
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Refining protein amide I spectrum simulations with simple yet effective electrostatic models for local wavenumbers and dipole derivative magnitudes†
Cesare M.
Baronio
 and
Andreas
Barth
*
and
Andreas
Barth
*
Department of Biochemistry and Biophysics, Stockholm University, Stockholm, Sweden. E-mail: barth@dbb.su.se
First published on 15th December 2023
Abstract
Analysis of the amide I band of proteins is probably the most wide-spread application of bioanalytical infrared spectroscopy. Although highly desirable for a more detailed structural interpretation, a quantitative description of this absorption band is still difficult. This work optimized several electrostatic models with the aim to reproduce the effect of the protein environment on the intrinsic wavenumber of a local amide I oscillator. We considered the main secondary structures – α-helices, parallel and antiparallel β-sheets – with a maximum of 21 amide groups. The models were based on the electric potential and/or the electric field component along the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond at up to four atoms in an amide group. They were bench-marked by comparison to Hessian matrices reconstructed from density functional theory calculations at the BPW91, 6-31G** level. The performance of the electrostatic models depended on the charge set used to calculate the electric field and potential. Gromos and DSSP charge sets, used in common force fields, were not optimal for the better performing models. A good compromise between performance and the stability of model parameters was achieved by a model that considered the electric field at the positions of the oxygen, nitrogen, and hydrogen atoms of the considered amide group. The model describes also some aspects of the local conformation effect and performs similar on its own as in combination with an explicit implementation of the local conformation effect. It is better than a combination of a local hydrogen bonding model with the local conformation effect. Even though the short-range hydrogen bonding model performs worse, it captures important aspects of the local wavenumber sensitivity to the molecular surroundings. We improved also the description of the coupling between local amide I oscillators by developing an electrostatic model for the dependency of the dipole derivative magnitude on the protein environment.
O bond at up to four atoms in an amide group. They were bench-marked by comparison to Hessian matrices reconstructed from density functional theory calculations at the BPW91, 6-31G** level. The performance of the electrostatic models depended on the charge set used to calculate the electric field and potential. Gromos and DSSP charge sets, used in common force fields, were not optimal for the better performing models. A good compromise between performance and the stability of model parameters was achieved by a model that considered the electric field at the positions of the oxygen, nitrogen, and hydrogen atoms of the considered amide group. The model describes also some aspects of the local conformation effect and performs similar on its own as in combination with an explicit implementation of the local conformation effect. It is better than a combination of a local hydrogen bonding model with the local conformation effect. Even though the short-range hydrogen bonding model performs worse, it captures important aspects of the local wavenumber sensitivity to the molecular surroundings. We improved also the description of the coupling between local amide I oscillators by developing an electrostatic model for the dependency of the dipole derivative magnitude on the protein environment.
Introduction
The amide I vibrations of proteins give rise to the most analyzed absorption band in biological infrared spectroscopy. These vibrations comprise mainly the CO stretching vibration of the polypeptide backbone. The amide I vibration of a particular amide group is influenced by the electrostatic environment and thus by the strength and pattern of hydrogen bonding to this amide group.1,2 In addition, it couples to the amide I vibrations of other amide groups,3,4 which gives rise to collective and delocalized amide I normal modes. These effects make the amide I absorption sensitive to the secondary structure of the protein backbone, which explains its importance for the characterization of proteins.
The theoretical description of the amide I absorption reaches back several decades.5–8 At present, it is able to explain the structural sensitivity in qualitative terms but is still insufficient for a quantitative analysis. Thus, further improvement is desirable in order to extract more structural information from the experiments by computing spectra of structural models. This will be beneficial for the interpretation of absorption spectra as well as of difference spectra of protein reactions, where the interest is to localize and characterize the protein backbone segments that generate the observed conformational changes.
Current approaches9 to model the amide I spectrum of proteins include quantum chemical calculations of small structures10,11 and parameter transfer to larger structures,12–16 as well as Fourier transformation of the dipole autocorrelation function.17,18 However, because quantum chemical calculations are too time-consuming for ordinary proteins, most calculations are based on the floating oscillator mechanism,8,19–22 in which each amide group hosts an amide I oscillator that is characterized by its local wavenumber and its coupling with the oscillators of other amide groups. Coupling results in collective amide I normal modes, to which individual amide I oscillators contribute to varying degrees. Nearest neighbor coupling constants are often taken from quantum chemical calculations23–27 and include mechanical through-bond interactions, whereas interactions between non-nearest neighbor amide I oscillators can be described successfully by transition dipole coupling (TDC)3,4,7,28–30 or transition charge coupling.26,31 Our previous work32 has optimized the parameters for TDC for the main secondary structures – α-helices, parallel and antiparallel β-sheets – by comparison with DFT calculations of the amide I infrared absorption.
If there were no vibrational coupling, each amide group would vibrate independently from the other amide groups with its local wavenumber, which depends on the environment of the amide group.1,2,24,33,34 This environmental influence is typically divided into two components: (i) the contribution from the two closest amide group neighbors within the peptide chain and (ii) the contribution from the rest of the protein and from the water environment. The first influence depends on the local conformation of the protein backbone and is thus termed local conformation effect. It alters the potential energy surface of a specific amide group through a combination of electrostatic interactions and electronic polarization effects.35 In the floating oscillator approach, this influence is usually implemented by applying wavenumber shifts to the local wavenumbers that were obtained from a series of density functional theory (DFT) calculations of diamides with different backbone dihedral angles.24–27 The shifts induced by the preceding and following neighbors of a particular amide group are thought to be additive.
The second influence on the local wavenumber has been modeled differently in calculations of protein spectra: either by parameter transfer from DFT calculations of small structures,36 by local hydrogen bonding models,20,32,37–39 or by electrostatic models that relate the electric field, field gradients and/or the electric potential at the amide group atoms to the local wavenumber.33,35,40–48 Field and potential influence the local wavenumber because they change the electrostatic energy during a molecular vibration – the former couples to electrons that follow the vibrations of the nuclei, whereas the latter couples to those that participate in an interatomic charge flux.49
Most of the electrostatic models were developed from simulations of an amide group that interacts with water molecules. In a different approach, the model was obtained from experimental data.50 To the best of our knowledge, the susceptibility of the local wavenumber on the surrounding protein environment has not yet been modeled from DFT calculations of peptide structures. There is also a need to optimize the set of charges used to describe the spectroscopic effects.21,51 This has been done for a set of diamides and NMA dimers using an electrostatic model developed for the interaction with water (2H2O),35 resulting in a large negative charge on the amide oxygen, a positive charge on the nitrogen and a negative charge on the hydrogen, which differs from common force fields.
Here, we present a combined approach where charges and parameters were optimized for a set of common secondary structures with the aim to describe the effect of the electrostatic environment on the local wavenumber of each amide group. We find that a model considering the electric field at the positions of the O, N, and H atoms is a good compromise between the quality of the description, the number of free parameters, and the robustness of the model. This model is superior to a description by a local hydrogen bonding model.
A further quantity considered in this work is the local dipole derivative, which influences the calculated spectrum in two ways:
(i) It determines the strength of TDC coupling between non-nearest neighbors. Thus, correct dipole derivatives are important for example to calculate the correct splitting between the high and low wavenumber bands of antiparallel β-sheets.
(ii) The square of the dipole derivative is proportional to the dipole strength, which is proportional to the integrated absorbance of an absorption band. Correct dipole derivatives are therefore a prerequisite for the correct calculation of the relative intensities of the normal modes.
It is known that the dipole derivative magnitude is increased by hydrogen bonding by a factor of ∼1.4, corresponding to a change in dipole strength by a factor of ∼2,2,52,53 and it was found to differ by 20% for different amide groups in a β-hairpin.54 Nevertheless it is usually approximated by a constant in protein spectrum calculations. In order to consider the effect of the molecular environment on the dipole derivative magnitude, we have previously presented a model that is based on the strength of hydrogen bonding to the carbonyl group of the considered amide group.32 Here, we present instead an electrostatic model for this purpose and find that it describes the TDC coupling constants better. Both improvements discussed in this work are steps towards a quantitative description of the protein absorption in the amide I range, for which the theory-based parameters eventually need to be recalibrated by comparison to experimental data.
Methods
Structures
Our DFT calculations were done on poly-L-alanine model structures (Ac–(Ala)n–NH–CH3) for parallel and antiparallel β-sheets and for α-helices, as described previously.32 Medium and large structures contained 11 or 21 amide groups for the α-helices and two or four strands with five amide groups each for the sheets. Small structures contained three amide groups for the α-helix and five amide groups for individual β-strands. These nine structures were used to optimize and assess the electrostatic models for the local wavenumber shifts and contain together 105 amide groups, of which 71 are inner amides with an N- and C-terminal sequence neighbor.Additionally, we did DFT calculations on di-amides Ac–Ala–NH–CH3 (2 amide groups) with the same secondary structures and trans N-methylacetamide (NMA). The dihedral angles for all structures were: ϕ = −138.6°, φ = 134.5° for the antiparallel β-strands; ϕ = −121.0°, φ = 114.8° for the parallel β-strands; and ϕ = −57.0°, φ = −47.0°, ω = 180.0° for the α-helices.32 Selected structures are shown in Fig. S1 and S2 of the ESI† and pdb files of the structures are contained in a supplementary data set that is available on figshare (https://doi.org/10.17045/sthlmuni.24324886).
DFT calculations
For optimizing the electrostatic models we used the Gaussian 0955 DFT calculations with density functional BPW9156–59 and 6-31G** basis set from our previous article,32 which also contained a motivation of the choice of functional and basis set. The structures were optimized with frozen ϕ, φ, and – for the helices – also ω angles. Freezing ω was not necessary for the sheets because ω was close to 180° after geometry optimization. Molden was used to store the optimized structure in Cartesian coordinates, which served as input structure for calculating the vibrational properties at the same level of theory. The additional DFT calculations of diamides and NMA done for this work were performed in the same way. The log files of the frequency calculations are contained in the supplementary data set (https://doi.org/10.17045/sthlmuni.24324886).The F-matrix was obtained using Hessian matrix reconstruction60 by transforming the wavenumber eigenvalue matrix from the DFT calculations into a reconstructed Hamiltonian that contains the local wavenumbers in the diagonal and the coupling constants in cm−1 outside the diagonal (available for all structures in the supplementary data set at https://doi.org/10.17045/sthlmuni.24324886). The former are termed local DFT wavenumbers ṽDFT. Finally, this matrix was converted into an F-matrix32 using published conversion factors19,61,62 and made symmetrical by averaging the matrix elements that refer to the interaction between the same two amide groups. The result is termed DFT F-matrix.
Electrostatic models for the local wavenumber
The electric potential Φ (eqn (1)) and the electric field E (eqn (2)) were calculated at the locations of the four amide atoms C, O, N, and H of each amide group from partial charges on all other amide groups using. For this we used several charge sets, which are specified in the suplementary data set on figshare. The most relevant sets are compiled in Table S1 (ESI†). Atoms other than the four amide atoms were uncharged.| Φi = Σcj/rij | (1) |
Ei = ΣeCO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) eijcj/rij2 eijcj/rij2 | (2) |
Bold print indicates vectors. Φi is the electric potential (unit: Eha0−1), Ei the electric field component parallel to the CO bond (unit: Ehe−1a0−1) at the position of atom i of the amide group of interest, cj the partial charge (unit: elementary charge e) of atom j in an amide group outside the considered amide group, rij the scalar distance (unit: Bohr radius a0) between atoms i and j, eij the unit vector along the direction from atom j to atom i, eCO the unit vector from the C-atom to the O-atom of the amide group of interest and the sums are over all amide atoms j outside the amide group of interest. Ei is positive when the electric field component points from the C to the O atom.
The electrostatic models describe the effect of the electrostatic environment on the local amide I wavenumber ṽ of a particular amide group. This influence is expressed by a shift ΔṽESM (where ESM stands for electrostatic model) of the local amide I wavenumber ṽ from the wavenumber ṽ0 of an isolated amide group.
| ṽ = ṽ0 + ΔṽESM | (3) |
The most general model considered the electric field E and electric potential Φ at the locations of all four amide atoms to calculate ΔṽESM
| ΔṽESM = Σ(diEi + liΦi) | (4) |
O bond. This model is termed 4P4F8 because it evaluates the electric potential and the electric field at 4 atoms and has eight free parameters that relate the electrostatics to the wavenumber shift. We tested also one variant of this model, suggested by Torii,49,63 in which one d and one l parameter is dependent on the other parameters as described in detail in ESI.† Therefore it has only six free parameters and we term it 4P4F6.
The other electrostatic models are simplifications of the general 4P4F8 model and consider a subset of atoms and/or either only the fields or the potentials. The number of free parameters in the simpler models is given by the number of atoms at which the electric field and the electric potential are evaluated and is therefore not additionally stated as a subscript. For example, a model which considers the electric field at three atoms is named 3F and contains three independent parameters. The considered atoms are given as subscripts, e.g. 3FONH considers the field at the O, N, and H atom of the amide group for which the wavenumber shift ΔṽESM is calculated.
Short-range hydrogen bonding model
As an alternative to the electrostatic models, the local environment can also be described by a short-range model that considers only the hydrogen bonds of the amide group of interest.37,39,48 It relates the local wavenumber shift of a particular amide group to the Kabsch–Sander energies64 (EKS in units of kJ mol−1) of its hydrogen bonds.| ΔṽKS = ξOEOKS + ξHEHKS | (5) |
The parameters ξO and ξH describe the shifts due to hydrogen bonding to the amide oxygen and the amide hydrogen, respectively, and their values are ξO = 2.4 cm−1 mol kJ−1 and ξH = 1.0 cm−1 mol kJ−1.37 Note that the wrong units were stated in our previous publication.39
Parameter optimization for the local wavenumber shift
We used the small (3 or 5 amides), intermediate (10 or 11 amides) and large models (20 or 21 amides) for the three secondary structures to optimize the parameters and the charge set of the different electrostatic models. The shifts ΔṽESM calculated with the various electrostatic models were compared to shifts ΔṽDFT obtained from the DFT calculations, which were calculated as follows: the DFT amide I wavenumber of NMA (ṽNMA = 1735.0 cm−1) was subtracted from the local DFT wavenumbers ṽDFT obtained in the process of reconstructing the DFT F-matrix:| ΔṽDFT = ṽDFT − ṽNMA | (6) |
Then these shifts were subtracted to obtain the shift difference ΔΔṽ between the DFT and the electrostatic calculations
| ΔΔṽ = ΔṽDFT − ΔṽESM | (7) |
Two quality measures for the performance of the considered electrostatic models were calculated: SSDiff is the sum of the squared differences ΔΔṽ for the considered amide groups.
| SSDiff = ΣΔΔṽi2 | (8) |
The sum is either over all 105 amide groups in our set of nine structures, or – when mentioned – over the 71 inner amide groups, i.e. those that have an N- and a C-terminal neighbor. SSDiff can give large values even when the variations between the local wavenumbers within a given structure are well reproduced, but when there is a general difference to the DFT shifts for all amide groups in this structure. Such a general difference could for example be caused by an effect of the local conformation which will have a strong impact on the parameters of the model because the effect is present for all amide groups. Also, groups with only a single hydrogen bond are relatively few and might not be modeled well by only minimizing SSDiff, which could result in a poor modeling of the local wavenumber variations. This however, should be avoided because the groups within a secondary structure element are those that couple most strongly due to their closeness and the effect of coupling depends on the variation of their local wavenumbers. Therefore we aimed to increase the importance of the variations in the optimization and to reduce the effect of a general difference to the DFT shifts by introducing the second quality measure: SSDev.
For its calculation (eqn (9)), first an average shift difference ΔΔṽj between DFT and electrostatic shifts was calculated for each of the nine structures j. The average shift difference was calculated either from all amide groups of a given structure, or from only the inner amide groups when SSDev of the inner groups was of interest. Then, the deviation ΔΔṽi − ΔΔṽj of the individual shift differences from the average shift difference for the respective structure was calculated, squared and summed over all amide groups or over the inner groups.
| SSDev = Σ(ΔΔṽi − ΔΔṽj)2 | (9) |
To minimize the shift differences and to optimize the parameters, we used the Matlab function lsqnonlin where the SSDiff and SSDev values for each of the nine structures constituted the vector components of the vector-valued function used for minimization, which gives a total of 18 vector components. Most optimizations were done in this way, which is termed optimization method 1. However, we also minimized SSDiff and SSDev directly for a subset of models and charge sets, which is termed optimization method 2. Both optimizations produced similar results, with generally slightly smaller SSDiff and SSDev values for optimization method 2. For the individual large structures however, it also generated the worst RMSDiff and RMSDev values with the most relevant electrostatic models 3FONH (charge set Set-3FONH) and 4P4F8 (charge set Set-4P4F8) as well as the worst RMSDiff with a combination of local conformation effect and local hydrogen bonding model. Anticipating that the large structures represent protein structures better than the smaller models, we focus therefore on optimization method 1.
It turned out that the inclusion of SSDev in the parameter optimization improved its value considerably with only little increase of SSDiff. The parameters of the short-range hydrogen bonding model were optimized in the same way using ΔṽKS instead of ΔṽESM.
Optimization of charge set
Optimizing the charge set together with the model parameters failed because of the large number of parameters to be optimized. Therefore the charge set was pre-selected in all parameter optimizations. In one case – for the 4P4F6 model – we optimized the partial charges with fixed model parameters. This was done in the same way as described above for the parameter optimizations.Local conformation effects
For quantifying the local conformation effect, we used the local wavenumbers from our diamide DFT calculations. As for the other structures, these were converted into local wavenumber shifts by subtracting the reference wavenumber of NMA (1735.0 cm−1) from the local wavenumbers of the diamides.We used also previous results on the local conformation effect for the comparison with our diamide data. Choi & Cho,24 (indicated by C in our tables) tabulated local wavenumbers for different secondary structures (their Table 1), which we used without modification although the dihedral angles for the parallel β-sheet differ by 2 degrees from our values.
| Model | Number of charge setsa | Best charge set for | Name of charge setb | RMSDiff (cm−1) | RMSDev (cm−1) |
|---|---|---|---|---|---|
| a For each model, the number of studied charge sets is listed. b The charge sets are specified with a shorthand notation: the partial charges in e are multiplied with 100 and listed in the order C/O/N/H. The minus sign for negative partial charges is omitted and they are indicated by italic type instead. In some cases a good compromise is also listed. The best charge sets of the most relevant models were given names. c LCE refers to parameter optimizations where the local conformation effect was explicitly accounted for (see text). d For some models, two values are stated for RMSDiff and RMSDev: the first value relates to all amide groups, whereas the second in italics relates only to the inner amide groups, i.e. those that have an N-terminal and C-terminal neighbor. e The model named Zero refers to a calculation in which no wavenumber shift was applied, i.e. all amides had the NMA wavenumber and were compared to the DFT wavenumbers. | |||||
| 4P4F8 | 41 | RMSDev | 40/55/05/10 (set-4P4F8) | 4.39 | 3.57 |
| RMSDiff | 50/60/00/10 | 4.35 | 3.68 | ||
| 4P4F8 and LCEc | 8 | RMSDev and RMSDiff | 35/55/05/15 | 2.44 | 2.19 |
| 4P4F6 | 41 | RMSDev | 70/90/00/20 | 5.07 | 3.87 |
| Compromise | 50/60/10/20 | 4.80 | 3.97 | ||
| RMSDiff | 60/60/10/10 | 4.62 | 4.19 | ||
| 4P | 41 | RMSDev | 40/40/10/10 | 5.23 | 4.50 |
| RMSDiff | 42/42/10/10 | 5.23 | 4.50 | ||
| 4F | 41 | RMSDev | 40/55/00/15 (set-3FONH) | 5.18 | 3.84 |
| Compromise | 30/40/00/10 | 5.14 | 3.85 | ||
| RMSDiff | 50/65/00/15 | 5.13 | 3.87 | ||
| 3FONH | 36 | RMSDev | 40/55/00/15 (set-3FONH) | 5.27, 4.61d | 3.88, 3.26d |
| RMSDiff | 50/70/00/20 | 5.27, 4.59d | 3.88, 3.24d | ||
| Compromise for inner amides | 50/70/10/30 | 5.44, 4.60d | 3.96, 3.15d | ||
| 3FONH and LCEc | 9 | RMSDev | 35/50/00/15 | 4.68 | 4.31 |
| Compromise | 40/55/00/15 (set-3FONH) | 4.68 | 4.31 | ||
| RMSDiff | 45/60/00/15 | 4.67 | 4.32 | ||
| Zeroe | 38.5, 40.7d | 12.1, 10.2d | |||
Gorbunov et al.25 (indicated by G in our tables) presented maps of the local wavenumbers for the entire space of the dihedral angles Φ and Ψ using otherwise fully optimized gas phase DFT calculations in the middle panels of their Fig. 8. We identified amide group 1 with the N-terminal group and amide group 2 with the C-terminal group and ignored thus the footnote with reference list entry 40 in the original publication, which states the opposite numbering. Only our numbering provides agreement with our and other published data. The figures were digitized and the wavenumbers for specific backbone conformations extrapolated. Shifts of the local wavenumbers due to the local conformation effect were then obtained by scaling the wavenumbers with a factor of 0.9765 to improve the agreement with experiments and subsequent subtraction of our experimental reference wavenumber of 1707 cm−1 (see section Spectrum calculation).
La Cour Jansen et al.26,27 (indicated by LCJ in our tables) published tabulated shifts of the local wavenumbers, which were used as provided. When the absolute wavenumbers were of interest, the reference wavenumber was added to the shifts, which was the wavenumber of deuterated NMA in the gas phase (1717 cm−1)66 in the original publication.
Dipole derivative magnitude
Except for the nearest neighbor interactions, the non-diagonal elements in the F-matrix were calculated from TDC, which results from the electrostatic interaction of two transition dipole moments. The transition dipole moment for the transition between two vibrational states is proportional to the expectation value of the change of the molecular dipole moment upon a small change of the normal coordinate around the equilibrium position. This dipole derivative is a vector and therefore TDC calculations require the knowledge of the position, angle and magnitude of the dipole derivative, which can be expected to depend on the electrostatic environment. Here, we investigated whether its magnitude ∂μ/∂q can be described by an electrostatic model of the type| ∂μ/∂q = ∂μ0/∂q + ΣgiEi | (10) |
O bond at the locations of the four amide atoms i and the gi parameters relate the fields to a change in dipole derivative magnitude. We optimized these parameters, the dipole derivative angle, and its position using again the Matlab function lsqnonlin and considering the intermediate (10 or 11 amides) and large structural models (20 or 21 amides) as in our previous work.32 Thus, the vector of the input function for optimization consisted of six components – one for each structure. The component for each structure was the sum of its squared differences between non-diagonal F-matrix elements calculated by TDC and those from the DFT calculations. During optimization, the squared differences for the intermediate structures were multiplied by two in order to increase the weight of amide groups without complete hydrogen bonding. The quality of the model is then reported by the R value, which is simply the sum of the squared differences without multiplying the differences of the intermediate structures. It is calculated in the same way as the R values in our previous work32 and corresponds to SSDiff used to describe the optimization of the electrostatic models for the local wavenumber.
Spectrum calculation
The DFT spectra of the intermediate and large structures were published previously.32 All spectra presented in this work were obtained by diagonalizing the F-matrix with our Matlab program. We used different types of F-matrices to generate the spectra: (i) F-matrices containing only the diagonal elements (Fig. 2) either from the DFT F-matrix or calculated with the different local wavenumber models, (ii) DFT F-matrices, where all elements were obtained by Hessian reconstruction from the DFT results (Fig. 3, 4 and Fig. S5, S6, ESI†); (iii) F-matrices, which contained diagonal elements calculated with the local wavenumber models and the original non-diagonal elements from the DFT F-matrix (Fig. 3 and Fig. S5, S6, ESI†); and (iv) F-matrices, where the non-diagonal, non-nearest neighbor elements were calculated from TDC and the diagonal elements were obtained by the 3FONH model (Fig. 4). For all spectra except those in Fig. 2, the nearest neighbor elements were those of the DFT F-matrix.Before plotting the spectra, the diagonal elements of the DFT F-matrix were reduced to achieve a downshift of the local wavenumbers by 28 cm−1. Such a shift brings our calculated NMA gas phase wavenumber of 1735.0 cm−1 down to the wavenumber of trans-NMA in a N2 matrix (1706–1707 cm−1)67,68 and close to those in H2 (1710 cm−1)69 and Ar (1708 cm−1)68 matrices. We judge the NMA wavenumber in apolar media to be a reasonable starting wavenumber from which to apply the electrostatic shifts. Accordingly, the diagonal elements for the electrostatic or hydrogen bonding models were obtained after adding ṽ0 = 1707 cm−1 to the wavenumber shift derived from the models. The 28 cm−1 shift of the local wavenumbers for the DFT F-matrix and the choice of an unperturbed local wavenumber of 1707 cm−1 aim to bring the spectra in the experimentally observed spectral range. Using instead an unperturbed local wavenumber of 1735 cm−1, the electrostatic models simulate the original DFT calculations.
The intensity of a normal mode was calculated by first adding the dipole derivatives of individual amide oscillators weighted by their amplitude in the normal mode and then squaring the result. The intensity of a normal mode divided by its wavenumber is proportional to its dipole strength. If not mentioned otherwise, we used the dipole derivative properties optimized in our previous work:32 a dipole derivative position 1.043 Å away from the C-atom along the CO bond and 0.513 Å away from the C-atom along the CN bond, a dipole derivative magnitude ∂μ0/∂q of 2.2 D Å−1 u−1/2 for an isolated amide group, a dipole derivative angle of 22° from the CO bond towards the N-atom, and an A parameter of 0.01 to describe hydrogen bonding effects on the dipole derivative magnitude.
The spectra were generated from Lorentzian lines with 16 cm−1 full width at half maximum at the normal mode wavenumbers. The integral of each component band was equal to the intensity of the particular normal mode normalized to the number of amide groups in the structure.
Results and discussion
Performance of the electrostatic models
We tested a large number of electrostatic models and charge sets to describe the effect of the electrostatic environment on the local amide I wavenumber ṽ of a particular amide group. The models relate the electric potential and the electric field at the locations of the amide atoms of the considered group to a shift of its local amide I wavenumber. Potentials and fields were calculated with charges on all amide atoms of the other amide groups. The most general model 4P4F8 considered the electric field and the electric potential at all four amide atoms C, O, N, and H of the considered amide group and contained eight adjustable parameters (in the name 4P4F8, the number 4 indicates the number of amide atoms considered and P and F stand for electric potential and field, respectively). A further model – 4P4F6 – considered also field and potential on all amide group atoms, but two of the eight parameters were a function of the other parameters, which gives six adjustable parameter as explained in detail in ESI.† This model was developed to describe the effects of hydrogen bonding between an amide group and water49 but we use it here to describe the sensitivity of the local wavenumber on the protein environment, either with the original parameters or with parameters optimized in this work. These general models were then simplified to contain less free parameters down to models that considered only the field at two atoms of the amide group, where one atom was from the hydrogen bond accepting CO group and the other from the hydrogen bond donating NH group. For models that did not consider the electric field at all four amide atoms, the considered atoms are indicated by subscripts in the model name.The model parameters were optimized for each charge set and the performance of the models assessed using two quality parameters SSDiff and SSDev and the related RMSDiff and RMSDev as described in Methods. The performance depended on the charge set used to describe the partial charges on the amide atoms outside the considered group. Therefore, several charge sets were tested. The charge set with the smallest RMSDev after parameter optimization was considered to be the best for the respective model. In general this was also the charge set that resulted in the lowest or one of the lowest RMSDiff values. An exception is the 4P4F6 model, for which a compromise charge set is listed that performs relatively well on both quality measures.
For most models, the charge set is scalable, which means that a multiplication of all charges by a particular number can be compensated for by dividing the d and l parameters by the same number. We chose therefore to use charges that were close to the Gromos charges. The only non-scalable model is the 4P4F6 model because its parameters are related to a component of the dipole derivative.
For the most relevant models, the best charge sets are listed in Table 1 together with the quality parameters RMSDiff and RMSDev. The ESI† contains performance information for all tested models in Table S2 (ESI†), describes the performance of selected models for the inner amides, discusses the model parameters, and lists them in Table S3 (ESI†). Further information is contained in the supplementary data set available on figshare (https://doi.org/10.17045/sthlmuni.24324886).
The best electrostatic model was 4P4F8 with eight adjustable parameters, followed by 4P4F6 with six. However, the number of adjustable parameters can be reduced drastically to only three in 3FONH without much loss of performance. The main improvement that the larger 4P4F8 model provides seems to be a better prediction of the average wavenumber shift for each structure – included in RMSDiff – whereas the local wavenumber variations within each structure are relatively less improved – as indicated by the moderate decrease of RMSDev. Inspite of the slightly lower performance, the 3FONH model provides an excellent compromise between performance, number of parameters, and computational effort and we suggest that this model should be used for calculations of the amide I spectrum. A further simplification of the description in the 2F models reduced the performance. The best 2F models considered the electric fields on the O and N atoms.
All models performed better than a reference calculation without applying local wavenumber shifts. In this calculation all amide groups had the NMA wavenumber, which was compared to the local DFT wavenumbers for the calculation of RMSDiff and RMSDev. These results are listed in Table 1 under the model name Zero.
Comparison of the 4F and 4P models
We found an advantage of using electric fields instead of using only the electric potentials for describing the effect of the protein environment on the local wavenumber: the performance of the 4F model is better than the performance of the 4P model. However, the difference is not dramatic, which agrees with findings for NMA-water systems, where these two approaches were found to be equivalent.41 On the other hand, theoretical considerations suggest that the major part of the sensitivity of the diagonal force constant to the electrostatic environment is mediated by the electric field70 because it controls the electrical anharmonicity and a large part of the mechanical anharmonicity. This apparently contradicts the similar performance of the 4F and 4P models but theory and our model optimization can be reconciled because the electric potentials at close locations can be used to calculate the electric field as pointed out earlier.50 This can be seen in our 4P model, where the largest contributions to the local wavenumber shift ΔṽESM stem from the electric potential on the C and O atoms (Table S3, ESI†). These contributions are similar in magnitude but have the opposite sign, which means that they largely reflect the electric field along the CO bond.Comparison of our model parameters to those of published electrostatic models
Many electrostatic models or maps have been published40 that were mainly developed for the interaction of NMA with water molecules. They consider the electric potential, the electric field, and/or the field gradients at the site of various atoms of the molecule. The ESI† compares our model parameters to those of the former two model types. Models that include electric field gradients46,47,71 cannot be compared to our models. This discussion in the ESI† reveals that there is considerable parameter variation between the different models for NMA–water interactions, and there are differences between these and our models. We speculate that the latter differences arise because our electrostatic models were developed for typical interactions between amide groups in proteins and not for NMA–water interactions. Conceivable reasons for the differences are (i) that amide–amide interactions require different parameters than amide–water interactions – possibly because of different hydrogen bond geometries, (ii) that our optimization includes also longer distance effects for the large structures, and (iii) that it includes also local conformation effects (see below).Experiments with dipeptides at different pH values and thus with different electrostatic environments of the amide group established a strong correlation of the amide I wavenumber with the electric field component along the CO bond at the oxygen, followed by a good correlation for that field component at the carbon.72 The latter might seem surprising in the view of our results, where the 4F model and the 3F models that include the carbon atom result in small dC parameters (Table S3, ESI†) and a model not considering carbon – 3FONH – performs nearly equally well as the 4F model. This indicates little importance of the electric field on this atom for modeling the local amide I wavenumber. However, this does not need to contradict the correlation found by Reppert & Tokmakoff: the mentioned 4F and 3F models contained also the electric field on the oxygen and the small dC parameter may therefore simply reflect an only small benefit of including additionally also the field on the carbon in the model. That the field on the oxygen is more important for modeling is shown by the better performance of the 2FON model as compared to the 2FCN model (Table S2, ESI†) and agrees with the better correlation for the field on the oxygen found by Reppert and Tokmakoff.
Charge sets
The choice of charge set for the electrostatic calculations has an important influence on the spectrum calculated with a particular electrostatic model.51 In our work, the performance of the electrostatic models depended on the charge set for the partial charges of the amide atoms even though the model parameters were optimized for each charge set.Many models performed well for RMSDiff and RMSDev with the same charge set. These models are 4P4F8, 4P, 4F, 3FONH, 3FCON, and 2FOH. In contrast, 4P4F6, 3FCOH and most of the 2F models did not perform well on both quality parameters with the same charge set although both were minimized together in the optimization. Therefore these models need different charge sets to describe a secondary structure's average local wavenumber and its variation over the individual amide groups.
In the best charge sets, the charge on the oxygen is negative, that on the carbon and hydrogen positive and that on the nitrogen ranges from slightly negative to slightly positive. The CO group is negative for most models and the NH group positive. A few models have different charge requirements and perform best or relatively well with neutral CO and NH groups: 4P, 3FCOH, 2FOH, and 2FCN, of which 4P has the best performance. Further charge requirements for particular models are discussed in the ESI.† The results clearly demonstrate that the best charge set depends on the electrostatic model.
For the 4P model, a charge optimization has been carried out previously with parameters derived from NMA–water systems in order to model the local wavenumber shift of structures with two NMA molecules. The optimized charge set was 49/84/53/1835 which surprisingly has negative charge on the hydrogen atom. In our hands however, the best charge sets for the 4P model are electroneutral on the CO group and on the NH group and have relatively little charge on N and H. Moving positive charge from any amide atom to N decreased the performance of the model, but we never tested a charge set with negative H charge.
DSSP64 and Gromos73 charge sets do not satisfy the preference of most models for charged CO and NH groups. Instead they meet the requirement of the 4P model for electroneutral CO and NH groups. Nevertheless, they did not perform well for this model either because of their relatively large charges on the N and H atom. Other well-known charge sets, like those of Amber and Charmm, were not tested here because they have a net charge on the amide group. They have relatively large partial charges on the N (negative) and H (positive) atoms, which deteriorates the performance of the 3FONH and the more comprehensive models. In line with a previous suggestion,21,51,72 we conclude therefore that charge sets from common force fields are not necessarily well-suited to describe electrostatic effects on the amide I wavenumber of individual amide groups.
Local wavenumber shifts of individual amide groups
The local wavenumber shifts for the individual amide groups are compiled in the supplementary data set available on figshare. Fig. 1 shows such shifts for the 4P4F8 and the 3FONH model (blue and red bars, respectively). They are compared to the DFT values (golden bars) and to a simple indication of the hydrogen bonding to the considered amide group (gray bars): in the absence of hydrogen bonding the value is zero, hydrogen bonds to the NH hydrogen, the CO oxygen, and to both are indicated by shifts of −10, −20, and −30 cm−1, respectively. The choice of these numbers was guided by previous DFT calculations, which established a stronger effect of a hydrogen bond to the oxygen and the approximate additivity of the shifts of each hydrogen bond.1,68,74 | ||
| Fig. 1 Local wavenumber shifts of individual amide groups calculated by DFT (golden bars) and the 4P4F8 (blue bars) and the 3FONH model (red bars) using Optimization method 1 (Table 1). Top: Parallel β-strand and β-sheets (P), middle: antiparallel β-strand and β-sheets (A), bottom: helices (H). The horizontal axis displays the number of the amide groups, where 1 indicates the N-terminal amide group. Small, medium, and large structures are arranged from left to right. A simple indication of the hydrogen bonding type is shown by gray bars (0, −10, −20, and −30 cm−1 shift for hydrogen bonds to neither H nor O, to H, to O, and to H and O, respectively). | ||
The top and middle panels of Fig. 1 show the local wavenumber shifts for the β-strands and sheets. We focus first on the local DFT wavenumber shifts (golden bars) of the isolated strands on the left hand side of the top and middle panel, where the N- and C-terminal amide groups have less downshifted local wavenumbers than the inner amides. This indicates that end effects are important for the local wavenumber shifts and holds also for the larger sheets, where in particular the C-terminal amides have higher wavenumbers than inner amide groups with the same type of hydrogen bonding. The end effect can be explained by an interaction between neighboring amide groups in a β-strand, which are arranged such that their dipole–dipole interaction is attractive.60 The consequence of such an arrangement is that each neighbor of an amide group generates an electric field component that points from the oxygen to the carbon atom and thus moves the two atoms further apart. This elongates and weakens the CO bond which lowers the amide I wavenumber. The wavenumber decrease is stronger for the inner amide groups than for the terminal groups because the former are subjected to the added fields of two neighboring groups.60
In addition, the local DFT wavenumber shifts for the larger β-sheets are affected by hydrogen bonding, which is well-known from experiments2,75 and calculations:1,2,68,74 hydrogen bonding leads to downshifts of the amide I wavenumber in the range of several tenths of cm−1 with a stronger effect for hydrogen bonding to the amide oxygen than that to the hydrogen. This generates an alternating pattern of local wavenumber shifts for the two-stranded sheets, observed in Fig. 1, depending on whether the amide group is hydrogen bonded at the oxygen or the hydrogen atom. The same behavior is observed for the outer strands of the four-stranded sheets, whereas their two inner strands show a more uniform local wavenumber shift. These observations are in line with previous quantum chemical calculations of β-sheets.76 Two more observations are of interest: (i) an amide group with a single hydrogen bond to the oxygen can have an equally downshifted local wavenumber as one that has hydrogen bonds to both the oxygen and the hydrogen atoms, and (ii) the local wavenumber shifts do not seem to depend on the number of strands in a sheet as the shifts of the outer strand amides of the four-stranded sheet are comparable to those of the two-stranded sheets.
The local DFT wavenumber shifts of β-strands and β-sheets are well reproduced by the electrostatic models (blue and red bars in Fig. 1), including the alternating hydrogen bonding pattern of the two-stranded sheets and of the outer strands of the four-stranded sheets. The shifts for the antiparallel sheets are somewhat underestimated (not negative enough), while those of the parallel sheets are overestimated (too negative) by both models.
The bottom panel of Fig. 1 shows the results obtained for the three helix structures and we focus again first on the local DFT wavenumber shifts (golden bars). The small helix with three amide groups (bottom, left in Fig. 1) exhibits the highest wavenumber for the middle amide group – higher than the wavenumber of NMA. In contrast to β-sheets, the interaction between neighboring amide dipoles in a helix is repulsive.60 Here, an electric field component of neighboring amides points from the carbon to the oxygen of the considered amide group, which shortens the CO bond and increases the vibrational frequency. The wavenumber upshift is less for the terminal helix amide groups than for the inner amides because the former experience only the field of one neighbor. This results in a lower wavenumber of the terminal amides than of their nearest sequence neighbors, which can be seen in all helix structures. However, the effect is very small for the C-terminal group of the large helix.
The N-terminal amide exhibits the strongest downshift in all structures. This has been noted before29,77,78 and explained by particularly strong hydrogen bonding of its carbonyl group.78 In our helix structures, the N-terminal carbonyl (amide group 1) forms a hydrogen bond with the NH of amide group 4 (O⋯H distance 2.2 Å), which corresponds to the ordinary hydrogen bonding in α-helices between residues i and i + 4. Additionally, it forms a non-linear hydrogen bond with amide group 3 (O⋯H distance 2.4 Å). The respective distance for other amide carbonyls is considerably larger (2.9 Å for amide groups 2 and 3, 2.8 Å for the remaining groups). This additional hydrogen bond is found for all our helix structures, even for the smallest one with three amide groups and explains the low wavenumber of the N-terminal amide group (see Fig. S1d, ESI†).
The local DFT wavenumber shifts in the center of the larger helices show that hydrogen bonding to the oxygen (amides 2 and 3 at the N-terminus) causes stronger downshifts than hydrogen bonding to the hydrogen (last three amides at the C-terminus). Groups in the helix center with hydrogen bonds to both the oxygen and hydrogen atoms have lower wavenumbers than those with hydrogen bonds only to the oxygen. This contrasts with the β-sheets where such groups often have similar wavenumbers. Interestingly, there seems to be a helix length dependency on the local wavenumbers with longer helices having lower local wavenumbers. This can be explained by the interaction between hydrogen bonded amide groups being attractive (in contrast to that between sequence neighbors) leading to a wavenumber downshift. Lower local wavenumbers for longer helices are in line with the low wavenumber of the infrared absorption band of long helices that were experimentally observed for poly-L-lysine, poly-L-glutamic acid, and tropomyosin in D2O79,80 and H2O.81
Both electrostatic models (blue and red bars in Fig. 1) reproduce the DFT trends well, in particular the large downshift of the first amide group in the intermediate and large structures, the increase of the downshift from residue 2 to the center of these helices and its decrease toward the C-terminus, as well as the larger downshifts in the center of the long helix than in the center of the intermediate helix. Both electrostatic models calculate local wavenumber shifts that are slightly too negative for the central amide groups of the large helix.
Local conformation effect
Previous work has established that the intrinisic wavenumbers of diamides depend on the local backbone conformation.24–27,35 This effect was not considered explicitly in the results presented so far, which means that our electrostatic models included the effects of the local conformation. However, amide I calculations often treat the local conformation effect explicitly and independent from electrostatic effects by using DFT calculations of diamides. Accordingly, we performed our own diamide calculations using the same level of theory as for the larger structures. Table S4 (ESI†) compares the local wavenumbers of our calculations with those from the literature. After scaling, the wavenumber of the C-terminal groups of all structures are very similar for the different studies, which is true also for the N-terminal group of the antiparallel β-strand conformation. In contrast, our wavenumber of the N-terminal group of the helix is smaller than in the published work and that for the parallel β-strand conformation larger. All in all, the different DFT calculations agree quite well in spite of the different levels of theory used.In order to single out the local conformation effect, we subtracted the wavenumber of NMA (1735.0 cm−1) from the local DFT wavenumbers of our dipeptides. The obtained local wavenumber shifts are listed in Table 2 and compared to literature values in Table S5 (ESI†). We tested then how well these shifts are described by our two main electrostatic models (Table 2). The shifts predicted from the electrostatic models have the same sign as the DFT shifts, except for the small negative value of the N-terminal group in helical conformation, which has the wrong sign with the 3FONH model. They also predict correctly, which of the two groups has the larger shift in each structure. Therefore they capture aspects of the local conformation effect, which seems to be partly due to electrostatic interactions. This has been recognized before using a 4P model.65
| Structure | Group | Shift in cm−1a |
|||
|---|---|---|---|---|---|
| DFT | 3FONH (set-3FONH)b | 4P4F8 (set-4P4F8)b | 4P4F8 (40/50/00/10)b | ||
| a The wavenumber shifts were calculated by subtracting the NMA wavenumber (1735.0 cm−1) from the local wavenumbers of the diamides. b Charge set for the calculation. c N-terminal amide group. The shift of an N-terminal amide group describes how the local wavenumber of a particular amide group is influenced by the amide group that follows in the sequence. d C-terminal amide group. The shift of the C-terminal group reflects the effect of the preceding group. e The sum of the two shifts is the expected local wavenumber shift of an inner amide group in a protein, i.e. one that has a preceding and a following amide group. Discrepancies between the individual values for the N- and C-terminal amide group and their sums are due to rounding. f ABS: antiparallel β-sheet. g PBS: parallel β-sheet. h RMSDev is the root mean square deviation between the DFT shifts and the shifts obtained with the electrostatic models. RMSDev (N and C) refers to the shift deviations for the N- and the C-terminal amides. i RMSDev(Sum) refers to the deviations of the summed N- and C-terminal shifts. | |||||
| Helix | Nc | −2.0 | 2.1 | −0.7 | −1.9 |
| Cd | 10.6 | 10.9 | 12.8 | 10.4 | |
| Sume | 8.7 | 13.0 | 12.1 | 8.5 | |
| ABSf | Nc | −17.1 | −22.9 | −25.7 | −22.3 |
| Cd | −13.8 | −6.9 | −6.9 | −7.1 | |
| Sume | −30.8 | −29.9 | −32.6 | −29.4 | |
| PBSg | Nc | −14.4 | −23.7 | −24.5 | −21.4 |
| Cd | −11.5 | −7.2 | −6.8 | −6.9 | |
| Sume | −26.0 | −30.8 | −31.3 | −28.3 | |
| RMSDev (N and C)h | 5.8 | 6.5 | 4.9 | ||
| RMSDev (Sum)i | 3.8 | 3.8 | 1.6 | ||
The shifts from the electrostatic models differ from the DFT values with root mean square deviations of 5.8 cm−1 for the 3FONH with the best charge set Set-FONH and of 6.5 cm−1 for the 4P4F8 model with its charge set Set-4P4F8. Since the shift of the N-terminal group is due to the influence of the following C-terminal group and vice versa, the sum of the two shifts is the expected shift for an inner amide group that has both a preceding and a following amide group. The sums are better predicted by our models than the individual shifts with root mean square deviations of 3.8 cm−1 for both the 3FONH and the 4P4F8 model with their standard charge sets.
We explored a few other charge sets with the 4P4F8 model and found that the agreement with the DFT shifts can be improved to a root mean square deviation of 4.9 cm−1 for the shifts of the individual groups. The found charge set is particularly good in reproducing the shifts for the helical peptide and the summed shifts for all structures. This limited exploration might indicate that the best electrostatic model and its optimum charge set for describing the local conformation effect might be different from that for hydrogen bonding and long-range electrostatic effects.
Combining electrostatic models and DFT local conformation effect
We then tested whether our local wavenumber shifts for the N-terminal and C-terminal amides from Table 2 can be used to improve the performance of the electrostatic models. These shifts correspond to a map of the local conformation effect obtained at the same level of theory as that used in the DFT calculations for the larger structures. To explicitly consider the local conformation effect, we subtracted the N- and C-terminal diamide DFT shifts from the DFT local wavenumber shifts of the respective groups in larger structures and the sum of the diamide shifts from all other DFT shifts. We then optimized the parameters of the 4P4F8 and 3FONH models but without considering the nearest sequence neighbors in the calculations of the electric field and potential. This was because our diamide DFT calculations also reflect the electrostatic effects of an amide group on the local wavenumber of its nearest neighbor. Thus, including nearest neighbors in the field and potential calculations would have counted such effects twice.The performance of both models improved regarding RMSDiff for all charge sets tested (3FONH: 5.3 to 4.7 cm−1, 4P4F8:4.4 to 2.4 cm−1), which is better than a previous combination of a 4P model with the local conformation effect (RMSDiff: 5.9 cm−1).65 Regarding RMSDev, the 3FONH performance deteriorated for the better charge sets, but the 4P4F8 model improved considerably for all tested charge sets. However, this improvement of the 4P4F8 model came with a drastic change of its parameters when compared to the optimization without local conformation compensation as further discussed in the ESI.† The parameter changes result in large and opposite wavenumber shifts of the potential and field terms in a typical hydrogen bonding situation, which raises doubts regarding the robustness of this model (see ESI†). In contrast to the 4P4F8 model, the parameters of the 3FONH model change much less and do not change sign when the local conformation effect is explicitly considered. Thus this model seems to be less sensitive to different implementations of the amide I band calculations.
Fig. S3 in ESI† shows the local wavenumber shifts calculated for the 3FONH model with (light blue bars) and without (blue bars) explicit consideration of the local conformation effect. When the local conformation effect is considered, the agreement with the DFT shifts improves for nearly all inner amide groups of all structures, but not for the N-terminal helix groups and the C-terminal groups of the small helix and of the medium and large sheets. The improvement is moderate and the “pure” 3FONH model without an explicit consideration of the local conformation effect is still a good option. This avoids the need to choose a level of theory for calculating the local conformation map and to choose an appropriate reference wavenumber to calculate the shift induced by the local conformation effect.
Comparison to a short-range hydrogen bonding model
The electrostatic models are one way to describe the local wavenumber shifts due to the protein environment. They consider interactions that decay with r−1 and r−2, where r is the distance between the amide group of interest and other atoms in the structure. Thus, the models are sensitive also to long-range interactions. An alternative to this description are models that consider only short-range interactions due to direct hydrogen bonding. In the following we compare the two approaches. The short-range hydrogen bonding model detects hydrogen bonds to the O and H atoms of the amide group of interest, calculates their Kabsch–Sander energies64 and relates this to a local wavenumber shift.37,39,48 We combined this model with an explicit implementation of the local conformation effect and optimized its parameters as described in detail in the ESI.† The performance of the combined model is listed in Table S6 (ESI†). Its RMSDiff value turned out to be worse than that of most electrostatic models, whereas its RMSDev value was close to our main models 4P4F8 and 3FONH, but still worse. The RMSDev value indicates that short-range hydrogen bonding is a main reason for the local wavenumber variations within a given structure, but not sufficient for modeling – at least in its present implementation. On the whole, the model that combines local conformation effect and local hydrogen bonding performs worse than pure electrostatic models like 3FONH and 4P4F8, likely because it ignores long range electrostatic effects.Amide I spectra
Fig. 2 shows spectra calculated from only the diagonal elements of the F-matrix, or in other words the spectrum of the local wavenumbers of the uncoupled amide groups. Each row presents the calculations for a particular secondary structure and the different columns refer to the different sizes of the structural models. The “gold standard” spectrum is calculated from the diagonal elements of the DFT F-matrix and is shown in gold on the top of each panel. The other spectra were calculated with different models for the effect of the protein environment. The agreement with the DFT data is worst for a model (black curves) that uses a local hydrogen bonding model based on the Kabsch–Sander energy and the local conformation effect by a DFT based map from our diamide calculations, termed KS + LCEstd, where std indicates standard parameters. Deviations are particularly prominent for the helix calculations. This model can be improved by optimizing its parameters (gray curves, KS + LCEopt), but there are still considerable differences between the DFT diagonal spectrum regarding the number of component bands and their spectral positions. | ||
| Fig. 2 Spectra calculated from the local wavenumbers only. Top: Parallel β-sheets, middle: antiparallel β-sheets, bottom: helices. The local wavenumbers were obtained from the DFT F-matrix (DFT), from combining the local conformation effect from our diamide calculations with a local hydrogen bonding model based on the Kabsch–Sander energy (LCE + KS), either with standard parameters (std) or with optimized parameters (opt) according to Table S6 (ESI†), or from the electrostatic models 4P4F8 (with Set-4P4F8), 4P4F6 (with Set 70/90/00/20), 4F (with Set-3FONH), and 3FONH (with Set-3FONH) according to Table 1 and Table S3 (ESI†). Parameter optimization was done with Optimization method 1. The gray vertical lines indicate band positions in the spectrum obtained with the DFT F-matrix. | ||
The electrostatic models of this work perform better and produce similar spectral shapes. The deviations between them and the DFT spectra are largest for the small and medium helix spectra where the shapes are best calculated with the 4P4F8 model (dark blue lines), which nevertheless does not reproduce the spectral position of the component bands in the small helix spectrum. The spectra in Fig. 2 reproduce the deviations between the local wavenumbers calculated by DFT and by the 3F and 4P4F8 models (Fig. 1): for the medium and large parallel β-sheets as well as for the large α-helix, the shifts for most amide groups calculated by the electrostatic models (red and blue bars in Fig. 1) are too large when compared with the DFT shifts (golden bars in Fig. 1) and thus the spectra are downshifted too much (red and blue spectra in Fig. 2) with respect to those calculated with the DFT local wavenumbers (golden spectra in Fig. 2). The opposite is the case for the antiparallel β-sheet.
Fig. 3 reports “complete” spectra calculated from an F-matrix with the diagonal elements from DFT calculations or from the different models and the non-diagonal elements from the DFT F-matrix. Note that we do not use the original DFT spectra for the comparison because their normal mode intensities are calculated differently from our normal mode analysis. Thus they would not serve well to compare and test our calculations.
Generally, the models rank in the same way as discussed above for the local wavenumbers. However, it is interesting to observe that the electrostatic models perform more similar and that the simple 3FONH model reproduces the spectral shape at least as good as the 4P4F8 model (compare e.g. the small helix spectra). From a practical point of view therefore, there seems to be little benefit from using the larger 4P4F8 model.
In contrast to Fig. 2, the spectra of Fig. 3 include the effects of vibrational coupling between amide groups. For all spectra in Fig. 3 these are described by the non-diagonal elements of the DFT F-matrix. Comparing the spectra in Fig. 2 without vibrational coupling with those in Fig. 3 with coupling, it becomes clear that the β-sheet spectra have different shapes, whereas the α-helix spectra are similar. The peak positions of the large β-sheets are strongly downshifted by coupling, whereas only slight upshifts are seen for the small and medium helix. Thus, coupling has a strong effect on the β-sheet spectra, but not on the α-helix spectra.7,82–84 Therefore, an accurate calculation of local wavenumbers is particularly important for α-helical structures.
 | ||
| Fig. 3 Spectra calculated from complete F-matrices using local wavenumbers calculated by the models specified in Fig. 2. Top: Parallel β-sheets, middle: antiparallel β-sheets, bottom: helices. The diagonal elements were either from the DFT F-matrix or from the different models. The non-diagonal elements were always from the DFT F-matrix. The gray vertical lines indicate band positions in the spectrum obtained with the DFT F-matrix. | ||
The charge set has only a modest influence on the shape of the spectra when an electrostatic model is used with optimized parameters for each charge set. But deviations from the spectra calculated with the DFT F-matrix can be seen when the quality parameters RMSDiff and RMSDev are significantly worse than those of the better charge sets. This is shown in Fig. S5 and S6 (ESI†) for the 3FONH and the 4P4F8 model, respectively.
Dipole derivative parameters
Finally, we explored whether an electrostatic model can be used to improve also the non-diagonal matrix elements of the F-matrix, which depend on the dipole derivatives of the coupled amide groups. Except for the nearest neighbor interactions, the non-diagonal matrix elements are based on TDC. In our recent work32 we correlated the magnitude of the dipole derivative with the Kabsch–Sander energy of the hydrogen bond to the carbonyl oxygen. Here, we explored whether the dipole derivative can be described by an electrostatic model. Using the same approach as in our previous work,32 we minimized the difference between the F-matrix elements of the DFT F-matrix and those calculated by TDC and we considered only the medium and large structures. The optimum charge set for the 3FONH model (Set-3FONH) was used and only the effect of the field component parallel to the CO bond was tested (as in the electrostatic models for the shift of the local wavenumber). We did not consider the effect of the field at the H atom because our previous work32 did not indicate a significant effect of H bonding to the H atom. The best model to describe the magnitude of the dipole derivative considered the fields at the O, C and N atom with an R value of 5.6 × 10−4 mdyn2 Å−2 u−2 (R is used here to facilitate the comparison with our previous work. It corresponds to SSDiff used in other sections of this text). This model had three adjustable parameters that relate the fields to the dipole derivative magnitude. However, a nearly as good model could be obtained with only one field parameter using the sum of the fields on the O and C atom. It is termed DD(FCO) model (DD for dipole derivative) and determines the dipole derivative magnitude ∂μ/∂q according to| ∂μ/∂q = 2.1 − 30.8 (EO + EC) | (11) |
O bond on the oxygen and carbon atom calculated with the Set-3FONH charges. Table 3 lists all optimized dipole derivative parameters of this model, some of which were only slightly changed compared to our previous work. This model had an R value 5.9 × 10−4 mdyn2 Å−2 u−2, which is better than the R value of 7.5 × 10−4 mdyn2 Å−2 u−2 obtained in our previous work.32 In addition, the maximum dipole derivative (3.0 D Å−1 u−1/2) is larger than that obtained with the previous model (2.5 D Å−1 u−1/2) and thus in better agreement with the upper range of dipole derivative magnitudes reported in the literature.30,53,54 Using only the field on the oxygen atom made the model considerably worse with an R value of 6.7 × 10−4 mdyn2 Å−2 u−2.
| Reference | Position along COabc (Å) | Position along CNabc (Å) | ∂μ0/∂q (∂μmax/∂q)ad (D Å−1 u−1/2) | Angleac (degrees) | R (10−4 mdyn2 Å−2 u−2) |
|---|---|---|---|---|---|
| a The parameters were optimized using intermediate and large structural models. b The position is given by the distances from the C-atom along the specified bonds. c Angle and position of the dipole derivative are equal to those of the transition dipole moment. d ∂μ0/∂q is the dipole derivative magnitude in the absence of an electric field or of hydrogen bonding and ∂μmax/∂q (value in brackets) is the maximum dipole derivative calculated for the ensemble of structural models. | |||||
| DD(FCO), this work | 1.085 | 0.608 | 2.10 (3.04) | 22 | 5.9 |
| Baronio & Barth 202032 | 1.043 | 0.513 | 2.20 (2.51) | 22 | 7.5 |
In summary, we propose a new model to describe the dependency of the dipole derivative magnitude on the electrostatic environment. This model uses the average field at the C and O atoms and requires only a single field parameter. It indicates that the dipole derivative magnitude is largely determined by the CO group, in line with our previous finding.32
Fig. 4 shows spectra calculated with these models. The golden spectra were calculated with the DFT F-matrix elements and the black spectra with diagonal elements from the 3FONH model and the DFT non-diagonal elements. These spectra are also contained in Fig. 3 and are shown here for comparison. The blue and the brown spectra were calculated using 3FONH for diagonal elements, DFT for nearest neighbor elements, and TDC for all other non-diagonal elements. The dipole derivative magnitude was calculated according to the previous32 (blue lines) and the present model (brown lines) with the parameters listed in Table 3. It can be seen that the spectral shape is very similar in all cases. In particular, the spectra obtained with the same local wavenumbers (black, blue, and brown lines) show that both models for the dipole derivative magnitude generate spectra that are equivalent by visual inspection to those from the DFT non-diagonal elements (black lines), even though the new model describes the non-diagonal F-matrix elements better.
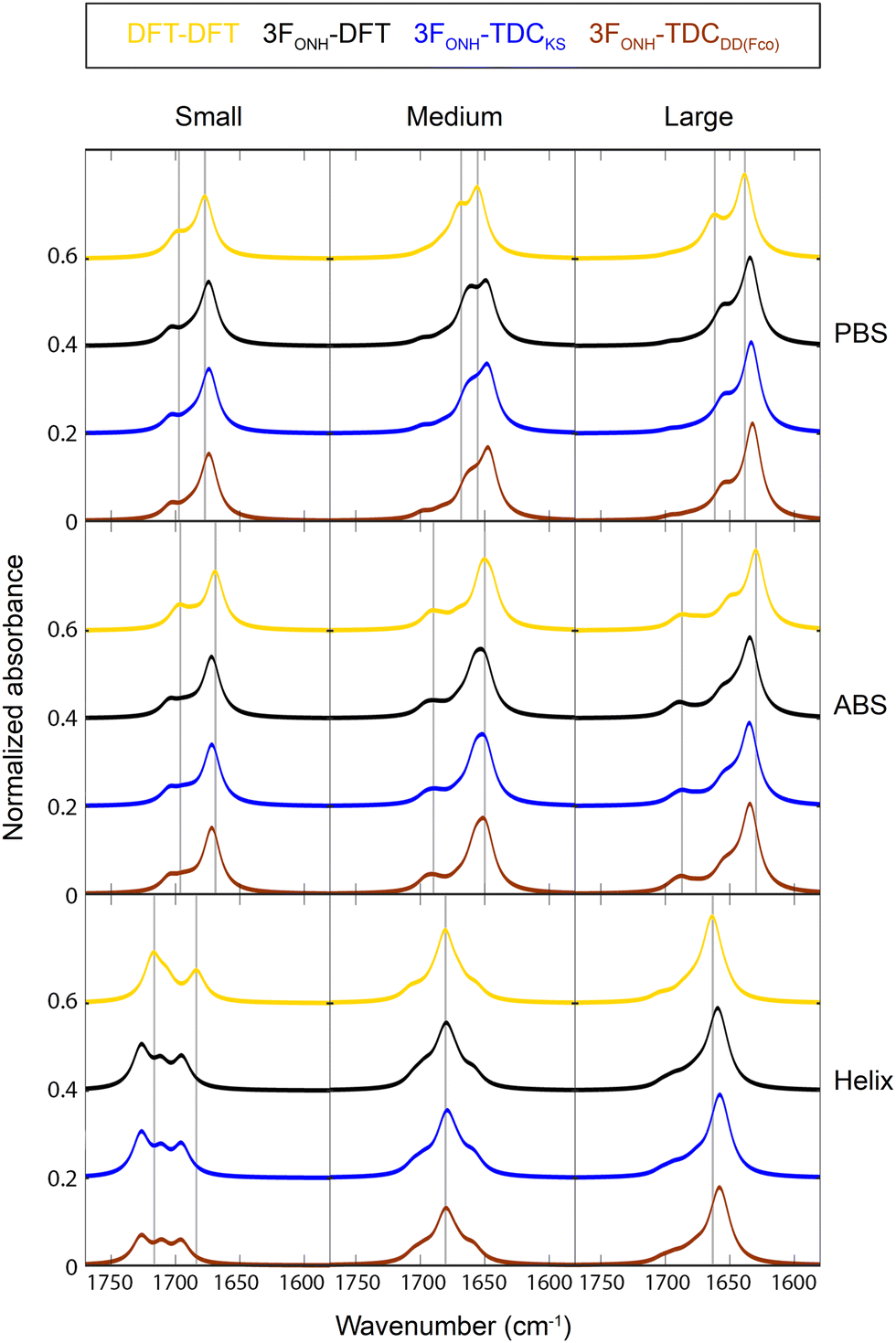 | ||
| Fig. 4 Spectra calculated from complete F-matrices. Top: Parallel β-sheets, middle: antiparallel β-sheets, bottom: helices. Gold (DFT-DFT): all F-matrix elements were from the reconstructed DFT F-matrix. Black (3FONH-DFT): diagonal elements from the 3FONH model and non-diagonal elements from the DFT F-matrix. Blue and brown: diagonal elements from the 3FONH model, nearest neighbor elements from the DFT F-matrix, all other elements from TDC calculations, either using an existing model for the dipole derivative magnitude32 (blue, 3FONH-TDCKS) or the present DD(FCO) model (brown, 3FONH-TDCDD(Fco)). The gray vertical lines indicate band positions in the spectrum obtained with the DFT F-matrix. | ||
Conclusions
This work developed a comprehensive set of electrostatic models for describing the local amide I wavenumber of amide groups that reside in the major secondary structures of proteins: parallel and antiparallel β-sheets and α-helices. The distinct advantage of employing these electrostatic models over higher-level computations lies in their drastically reduced calculation time. While the DFT frequency calculation of our large helix took 45 hours on a national supercomputer, our electrostatic models enable such calculations in less than 2 seconds on a standard office computer.The models were constructed by comparison to DFT calculations. The process involved optimization of both the model parameters and the charge set used for the electrostatic calculations. Even with fine-tuned parameters for each charge set, the charge set had a considerable impact on model performance and charge sets from common force fields were shown to underperform or are expected to do so. This highlights the need for developing specialized charge sets that are optimized for the spectroscopic property of interest as suggested earlier.21,51,72
The best compromise between accuracy and stability of the inherent parameters was found to be a model that considers the electric field on the three amide atoms O, N, and H (3FONH). Since it describes both the electrostatic effects and (partly) the local conformation effects, its parameters reflect a compromise between these two influences. Future work may disentangle these by using specialized models for each of them.
We developed also a simple electrostatic model for the dipole derivative magnitude. It is based on the average field at the C and O atoms, improves the description of the TDC coupling constants, and agrees better with literature values than our previous model.32
As the models were optimized by comparison to DFT calculations, their performance is tied to that of the DFT calculations. It can be anticipated that the model parameters eventually need to be fine-tuned for a best match with experimental spectra.
Data availability
The performance of the models with all tested charge sets, the individual local wavenumber shifts obtained, the DFT log files, and the reconstructed Hamiltonians are available on figshare (https://doi.org/10.17045/sthlmuni.24324886).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are indebted to Eeva-Liisa Karjalainen for the original version of our program for calculating the amide I spectrum and gratefully acknowledge valuable discussions with Prof. Hajime Torii (Shizuoka University, Japan). The DFT calculations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC, grants 2020/5-371, 2021/5-349, 2021/5-567), partially funded by the Swedish Research Council through grant agreement no. 2018-05973. This work was further supported by the Swedish Research Council (2021-04595), Hjärnfonden (FO2019-0127), Olle Engkvists stiftelse (211-0051), and Magn. Bergvalls stiftelse (2020-03624).References
- H. Torii, T. Tatsumi and M. Tasumi, J. Raman Spectrosc., 1998, 29, 537–546 CrossRef CAS.
- J. Kubelka and T. A. Keiderling, J. Phys. Chem. A, 2001, 105, 10922–10928 CrossRef CAS.
- Y. Abe and S. Krimm, Biopolymers, 1972, 11, 1817–1839 CrossRef CAS PubMed.
- Y. N. Chirgadze and N. A. Nevskaya, Dokl. Biophys., 1973, 208, 17–20 Search PubMed.
- T. Miyazawa, J. Chem. Phys., 1960, 32, 1647–1652 CrossRef CAS.
- S. Krimm and J. Bandekar, Adv. Prot. Chem., 1986, 38, 181–364 CrossRef CAS PubMed.
- S. Krimm and Y. Abe, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 2788–2792 CrossRef CAS PubMed.
- H. Torii and M. Tasumi, J. Chem. Phys., 1992, 96, 3379–3387 CrossRef CAS.
- A. Amadei, I. Daidone, A. Di Nola and M. Aschi, Curr. Opin. Struct. Biol., 2010, 20, 155 CrossRef CAS PubMed.
- N. A. Besley, Philos. Trans. R. Soc., A, 2007, 365, 2799 CrossRef CAS PubMed.
- P. Bour, J. Kubelka and T. A. Keiderling, Biopolymers, 2002, 65, 45–59 CrossRef CAS PubMed.
- P. Bouř, J. Sopková, L. Bednárová, P. Maloň and T. A. Keiderling, J. Comput. Chem., 1997, 18, 646–659 CrossRef.
- J. Kubelka and T. A. Keiderling, J. Am. Chem. Soc., 2001, 123, 12048–12058 CrossRef CAS PubMed.
- J. Kubelka, R. A. G. D. Silva and T. A. Keiderling, J. Am. Chem. Soc., 2002, 124, 5325–5332 CrossRef CAS PubMed.
- J. Kubelka, P. Bour and T. A. Keiderling, in Advances in Biomedical Spectroscopy, Biological and Biomedical Infrared Spectroscopy, ed. A. Barth and P. I. Haris, IOS Press, Amsterdam, 2009, vol. 2, pp. 178–223 Search PubMed.
- S. Ye, K. Zhong, J. Zhang, W. Hu, J. D. Hirst, G. Zhang, S. Mukamel and J. Jiang, J. Am. Chem. Soc., 2020, 142, 19071–19077 CrossRef CAS PubMed.
- C.-J. Feng, A. Sinitskiy, V. Pande and A. Tokmakoff, J. Phys. Chem. B, 2021, 125, 4620–4633 CrossRef CAS PubMed.
- J. Jeon, S. Yang, J.-H. Choi and M. Cho, Acc. Chem. Res., 2009, 42, 1280–1289 CrossRef CAS PubMed.
- H. Torii and M. Tasumi, in Infrared Spectroscopy of Biomolecules, ed. H. H. Mantsch and D. Chapman, Wiley-Liss, New York, 1996, pp. 1–18 Search PubMed.
- J. W. Brauner, C. R. Flach and R. Mendelsohn, J. Am. Chem. Soc., 2005, 127, 100 CrossRef CAS PubMed.
- M. Reppert and A. Tokmakoff, Annu. Rev. Phys. Chem., 2016, 67, 359–386 CrossRef CAS PubMed.
- F. S. Husseini, D. Robinson, N. T. Hunt, A. W. Parker and J. D. Hirst, J. Comput. Chem., 2017, 38, 1362–1375 CrossRef CAS PubMed.
- H. Torii and M. Tasumi, J. Raman Spectrosc., 1998, 29, 81–86 CrossRef CAS.
- J.-H. Choi and M. Cho, J. Chem. Phys., 2004, 120, 4383 CrossRef CAS PubMed.
- R. D. Gorbunov, D. S. Kosov and G. Stock, J. Chem. Phys., 2005, 122, 224904 CrossRef PubMed.
- T. la Cour Jansen, A. G. Dijkstra, T. M. Watson, J. D. Hirst and J. Knoester, J. Chem. Phys., 2006, 125, 044312 CrossRef PubMed.
- T. la Cour Jansen, A. G. Dijkstra, T. M. Watson, J. D. Hirst and J. Knoester, J. Chem. Phys., 2012, 136, 209901 CrossRef.
- S. Cha, S. Ham and M. Cho, J. Chem. Phys., 2002, 117, 740–750 CrossRef CAS.
- J. Kubelka, J. Kim, P. Bour and T. A. Keiderling, Vib. Spectrosc., 2006, 42, 63–73 CrossRef CAS.
- E. G. Buchanan, W. H. James, S. H. Choi, L. Guo, S. H. Gellman, C. W. Müller and T. S. Zwier, J. Chem. Phys., 2012, 137, 094301 CrossRef PubMed.
- P. Hamm and S. Woutersen, Bull. Chem. Soc. Jpn., 2002, 75, 985–988 CrossRef CAS.
- C. M. Baronio and A. Barth, J. Phys. Chem. B, 2020, 124, 1703–1714 CrossRef CAS PubMed.
- S. Ham, J.-H. Kim, H. Lee and M. Cho, J. Chem. Phys., 2003, 118, 3491–3498 CrossRef CAS.
- T. M. Watson and J. D. Hirst, J. Phys. Chem. A, 2002, 106, 7858–7867 CrossRef CAS.
- S. Ham and M. Cho, J. Chem. Phys., 2003, 118, 6915–6922 CrossRef CAS.
- D. R. Turner and J. Kubelka, J. Phys. Chem. B, 2007, 111, 1834–1845 CrossRef CAS PubMed.
- H. Maekawa, C. Toniolo, Q. B. Broxterman and N. Ge, J. Phys. Chem. B, 2007, 111, 3222–3235 CrossRef CAS PubMed.
- D. Paschek, M. Pühse, A. Perez-Goicochea, S. Gnanakaran, A. E. García, R. Winter and A. Geiger, Chem. Phys. Chem., 2008, 9, 2742–2750 CrossRef CAS PubMed.
- E.-L. Karjalainen, T. Ersmark and A. Barth, J. Phys. Chem. B, 2012, 116, 4831–4842 CrossRef CAS PubMed.
- C. R. Baiz, B. Błasiak, J. Bredenbeck, M. Cho, J.-H. Choi, S. A. Corcelli, A. G. Dijkstra, C.-J. Feng, S. Garrett-Roe, N.-H. Ge, M. W. D. Hanson-Heine, J. D. Hirst, T. L. C. Jansen, K. Kwac, K. J. Kubarych, C. H. Londergan, H. Maekawa, M. Reppert, S. Saito, S. Roy, J. L. Skinner, G. Stock, J. E. Straub, M. C. Thielges, K. Tominaga, A. Tokmakoff, H. Torii, L. Wang, L. J. Webb and M. T. Zanni, Chem. Rev., 2020, 120, 7152–7218 CrossRef CAS PubMed.
- J. R. Schmidt, S. A. Corcelli and J. L. Skinner, J. Chem. Phys., 2004, 121, 8887–8896 CrossRef CAS PubMed.
- Y.-S. Lin, J. M. Shorb, P. Mukherjee, M. T. Zanni and J. L. Skinner, J. Phys. Chem. B, 2009, 113, 592–602 CrossRef CAS PubMed.
- L. Wang, C. T. Middleton, M. T. Zanni and J. L. Skinner, J. Phys. Chem. B, 2011, 115, 3713–3724 CrossRef CAS PubMed.
- J. Choi, S. Ham and M. Cho, J. Phys. Chem. B, 2003, 107, 9132–9138 CrossRef CAS.
- P. Bouř and T. A. Keiderling, J. Chem. Phys., 2003, 119, 11253–11262 CrossRef.
- T. la Cour Jansen, J. Knoester and C. Jansen, J. Chem. Phys., 2006, 124, 044502 CrossRef PubMed.
- T. Hayashi, W. Zhuang and S. Mukamel, J. Phys. Chem. A, 2005, 109, 9747 CrossRef CAS PubMed.
- H. Maekawa and N. Ge, J. Phys. Chem. B, 2010, 114, 1434–1446 CrossRef CAS PubMed.
- H. Torii, J. Phys. Chem. B, 2018, 122, 154–164 CrossRef CAS PubMed.
- M. Reppert and A. Tokmakoff, J. Chem. Phys., 2015, 143, 061102 CrossRef PubMed.
- M. Reppert, A. R. Roy and A. Tokmakoff, J. Chem. Phys., 2015, 142, 125104 CrossRef PubMed.
- L. Ackels, P. Stawski, K. E. Amunson and J. Kubelka, Vib. Spectrosc., 2009, 50, 2–9 CrossRef CAS.
- J. Wang, Phys. Chem. Chem. Phys., 2009, 11, 5310–5322 RSC.
- S. Hahn, S. Ham and M. Cho, J. Phys. Chem. B, 2005, 109, 11789–11801 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revis. E.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- J. P. Perdew and W. Yue, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8800–8802 CrossRef PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- A. D. Becke, in Modern Electronic Structure Theory. Part II, ed. D. R. Yarkony, World Scientific, 1995, pp. 1022–1046 Search PubMed.
- S. Ham, S. Cha, J.-H. J.-H. Choi and M. Cho, J. Chem. Phys., 2003, 119, 1451–1461 CrossRef CAS.
- T. C. Cheam and S. Krimm, Chem. Phys. Lett., 1984, 107, 613–616 CrossRef CAS.
- J.-H. Choi and M. Cho, in Advances in Biomedical Spectroscopy, Biological and Biomedical Infrared Spectroscopy, ed. A. Barth and P. I. Haris, IOS Press, Amsterdam, 2009, vol. 2, pp. 224–260 Search PubMed.
- H. Torii, J. Phys. Chem. Lett., 2015, 6, 727–733 CrossRef CAS PubMed.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- R. D. Gorbunov and G. Stock, Chem. Phys. Lett., 2007, 437, 272–276 CrossRef CAS.
- L. C. Mayne and B. Hudson, J. Phys. Chem., 1991, 95, 2962–2967 CrossRef CAS.
- S. Ataka, H. Takeuchi and M. Tasumi, J. Mol. Struct., 1984, 113, 147–160 CrossRef CAS.
- H. Torii, T. Tatsumi, T. Kanazawa and M. Tasumi, J. Phys. Chem. B, 1998, 102, 309–314 CrossRef CAS.
- L. O. Paulson and D. T. Anderson, J. Phys. Chem. B, 2011, 115, 13659–13667 CrossRef CAS PubMed.
- H. Torii, J. Phys. Chem. A, 2004, 108, 7272–7280 CrossRef CAS.
- R. Bloem, A. G. Dijkstra, T. la, C. Jansen and J. Knoester, J. Chem. Phys., 2008, 129, 055101 CrossRef PubMed.
- M. Reppert and A. Tokmakoff, J. Chem. Phys., 2013, 138, 134116 CrossRef PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. Van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- N. A. Besley, J. Phys. Chem. A, 2004, 108, 10794–10800 CrossRef CAS.
- G. Eaton, M. C. R. Symons and P. P. Rastogi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 3257–3271 RSC.
- C. Lee and M. Cho, J. Phys. Chem. B, 2004, 108, 20397–20407 CrossRef CAS.
- J.-H. Choi, S. Hahn and M. Cho, Int. J. Quantum Chem., 2005, 104, 616–634 CrossRef CAS.
- J. Kubelka, R. Huang and T. A. Keiderling, J. Phys. Chem. B, 2005, 109, 8231–8243 CrossRef CAS PubMed.
- Y. N. Chirgadze and E. V. Brazhnikov, Biopolymers, 1974, 13, 1701–1712 CrossRef CAS PubMed.
- M. Jackson, P. I. Haris and D. Chapman, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1989, 998, 75–79 CrossRef CAS.
- S. Y. Venyaminov and N. N. Kalnin, Biopolymers, 1990, 30, 1259–1271 CrossRef CAS PubMed.
- N. A. Nevskaya and Y. N. Chirgadze, Biopolymers, 1976, 15, 637–648 CrossRef CAS PubMed.
- Y. N. Chirgadze and N. A. Nevskaya, Biopolymers, 1976, 15, 607 CrossRef CAS PubMed.
- Y. N. Chirgadze and N. A. Nevskaya, Biopolymers, 1976, 15, 627 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp02018e |
| This journal is © the Owner Societies 2024 |