PBSA. Dll values turned out to be negative under the specific force fields over times during molecular dynamics. In comparison, the average MM/PBSA values from the dynamic trajectories (violet points in Fig. 5(a)–(f)) are substantially lower than those from the static snapshots, indicating that the structural relaxation during molecular dynamics provide more reasonable structures. The results from the dynamic analysis highlight the differences between the c_2a and c_5a poses, as well as those between the c_2a and c_10a, boosting the number of positive samples from 68 to 84 and 80, respectively. Moreover, the structural relaxation leads to a much narrower gap between c_native and c_2a poses, consistent with the drastic increase in Pearson coefficient from 0.054 to 0.83 as shown in Fig. 6l. In contrast, relaxation increases the difference between c_5a and c_10a poses from 49:51 to 62:38. This difference could result from the fact that both c_5a and c_10a are metastable structures on the potential energy surface, which is insufficient to unambiguously determine the energetically favored pose. Additionally, it is reasonable to assume that two initial conformations differing remarkably in geometry will either evolve towards similar or different structures, thus leading to the energetic disparity. Next, we analyzed the interactions in terms of the decomposed energy (ELE, VDW, PBSUR, PBSOL, detailed explanations for these abbreviations is available in mm_pbsa.pl). Fig. 5(g)–(k) reveals that the dominant contributions for the differences come from ELE and VDW. Hence, the non-covalent interaction in vacuum can be inferred from ELE and VDW decomposed from the MM/PBSA binding affinities.
 | ||
| Fig. 6 MM/PBSA performance on trajectories at 2 ns intervals during the full 25 ns MD simulations; (a) the averaged MM/PBSA binding affinities for 4xtx; (b) the averaged MM/PBSA binding affinities for 5fov; (c) the averaged MM/PBSA binding affinities for all selected 100 complexes; (d) the Pearson correlation coefficient between experimental (binding affinity) values and MM/PBSA; (e) the number of complexes for which the averaged MM/PBSA binding affinity of the former trajectories larger than that of the latter for each pair among four initial poses. | ||
Subsequently, considering dynamic factors, we calculated the MM/PBSA binding affinities of 2500 snapshots from each of the 25 ns trajectories at 100 ps intervals. Fig. 5l shows the Pearson correlation coefficients for the binding affinities of different poses in the initial state and the averaged MM/PBSA binding affinities for the last 5 ns of the simulation, as well as the experimental values. The MM/PBSA binding affinity scores for the static poses generally exhibits a weaker correlation with the experimental values than the dynamic pose MM/PBSA binding affinities. For the static states, the highest Pearson correlation of 0.15 is observed with the c_2a, and similar for the corresponding dynamic poses, the highest Pearson correlation of 0.53 is achieved with the c_2a poses, closely followed by 0.45 from the c_native poses. These correlations far exceed the values obtained from the c_5a and c_10a poses, which are less than 0.25.
Furthermore, to investigate the evolution of protein and ligand flexibility over time, we divided the 2500 snapshots into 12 intervals of 2 ns each and calculated the averaged MM/PBSA scores during these periods. In similar fashion, we analyzed the trajectories of the 4xtx and 5fov complexes to observe the changes in MM/PBSA scores over time. From the 4xtx trajectories (Fig. 6(a)), the c_native (blue) and c_2a poses (sky blue) exhibit higher binding affinities compared to the c_5a (salmon) and c_10a poses (pink). In contrast, from the 5fov trajectories (Fig. 6(b)), the binding affinities in c_5a trajectories are stronger than those in the other three initial poses. These results indicate that even with an analysis employing dynamic trajectories, MM/PBSA does not always accurately distinguish the correct binding pose. Surprisingly, when considering all 100 complexes (Fig. 6(c)), the c_2a initial pose consistently outperform the other three initial poses, including the c_native pose in terms of Pearson correlation between MM/PBSA and experimental values (Fig. 6(d)). Although the difference between the c_native pose and c_2a pose is small, they both significantly outperform the c_5a and c_10a poses. Additionally, there is little fluctuation in the correlation values during the simulations. This is also reflected in the number of complexes where the averaged MM/PBSA binding affinity of pose1 is higher than that of pose2 among the four initial poses. These findings suggest that 25 ns of simulation time is not always sufficient to estimate binding affinities to experimental accuracy.
 | ||
| Fig. 7 Screening evaluation. (a) Screening performance of dynamic analyses combining averaged RMSD and averaged MM/PBSA during last 5 ns simulations. (b) Screening performance of dynamic analyses combining averaged RMSD and averaged MM/PBSA during last 5 ns simulations plus the lasting time of the strongest three interaction pairs among hydrophobic interactions, hydrogen bond interactions and water bridge interactions. | ||
2 Conclusion
In this study, we systematically investigated molecular dynamics trajectories for 25 ns simulatiom times for 100 different complexes staring from four initial conformations (c_native, c_2a, c_5a and c_10a) using both geometric and energetic arguments.From a geometric point of view, we analyzed the RMSD relative to the initial structure of the molecular dynamics simulation, as well as the decomposed interactions using PLIP. The c_native poses consistently exhibited higher stability compared to c_5a and c_10a. The average RMSD values of all 100 complexes gradually increased over time for all initial poses, indicating a wider range of configurations deviating from the initial structures being visited, aligning with the canonical distribution.
Regarding the energetic aspect, the remarkably high Pearson coefficient (0.86) between c_native and c_2a poses in terms of MM/PBSA binding affinities indicates that initial structures deviating slightly from one another tend to exhibit similar molecular dynamics trajectories within the accessible simulation time. Conversely, the low correlation between the c_native and the c_5a and c_10a poses illustrate the difficulty of initial structures with significant deviations in overcoming high energy barriers caused by factors like torsion angles. Moreover, in terms of binding affinity correlations, the c_2a initial conformations exhibits a significant energetic advantage over the c_5a and c_10a poses, even slightly outperforming the c_native poses.
Furthermore, simulation time shows similar patterns in the rankings of the four poses in terms of both RMSD and MM/PBSA binding affinities, although longer simulation times did not necessarily improve the results. Overall, the combination of these techniques improves the accuracy of identification of native poses compared to relying solely on single static poses.
In conclusion, the Moira workflow provides a comprehensive paradigm for the exploration of ligand-protein interactions based on classical molecular dynamics, including both the simulations and their analysis. Thereby, the workflow facilitates convenient investigation of ligand–protein interactions on a general basis, important for the further advancement of drug design methodologies.
3 Methods
3.1 Workflow
The Molecular dynamics trajectory analysis (Moira) package is comprised of four modules (Dock_suite, Md_suite, MM/PBSA_suite and PLIP_suite). It integrates several classical software packages and scripts, including software for simulations (AutoDock Vina v.1.1.2,35 Amber2036), analysis (isoRMSD.py script from https://github.com/0ut0fcontrol/isoRMSD, mm_pbsa.pl script in Amber20 and PLIP v.2.1.737) and visualization tools (pymol v.2.5.2,38 jupyter notebook v.6.0.139). When provided with an initial 3D complex structure as input, for instance as a pdb file, the workflow automatically implements all the calculations to generate the MD trajectories, executes the analysis of the latter, as well as their visualization (Fig. 8). | ||
| Fig. 8 Workflow of our methodology. | ||
As shown in Fig. 1, docked protein–ligand structures were first generated using RDKit to provide the initial 10 ligand conformations. Then, by using Vina to make the docked structures while keeping the best 10 of each initial ligand conformation, it demonstrated that these conformations are at least reasonable as docked ligand structures. Secondly, taking the deviation from native structures into consideration, c_2a (usually regarded as the native structure with trivial deviation), c_5a (slight deviation), c_10a (drastic deviation) structures from molecular docking plus native structure were selected as the initial structures for molecular dynamics runs to generate the 25 ns trajectories, which were split into 2 fs intervals. Finally, the resulting trajectories were comprehensively analysed.
3.2 Data cleaning
All experimental crystal structures are acquired from the pdbbind-CN database version 2019 (https://www.pdbbind.org.cn), which gathers X-ray Free Electron Laser (X-ray), Nuclear magnetic resonance (NMR), or Single-particle electron cryo-microscopy (cryo-EM) data. Until now, the database has collected 17679 protein–ligand crystal structures with binding affinities in terms of the equilibrium dissociation (Kd) and the inhibition (Ki) constants or the half maximal inhibitory concentration values (IC50). Among them, 4852 entries are considered high quality. We analyze the database in terms of the following aspects: (1) the SMILES of the ligands, (2) the protein sequence, (3) the uniprot ID of the proteins, (4) the number of chains, (5) the number of metal ions, and (6) the number of non-standard amino acids. For multiple chains, we select one chain. A ligand interacting only with a single chain without direct contact with other chains will have a negligible effect on the protein–ligand interactions in total. On the other hand, if the ligand is surrounded by multiple chains, it will dramatically influence the interaction pattern between the ligand and the protein.
Thus, we randomly selected 100 compounds (shown in Table S1, ESI†) from the high-quality set in the pdbbind-CN database, which were subsequently filtered on the basis of the six aspects described above to avoid multiple chains, metal ions and non-standard amino acids in our complexes. We thereby simplified our analysis by reducing the influence from the aforementioned confounding factors.
3.3 Molecular docking
Molecular docking was performed on 17679 complexes with Autodock Vina to generate ligand docking poses.
For the receptor, the protein pdb file was pre-processed to remove solvent molecules, multiple chains and metal ions using PyMOL. For the ligand, it is known that the number of possible conformers is exponentially correlated to the number of rotatable bonds. To obtain multiple highly different conformers, we generated 3D conformations from SMILES.
Step 1: convert ligand mol2 file to SMILES with allBondsExplicit and allHsExplicit true as well as sanitize False based on RDKit package.
Step 2: employ AllChem.EmbedMultipleConfs module to generate 10 conformations with pruneRmsThresh of 1 and maxAttempts of 500 from SMILES, which will be taken as initial conformers in the format of pdb file for docking.
Step 3: convert the receptor and ligand pdb files to the pdbqt format required by Autodock Vina using the MGLTools program of the AutoDockTools package. All rotatable bonds are allowed to relax during docking, while receptor atoms were all kept rigid.
Step 4: define the docking parameters. The geometric center of the native ligand atoms was defined as the center grid of the docking cubic box, and the box grid was set as 60 Å × 60 Å × 60 Å with 0.375 Å per grid point. The 10 best scoring poses were retained each time. The maximum allowed score difference between the lowest and the highest scores was set to 10 during docking, and default values were used for other parameters.
In consideration of some sophisticated situations at different stages, we removed complexes which failed to generate conformers by RDKit, or those that failed during docking or RMSD calculation in subsequent steps. In the end, 13424 complexes passed successfully through all of the steps in the procedure, generating 1044819 conformers in total. The isoRMSD.py script was used to calculate the RMSDs between the docked and the native ligand poses without translation and rotation after aligning the protein. Due to the experimental uncertainty of the hydrogen positions, only heavy atoms are considered. The docking and native ligand RMSD values range from 0 to 20 Å, with a spread approximating a normal distribution.
3.4 Molecular dynamics
All molecular dynamics simulations were implemented using the pmemd module of the Amber 20 program. For the preparation of input files, ligands were parametrized with restrained electrostatic potential charges generated by AM1-BCC methodology, while proteins were described based on the Amber ff14SB. Then, the complex was solvated in a TIP3P cuboid water box with a distance of at least 12 Å from any protein atoms. Na+ or Cl− were used as counter ions to neutralize the system charges for the simulations. Particle mesh Ewald (PME) was employed to treat the long-range electrostatic interactions. The dielectric constant was set to 1.0, and The nonbonded interactions are truncated to within 8.0 Å.For the simulations, there are four stages: (1) minimization: a total of 1000 steps of minimization, the box was initially minimized by 500 steepest descent steps, followed by 500 conjugate gradient steps. (2) Heating: the box was then gradually heated up to 300 K from 0 K and was relaxed within 50 ps by a simulation in the NVT ensemble (constant number of atoms, volume, and temperature). (3) Equilibrium: the system was further relaxed in an NPT ensemble (constant number of atoms, pressure, and temperature) simulation of 50 ps. For the first three stages above, weak restrains with 2.0 kcal mol−1 Å−2 was assigned to all heavy atoms of the complex for first three stages. (4) Production: the molecular dynamics simulation of 25.5 ns was carried out under NPT ensemble with all atoms relaxed. Due to the expensive computational demand, only one single MD runs were performed for each docking pose, despite the influence of the initial velocities assigned in the simulations.40,41
3.5 RMSD
All RMSD calculations are done considering only heavy atoms. High RMSD values suggest large deviation from reference structure, implying a distinct change. If the RMSD of a ligand was larger than the given threshold (2.0 Å), the docked pose was considered highly deviant (static) or unstable (dynamic). For docking, the absolute RMSD measures the distance of a generated docking pose relative to the experimental native ligand conformation. For molecular dynamics, an additional alignment step of the proteins was carried out initially to avoid including translation effects. In the MD trajectories, the RMSD measures the distance of poses relative to the initial ligand conformations.3.6 PLIP-interaction analysis
Geometric criteria with default values (distance and angle thresholds) in PLIP were used to define various interactions, and the definitions of interaction patterns are as follows (more details is available in the PLIP website. https://github.com/pharmai/plip/blob/master/plip/basic/config.py).3.7 MM/PBSA
In MM/PBSA, the binding free energy (ΔGbind) between a ligand (L) and a receptor (R) to form a complex RL is calculated as:| ΔGbind = ΔH − TΔS = ΔEMM + ΔGsol − TΔS |
| ΔEMM = ΔEinternal + ΔEelectrostatic − ΔEvdw |
| ΔGsol = ΔGPB + ΔGSA |
| ΔGSA = γ × SASA + b |
| ΔGbind = ΔGcomplex − ΔGprotein − ΔGligand |
Data and software availability
All information including 3D structures and binding affinities of the complexes were downloaded from PDBbind-CN database version 2019 (https://www.pdbbind.org.cn/). The original poses and analyses can be obtained on the Zenodo repository at https://doi.org/10.5281/zenodo.7823237. All calculations and analysis software or scripts in this work include AutoDock Vina v.1.1.2,35 Amber20,36 isoRMSD.py script from https://github.com/0ut0fcontrol/isoRMSD, mm_pbsa.pl script in Amber20 and PLIP v.2.1.737) as well as visualization tools (pymol v.2.5.2,38 jupyter notebook v.6.0.139).Code availability
The source codes and examples of Moira are available on the GitHub repository at https://github.com/Meganwu/moira.Author contributions
N. W. conceived the concept. N. W. prepared and processed all data as well as analyzed the results. N. W. and R. Z implemented the model and performed computational experiments. N. W. drew all figures and wrote the manuscript. R. Z., X. P., L. F., K. C. and J. S. J. participated in discussions and modifications for language, figures and results in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the National Key Research and Development Program of China (2021YFF1201300), the National Natural Science Foundation of China (61872216). Thanks for valuable supports and discussions from Jianyang Zeng at Tsinghua university.References
- P. Śledź and A. Caflisch, Protein structure-based drug design: from docking to molecular dynamics, Curr. Opin. Struct. Biol., 2018, 48, 93–102 CrossRef PubMed.
- E. H. B. Maia, L. C. Assis, T. A. De Oliveira, A. M. Da Silva and A. G. Taranto, Structure-based virtual screening: from classical to artificial intelligence, Front. Chem., 2020, 8, 343 CrossRef CAS PubMed.
- J. Bao, X. He and J. Z. Zhang, DeepBSP—a machine learning method for accurate pre574 diction of protein–ligand docking structures, J. Chem. Inf. Model., 2021, 61(5), 2231–2240 CrossRef CAS PubMed.
- J. Jiménez, M. Skalic, G. Martinez-Rosell and G. De Fabritiis, K deep: protein–ligand absolute binding affinity prediction via 3d-convolutional neural networks, J. Chem. Inf. Model., 2018, 58(2), 287–296 CrossRef PubMed.
- D. Jones, H. Kim, X. Zhang, A. Zemla, G. Stevenson and W. D. Bennett, et al., Improved protein–ligand binding affinity prediction with structure-based deep fusion inference, J. Chem. Inf. Model., 2021, 61(4), 1583–1592 CrossRef CAS PubMed.
- C. Shen, J. Ding, Z. Wang, D. Cao, X. Ding and T. Hou, From machine learning to deep learning: Advances in scoring functions for protein–ligand docking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10(1), e1429 CAS.
- J. Åqvist, C. Medina and J. E. Samuelsson, A new method for predicting binding affin586 ity in computer-aided drug design, Protein Eng., Des. Sel., 1994, 7(3), 385–391 CrossRef PubMed.
- S. Genheden and U. Ryde, The MM/PBSA and MM/GBSA methods to estimate ligand589 binding affinities, Expert Opin. Drug Discovery, 2015, 10(5), 449–461 CrossRef CAS PubMed.
- E. Wang, H. Sun, J. Wang, Z. Wang, H. Liu and J. Z. Zhang, et al., End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design, Chem. Rev., 2019, 119(16), 9478–9508 CrossRef CAS PubMed.
- H. Sun, Y. Li, S. Tian, L. Xu and T. Hou, Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA method595 ologies evaluated by various simulation protocols using PDBbind data set, Phys. Chem. Chem. Phys., 2014, 16(31), 16719–16729 RSC.
- T. Tuccinardi, What is the current value of MM/PBSA and MM/GBSA methods in drug discovery?, Expert Opin. Drug Discovery, 2021, 16(11), 1233–1237 CrossRef CAS PubMed.
- L. Wang, J. Chambers and R. AbelProtein–ligand binding free energy calculations with FEP+, Biomolecular Simulations, Springer, 2019, pp. 201–232 Search PubMed.
- L. F. Song and K. M. Merz Jr, Evolution of alchemical free energy methods in drug discovery, J. Chem. Inf. Model., 2020, 60(11), 5308–5318 CrossRef CAS PubMed.
- W. Jiang and B. Roux, Free energy perturbation Hamiltonian replica-exchange molecular dynamics (FEP/H-REMD) for absolute ligand binding free energy calculations, J. Chem. Theory Comput., 2010, 6(9), 2559–2565 CrossRef CAS PubMed.
- J. Chen, X. Wang, L. Pang, J. Z. Zhang and T. Zhu, Effect of mutations on binding of ligands to guanine riboswitch probed by free energy perturbation and molecular dynamics simulations, Nucleic Acids Res., 2019, 47(13), 6618–6631 CrossRef CAS PubMed.
- M. Li, X. Liu, S. Zhang, S. Liang, Q. Zhang and J. Chen, Deciphering the binding mechanism of inhibitors of the SARS-CoV-2 main protease through multiple replicaaccelerated molecular dynamics simulations and free energy landscapes, Phys. Chem. Chem. Phys., 2022, 24(36), 22129–22143 RSC.
- D. Plewczynski, M. Łaźniewski, R. Augustyniak and K. Ginalski, Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database, J. Comput. Chem., 2011, 32(4), 742–755 CrossRef CAS PubMed.
- N. S. Pagadala, K. Syed and J. Tuszynski, Software for molecular docking: a review, Biophys. Rev., 2017, 9(2), 91–102 CrossRef CAS PubMed.
- K. Crampon, A. Giorkallos, M. Deldossi, S. Baud and L. A. Steffenel, Machine-learning methods for ligand–protein molecular docking, Drug Discovery Today, 2021, 27(1), 151–164 CrossRef PubMed.
- J. A. Erickson, M. Jalaie, D. H. Robertson, R. A. Lewis and M. Vieth, Lessons in molecular recognition: the effects of ligand and protein flexibility on molecular docking accuracy, J. Med. Chem., 2004, 47(1), 45–55 CrossRef CAS PubMed.
- R. D. Taylor, P. J. Jewsbury and J. W. Essex, A review of protein-small molecule docking methods, J. Comput.-Aided Mol. Des., 2002, 16(3), 151–166 CrossRef CAS PubMed.
- G. M. Verkhivker, D. Bouzida, D. K. Gehlhaar, P. A. Rejto, S. Arthurs and A. B. Colson, et al., Deciphering common failures in molecular docking of ligand-protein complexes, J. Comput.-Aided Mol. Des., 2000, 14(8), 731–751 CrossRef CAS PubMed.
- M. Ogrizek, S. Turk, S. Lešnik, I. Sosič, M. Hodošček and B. Mirković, et al., Molecular dynamics to enhance structure-based virtual screening on cathepsin B, J. Comput.-Aided Mol. Des., 2015, 29(8), 707–712 CrossRef CAS PubMed.
- T. Sakano, M. I. Mahamood, T. Yamashita and H. Fujitani, Molecular dynamics analysis to evaluate docking pose prediction, Biophys. Physicobiol., 2016, 13, 181–194 CrossRef CAS PubMed.
- T. Young, R. Abel, B. Kim, B. J. Berne and R. A. Friesner, Motifs for molecular recognition exploiting hydrophobic enclosure in protein–ligand binding, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(3), 808–813 CrossRef CAS PubMed.
- N. T. Southall, K. A. Dill and A. Haymet, A view of the hydrophobic effect, ACS Publications, 2002 Search PubMed.
- M. Maurer and C. Oostenbrink, Water in protein hydration and ligand recognition, J. Mol. Recognit., 2019, 32(12), e2810 CrossRef CAS PubMed.
- S. K. Panigrahi and G. R. Desiraju, Strong and weak hydrogen bonds in the protein–ligand interface, Proteins: Struct., Funct., Bioinf., 2007, 67(1), 128–141 CrossRef CAS PubMed.
- F. Y. Lin and A. D. MacKerell Jr, Do halogen–hydrogen bond donor interactions dominate the favorable contribution of halogens to ligand–protein binding?, J. Phys. Chem. B, 2017, 121(28), 6813–6821 CrossRef CAS PubMed.
- K. Liu, E. Watanabe and H. Kokubo, Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations, J. Comput.-Aided Mol. Des., 2017, 31(2), 201–211 CrossRef CAS PubMed.
- S. Zhou and L. Wang, Unraveling the structural and chemical features of biological short hydrogen bonds, Chem. Sci., 2019, 10(33), 7734–7745 RSC.
- D. C. Thompson, C. Humblet and D. Joseph-McCarthy, Investigation of MM-PBSA rescoring of docking poses, J. Chem. Inf. Model., 2008, 48(5), 1081–1091 CrossRef CAS PubMed.
- H. Sahakyan, Improving virtual screening results with MM/GBSA and MM/PBSA rescoring, J. Comput.-Aided Mol. Des., 2021, 35(6), 731–736 CrossRef CAS PubMed.
- G. Landrum, et al., RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling, Greg. Landrum, 2013, 8, 31 Search PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31(2), 455–461 CrossRef CAS PubMed.
- D. A. Case, T. E. Cheatham III, T. Darden, H. Gohlke, R. Luo and K. M. Merz Jr, et al., The Amber biomolecular simulation programs, J. Comput. Chem., 2005, 26(16), 1668–1688 CrossRef CAS PubMed.
- M. F. Adasme, K. L. Linnemann, S. N. Bolz, F. Kaiser, S. Salentin and V. J. Haupt, et al., PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA, Nucleic Acids Res., 2021, 49(W1), W530–W534 CrossRef CAS PubMed.
- W. L. DeLano, et al., Pymol: An open-source molecular graphics tool, CCP4 Newsl. Protein Crystallogr., 2002, 40(1), 82–92 Search PubMed.
- T. Kluyver, B. Ragan-Kelley, F. Pérez, B. E. Granger, M. Bussonnier, J. Frederic, et al., Jupyter Notebooks-a publishing format for reproducible computational workflows, 2016, vol. 2016 Search PubMed.
- S. A. Hollingsworth and R. O. Dror, Molecular dynamics simulation for all, Neuron, 2018, 99(6), 1129–1143 CrossRef CAS PubMed.
- K. Kang and W. Cai, Size and temperature effects on the fracture mechanisms of silicon nanowires: Molecular dynamics simulations, Int. J. Plast., 2010, 26(9), 1387–1401 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp03492e |
| ‡ These authors contributed equally. |
| This journal is © the Owner Societies 2024 |