
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling the hierarchical structure of saturated monoacid triglycerides during crystallization – a comprehensive time-resolved X-ray scattering study†
Ivana A.
Penagos
 a,
Fien
De Witte
a,
Tom
Rimaux
b,
Koen
Dewettinck
a and
Filip
Van Bockstaele
*a
a,
Fien
De Witte
a,
Tom
Rimaux
b,
Koen
Dewettinck
a and
Filip
Van Bockstaele
*a
aFood Structure and Function Research Group (FSF), Ghent University, Coupure Links 653, 9000 Ghent, Belgium. E-mail: filip.vanbockstaele@ugent.be
bVandemoortele R&D Centre, Prins Albertlaan 79, 8870 Izegem, Belgium
First published on 2nd October 2024
Abstract
Time-resolved studies are crucial for capturing the dynamic evolution of triglyceride structures during crystallization, providing insights into the self-assembly of diverse triglyceride structures across various length scales. While previous research has characterized triglyceride's polymorphisms and long spacings using wide-angle and small-angle X-ray techniques, time-resolved studies on the mesoscale of triglycerides have been limited, particularly in the study of crystalline nanoplatelets (CNPs) and their aggregates. This study pioneers the application of ultra-small-angle X-ray scattering (USAXS) for time-resolved analysis of commercial sources of saturated monoacid triglycerides. The methodology presented here integrates real-time synchrotron X-ray scattering (XRS) with differential scanning calorimetry (DSC), phase contrast microscopy (PCM), and cryo-scanning electron microscopy (Cryo-SEM) to investigate the crystallization behavior of a 30% dilution of palm stearin (PS) in high oleic sunflower oil (HOSO) and a 30% dilution of fully hydrogenated rapeseed oil (FHRO) in HOSO. Covering several orders of magnitude, this comprehensive multiscale approach enhances our kinetic understanding of triglyceride structural transformations. Both systems followed a polymorphic transition from α to β, with a transition from 2L(α) to 2L(β) lamellar stacking configuration. Our results suggest that structural evolutions in non-pure triglyceride systems advance gradually over extended timescales post-crystallization. While lamellae and CNP thicknesses decreased with the polymorphic transition, CNP size and aggregation changed in four distinct phases. We believe that the methodology presented here will greatly improve our understanding of the self-assembly of lipids, getting us closer to bottom-up optimization of triglyceride-based formulations in food.
Introduction
The compositional features of triglycerides (TAGs) are well-established within the domain of standard structural organic chemistry. However, transitioning from understanding these compositional features to predicting macroscopic performance, such as texture, rheology, and oil binding capacity, remains a significant challenge and an eagerly pursued research goal in the field of lipid structuring.1 The difficulty arises because TAG fatty acid chains are flexible, which allows an identical set of molecules to form different structural arrangements.1–3 In fact, TAGs can self-assemble in various ways depending on the crystallization conditions; therefore, the same set of molecules may result in different macroscopic properties if processed differently over time. The non-triviality of predicting the macroscopic performance of TAGs thus arises from the fact that the processing history strongly influences the final structure.1 With this in mind, time-resolved studies become relevant as they capture the evolution of triglyceride structures during their formation. Observing modifications during crystallization makes it possible to identify differences in the self-assembly behavior of TAGs, thus improving the understanding of the dynamics that dominate the system under non-equilibrium conditions.Getting to our current understanding of the hierarchical structure of triglyceride systems has not been an easy task and has required the efforts of various researchers.4–7 A visual representation of the units composing TAG systems can be found elsewhere.8 TAGs are esters of glycerol and three fatty acids (propane-1,2,3-triol),9,10 widely recognized for exhibiting polymorphism and monotropic transformations. At the sub-nanometric level, polymorphisms of saturated monoacid TAGs can be categorized into three main sub-cell packings, namely α, β′, and β. These are characterized by the cross-sectional packing of aliphatic chains, producing hexagonal (α), orthorhombic (β′) or triclinic (β) configurations.11,12 Besides polymorphic form, the stacking of TAGs can also be described by the number of repetitive hydrocarbon chains forming a unit lamella.13 Double chain length (2L) and triple chain length (3L) are the most frequently occurring arrangements.
Due to their importance in practical applications, such as the tempering of chocolate or the formation of granular crystals in margarine, time-resolved polymorphic transformations in TAGs have been extensively studied in model systems.9,10,14–18 These studies have specifically focused on short and long spacings, where X-ray techniques, such as X-ray diffraction (XRD) and X-ray scattering (XRS) are commonly used. Since short spacings are identified between q = 5 nm−1 and q = 25 nm−1, wide-angle techniques such as wide-angle X-ray scattering (WAXS) or wide-angle X-ray diffraction (WAXD) are predominantly used for polymorphic identification. In contrast, small-angle techniques capture the long spacings of TAGs, and therefore, small-angle X-ray scattering (SAXS) and small-angle X-ray diffraction (SAXD) are used.
To the best of our knowledge, to this date, only our group has performed XRS time-resolved studies on crystalline nanoplatelets (CNPs) of TAGs.19,20 In fact, CNPs are thought to form as lamellae stack on top of each other. These form platelets that then aggregate into higher level structures. The first confirmation of the existence of CNPs was reported in 2010, when Acevedo and Marangoni7 presented a series of TEM images, revealing high anisotropic platelet-like crystals of nanometric dimensions. Since then, continuous efforts have been put in place to quantify these units by microscopy2,4,21–26 and Ultra Small-Angle X-ray Scattering (USAXS).2,24,26–33 However, time-resolved studies of these units remains a significant challenge.
On the one hand, while techniques like TEM and SEM can visualize CNPs, they often require lengthy deoiling protocols, making electron microscopy unsuitable for time-resolved studies. On the other hand, until recently, time-resolved studies with USAXS were not feasible due to instrumentation and data analysis constraints. Only with the development of extremely brilliant X-ray sources in 2016,34 which offer high time and reciprocal-space resolutions simultaneously,35 time-resolved studies in the USAXS range became feasible. A major upgrade during 2013–2014 of the ID02 beamline at the ESRF extended the sample-to-detector distance to 31 m, enabling real-time measurements on the USAXS range.35,36 As such, this expansion solved the instrumentation challenges of measuring in real time the fat crystal structure at the mesoscale. In addition, the recent improvements on models for the interpretation of time-resolved USAXS data of TAGs were necessary to process the data.8
In this article, we present, for the first time, a time-resolved multiscale analysis on commercially available saturated monoacid TAGs during crystallization. For this, two commercial triglyceride systems, palm stearin (PS) and fully hydrogenated rapeseed oil (FHRO) diluted in high oleic sunflower oil (HOSO), were analyzed through XRS. Results span four orders of magnitude (from Å to μm). In the setup proposed, time-resolved synchrotron WAXS (1–10 Å), SAXS (1–10 nm) and USAXS (0.1–1 μm) were used to study the crystallization behavior of a 30% wt PS in HOSO and 30% wt FHRO in HOSO. The XRS results are accompanied by time-resolved microscopy studies, more specifically, phase contrast microscopy (PCM). Cryo-SEM was used to visualize the final CNP configuration. Finally, differential scanning calorimetry was also used to identify changes in the thermal behavior of the sample.
Materials and methods
Materials
Fully hydrogenated rapeseed oil (FHRO), palm stearin (PS), and high oleic sunflower oil (HOSO) were kindly supplied by Vandemoortele Lipids NV (Izegem, Belgium). The primary fatty acid in FHRO was stearic acid (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0) at approximately 90.2%, with minor amounts of palmitic acid (16:0) and arachidic acid (20:0) at around 5.4% and 1.8%, respectively. For PS, the main fatty acids were palmitic acid (16:0) at 77.9%, stearic acid (18:0) at 5.7%, and oleic acid (18:1c) at 11.4%. Acetone (>99%, CAS 67-64-1), n-hexane (>99%, CAS 110-54-3), and isobutanol (>99%, CAS 78-83-1) were obtained from ChemLab (Zedelgem, Belgium).
:0) at approximately 90.2%, with minor amounts of palmitic acid (16:0) and arachidic acid (20:0) at around 5.4% and 1.8%, respectively. For PS, the main fatty acids were palmitic acid (16:0) at 77.9%, stearic acid (18:0) at 5.7%, and oleic acid (18:1c) at 11.4%. Acetone (>99%, CAS 67-64-1), n-hexane (>99%, CAS 110-54-3), and isobutanol (>99%, CAS 78-83-1) were obtained from ChemLab (Zedelgem, Belgium).
Dilution preparation
A 30% (w/w) dilution of FHRO in HOSO was carefully weighed (200 g) in a 250 ml beaker (here named 30% FHRO). The sample was heated to 80 °C for at least 15 minutes until fully melted. This procedure was then repeated for a second dilution, consisting of 30% (w/w) PS in HOSO, which is here named 30% PS.Thermal behavior (DSC)
Measurements were conducted using a DSC Q1000 (TA Instruments, New Castle, DE, USA). The samples were placed on Tzero aluminum pans, filled with 3–12 mg of sample, closed with Tzero aluminum lids, and hermetically sealed. To evaluate crystallization behavior, the samples were first heated to 80 °C for at least 15 minutes to erase any crystal memory. Subsequently, a cooling ramp was applied with rates varying between 1 °C min−1 and 10 °C min−1, followed by an isothermal hold at 10 °C or 15 °C for 60 minutes. To evaluate their melting behavior, the samples were then reheated to 80 °C at a rate of 20 °C min−1. Each sample was prepared in triplicate, and the results were analyzed using the TRIOS Software (TA Instruments, version 5.1).Phase Contrast Microscopy (PCM)
For phase contrast microscopy, a Leica DM2500 LED microscope (Wetzlar, Germany) equipped with a Leica K3C color camera was used. A small amount of sample was placed between a glass slide and a coverslip and statically crystallized using a Linkam PE120 Peltier system with a T95 system controller. Each sample was first heated to 80 °C for 15 minutes to erase any crystal memory. Crystallization occurred as the sample was cooled from 80 °C to 15 °C at a rate of 10 °C min−1. The sample was then held isothermally at 15 °C for 15 minutes. Images were captured using either an HC PL FLUOTAR 40×/0.80 PH2 or an HC PL FLUOTAR 20×/0.55 PH2 objective and the Leica LAS X software. Time-resolved images were captured every 3 s. Videos were reconstructed using 24 frames per second using LAS X software. Brightness and contrast were modified whenever necessary to improve quality of the visuals. The experiment was replicated at least twice to ensure repeatability.Cryogenic Scanning Electron Microscopy (Cryo-SEM)
Visualization of CNPs was conducted using cryogenic electron scanning microscopy (cryo-SEM). The procedure was the same as the one described by Penagos, et al.8 Samples were statically crystallized in a Linkam shear cell (CSS450, Linkam, Salfords, UK). For this, samples were heated to 80 °C for 15 minutes to eliminate any crystal memory, followed by a cooling ramp at 10 °C min−1 to 15 °C. The sample was then maintained isothermally at this temperature for a minimum of 15 minutes, and subsequently transferred to a pre-tempered glass vial for deoiling (15 °C). A small portion of the crystallized fat sample was dissolved in a pre-cooled (15 °C) solvent mixture of 80/20% v/v hexane/acetone. The solvent was exchanged twice every 12 hours, to ensure complete removal of liquid oil and disruption of the supra-CNP network. A drop of the solvent suspension was placed on a double-sided adhesive conductive carbon disc (SPI Supplies, West Chester, USA) attached to an SEM aluminum stub, and the excess solvent was allowed to evaporate for at least 4 hours. For visualization, the stub was vitrified in liquid nitrogen slush (−210 °C) and transferred to a PP30100T cryo-transfer system (Quorum Technologies Ltd., East Sussex, UK) maintained at −140 °C. The sample underwent sublimation at −70 °C for 90 minutes, followed by sputter-coating with platinum using argon gas. The visualization was performed using a Jeol JSM-7100F TTLS LV TFEG-SEM (Jeol Europe BV, Zaventem, Belgium) under high-vacuum conditions (1 × 10−6 mbar) at −140 °C with an accelerated voltage of 3 keV. To ensure the representativeness of the results, at least 50 images were taken from different locations of the sample. Brightness and contrast were modified whenever necessary to improve quality of the visuals.X-ray scattering
In each acquisition, two dimensional patterns were recollected and normalized to an absolute intensity scale using the standard procedure (detector dark-image subtraction, flat field division and intensity normalization). The two-dimensional images were then reduced to 1D profiles by means of an azimuthal integration. The scattering intensity I(q) was plotted against the scattering vector q. The 5 acquisitions per cycle were then averaged to obtain one-dimensional scattering profile per time-frame. An empty capillary was subtracted to each acquisition to correct for sample holder and background (empty beam, capillary and air gap). Samples were normalized by sample thickness. The software SAXSutilities37 was utilized for averaging, subtracting and normalizing. Whenever necessary, eqn (1) was applied to convert from the inverse scattering vector q (nm−1) to real space spacings d (nm):
 | (1) |
In addition, from the SAXS region, the CNP thickness could also be obtained. For this, two alternative methods were utilized. First, the average crystallite size (DSchr) was derived using the Scherrer equation (eqn (2)). In this equation, FWHM001 represents the full width at half maximum of the 001 SAXS peak (in inverse length), and K is the Scherrer constant, assumed to be 0.9 in this context.
 | (2) |
 | (3) |
Results
Thermal behavior
Fig. 1 presents the DSC results of the crystallization and melting behavior of samples 30% FHRO and 30% PS. During the cooling phase (Fig. 1A), a single crystallization event was evidenced for both samples. The onset crystallization temperature (Tc) was found to be around 41.64 ± 0.24 °C for the 30% FHRO sample, with an exothermic peak located at 40.74 ± 0.24 °C. As expected based on the composition, lower crystallization temperatures were reported for the 30% PS system, reporting a Tc at 28.56 ± 0.07 °C, with a crystallization peak at 27.75 ± 0.07 °C. The enthalpy of crystallization (ΔHc) was measured to be 55.50 ± 3.15 J g−1 for the 30% FHRO system and 24.29 ± 1.90 J g−1 for the 30% PS system.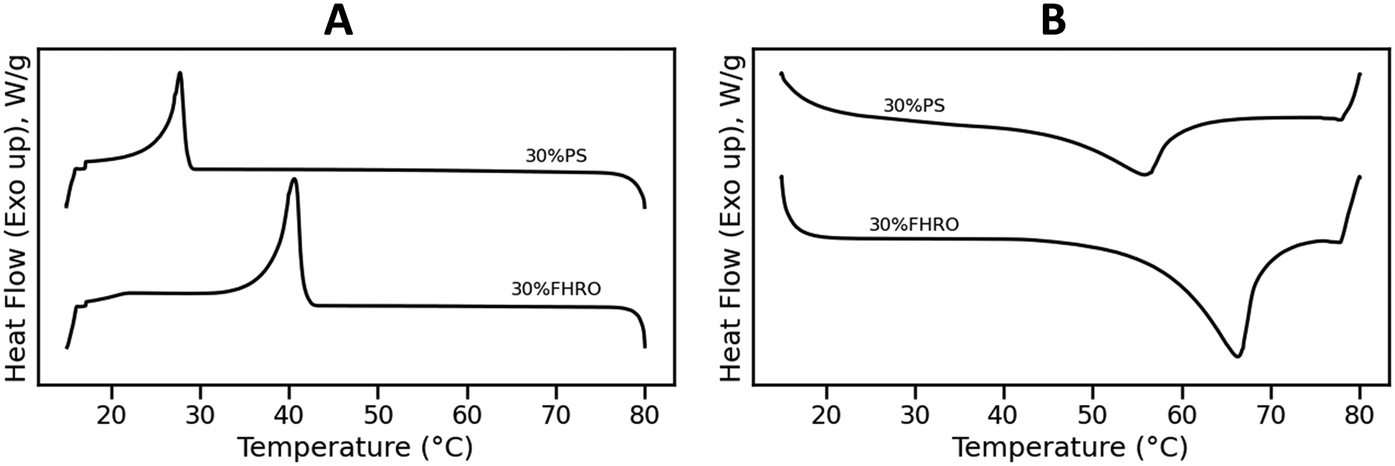 | ||
| Fig. 1 Crystallization (A) and melting (B) curve of a 30% FHRO dilution in HOSO (denoted as 30% FHRO) and a 30% dilution of PS in HOSO (denoted as 30% PS). The cooling rate was set to 10 °C min−1 while the melting rate was set to 20 °C min−1. | ||
Fig. 1B presents the melting behavior. An endothermic peak (Tm) was located at 66.17 ± 0.48 °C for the 30% FHRO system, while a melting peak was observed at 55.52 ± 0.35 °C for the 30% PS system. In both cases, a single melting peak is evidenced. The melting points of pure tristearin (SSS) and pure tripalmitin (PPP) in the β phase are 66.0 °C and 72.8 °C respectively.9 The reduction in melting temperature is attributed to the solubility of the material. In fact, similar results were reported by Pellegrino, et al.17 for a 30% PPP in OOO system as well as by Acevedo & Marangoni (2018), for a 30% dilution of fully hydrogenated canola oil in HOSO.21
Microscopy
Fig. 2(A–F) illustrates the time-resolved microscopy results for the stearic system. Initially, multiple nucleation sites are observed as the sample starts crystallizing (Fig. 2A). Subsequently, crystal growth occurs at these sites, eventually leading to a complete space-filling network (Fig. 2F). The crystal growth process is gradual, with the transition decelerating as solid TAGs in the solution are depleted. The polymorphic transition is not visualized as a sharp transition, but rather as a progressive transformation (see Video S1†). Fig. 2(G–L) displays the microscopy results for the 30% PS system, showing a similar behavior (see Video S2†). A notable distinction lies in the spherulite morphology: 30% PS forms spherulites with a looser conformation compared to the 30% FHRO. | ||
| Fig. 2 Time-resolved phase contrast micrographs showcasing the crystallization of a 30% FHRO dilution in HOSO (A–F) and a 30% PS dilution in HOSO (G–L). Different letters indicate different acquisition times. Videos S1 and S2† showcase the polymorphic transition. The scale bar is approximately 50 μm. | ||
Cryo-SEM and PCM were combined to visualize the final crystalline conformation after 15 minutes of isothermal crystallization time. Fig. 3 shows the resulting microscopies, highlighting the different aggregation units at various length scales. The lamellar structures are observed as extremely thin, paper-like layers. These units stack on top of each other forming parallelepiped-like structures, here called crystalline nanoplatelets (CNPs). In the stearic sample, these CNPs appear thicker, better defined, and exhibit less polydispersity. Conversely, in the palmitic sample, the CNPs seem less rigid and more flexible, showing a greater propensity for bending. At the crystallite level (CNP aggregates), the stearic samples exhibit more rigid aggregation, whereas the palmitic samples tend to form more pliable aggregates, where bending of CNPs seems to be possible. This difference extends to the formation of crystal clusters and flocs. As previously mentioned, a significant distinction is observed in the spherulite morphology, where 30% PS forms spherulites with a seemingly looser conformation compared to the 30% FHRO (as shown in Fig. 2).
 | ||
| Fig. 3 Multiscale structure analysis of stearic- and palmitic-based fat blends (30% FHRO and 30% PS in HOSO) visualized through cryo-SEM highlighting lamellar structures, crystalline nanoplatelets (CNPs), CNP aggregates and crystal clusters, and phase contrast microscopy showing floc morphology and aggregation. The iconography to the left is based on Penagos, et al.8 | ||
X-ray scattering
Time-resolved WAXS was employed to study the polymorphic transitions. Fig. 4A presents the obtained WAXS profile for a 30% FHRO dilution in HOSO. Initially, the samples showed no crystalline structures, as indicated by the characteristic broad oil bump in the scattering pattern (q = 13.51 nm−1). Progressively, the oil bump reduces, and a single peak at q = 15.12 nm−1 emerges, attributed to the formation of the α phase. Further, a polymorphic transition is observed, where a progressive decrease in the α peak is visible, while three characteristic peaks slowly increase (q = 13.79, 16.18, and 16.86 nm−1), suggesting the formation of the β polymorph (d = 4.58 nm). A similar trend could be observed for the palmitic system (Fig. 4C), as the system transitioned from the oil (q = 13.51 nm−1) to the α phase (q = 15.18 nm−1) and subsequently the β phase (q = 13.73, 16.05, and 16.56 nm−1). | ||
| Fig. 4 WAXS results. In panels A and C, surface plots of the WAXS profiles. In panels B and D, the orthogonal projection of the data expanding on the polymorphic transition. A vertical shift is performed to allow better visualization of each profile. Labels to the left indicate temperature. In panels A and B, a dilution of 30% FHRO in HOSO is presented. In panels C and D, a 30% dilution of PS in HOSO is shown. | ||
In monotropic phase transitions, it is conventionally presumed that polymorphic transformations occur following a pre-determined order, where α transforms into β′, which later forms β. As evidenced in Fig. 4A and C, the α-phase seems to transform directly into the more stable β-phase. The transition is better visualized in Fig. 4B and D. Although we do not exclude the formation of an intermediate β′ phase, experimentally, the β formation occurs rapidly, and we have no compelling evidence of the existence of an intermediate β′ phase. In contrast, experimental results suggest the coexistence of both α and β phases. The direct transition from the α-phase into the β polymorphic has been experimentally described in literature by previous authors for monoacid saturated TAGs. Kellens, et al.6 first reported the direct transformation of the α phase into the β form. The same experimental conclusions were later obtained by Sato14 and, more recently, by Seilert, et al.16
Fig. 5 presents the SAXS results. Interestingly, while the WAXS profile of the stearic sample appears to undergo several transitions, the SAXS profile only shows the formation of a single peak at q = 1.25 nm−1 (see Fig. 5A), attributed to the formation of 2L(α) domains (double chain length packing). The 2L(α) domains exhibit an average lamellar thickness of 5.02 nm. Over time, the peak slowly reduces intensity and progressively shifts to slightly lower q-values (better visualized in Fig. 5C). The sample does not transition to the 2L(β) form during the 15 minute isothermal measurement period.
 | ||
| Fig. 5 SAXS results. In panels A and B, surface plots of the SAXS profiles. In panels C and D, the orthogonal projection of the data expanding on the long-spacing transition. | ||
In contrast, the transition in the palmitic sample is more evident. Due to the shorter fatty acid chain length, the initial peak forms at higher q-values (q = 1.37 nm−1), still corresponding to a 2L(α) lamellar phase with a thickness of 4.58 nm. The peak progressively decreases in intensity and shifts over time to higher q-values, suggesting a reduction in the size of the lamellar domains. This shift is attributed to the formation of an angle of tilt, indicating the transition to 2L(β). The final recorded lamellar thickness is approximately 4.25 nm, still indicative of double chain packing, characterized by an angle of tilt around 65.5°.5
In the USAXS area, the behavior of CNP and CNP aggregates can be observed. Fig. 6(A and C) presents the surface plots of USAXS data, providing a detailed view of the scattering intensity variations. Before crystallization occurs (at times below 200 seconds), the scattering of the oil phase remains low in intensity. No crystalline structures are present that provide enough scattering signal to generate a well-defined profile. As the onset of crystallization is reached, the intensity rapidly increases, indicating the formation of crystalline domains in the mesoscale. The system then rapidly transforms, as evidenced by variations in intensity at diverse q-values.
 | ||
| Fig. 6 USAXS results. In panels A and B, results for a 30% FHRO dilution in HOSO. In panels C and D, USAXS results for a 30% dilution of PS in HOSO. In A and C, surface plots are shown. Panels B and D present a Kratky representation of the data, removing the scattering profiles where only the oil is visible. The data is presented as q2I vs.q. The arrow presented in panel B corresponds to a hot pixel, hence, this feature does not represent the sample's behavior. | ||
A more insightful representation of the results is achieved through a Kratky transformation, in which the data is converted from I vs.q to q2I vs.q (Fig. 6). This transformation allows for the formation of a bump in the high-q range, which can be attributed to the scattering from the cross-section of the CNPs. In other words, using a Kratky plot allows for a clearer visualization of the curvature of the bend. In this representation, the Guinier inflection is visualized as a peak, which helps in analyzing the conformation of the CNPs. The position of this peak offers insight into the size of the CNP cross-section. If the maximum intensity is placed at higher q-values, this indicates smaller CNP cross-sections. In contrast, a bump located at lower q-values suggests larger CNP cross-sections. The arrow in the plot identifies a hot pixel. From Fig. 6, panels B and D, we can identify similar behaviors for both palmitic and stearic samples: the Kratky bump is initially formed rapidly at lower q-values, and slowly shifts towards higher q-values, indicating the rapid formation of bigger CNPS, which then transform into smaller CNPs as the crystallization evolves.
Discussion
Fig. 7 presents a comprehensive compilation of structural information derived from XRS results (panels C to H), alongside the corresponding time and temperature profile (panel A) and DSC results (panel B) for the stearic sample. The time is counted from the beginning of the cooling ramp (at 80 °C). Only data with enough scattering intensity is here presented, as enough intensity is necessary to fit the models. | ||
| Fig. 7 Summary overview of the structural transitions occurring at the nano and mesoscale for a 30% FHRO dilution in HOSO (A–G) and a 30% PS dilution in HOSO (H–N). In panel A and H, the time–temperature profile are shown (for both DSC and XRS experiments). The DSC results are presented in panel B and I, where the heat flow is presented as a function of time. In panel C and J, the intensity over time of the α (q = 15.14 nm−1) and the β (q = 13.65 nm−1) phases. In panel D and K, lamellar thickness over time. In panel E and L, CNP thickness was calculated by applying the Scherrer equation in the d001 SAXS peak (eqn (2)), by applying the BWA method and by modeling the USAXS range (eqn (3)). In panel F and M, CNP width and in panel G and N, power law value P, indicative of aggregation dynamics (eqn (3)). Labels (I)–(IV) indicate four distinct moments identified in the transformation of CNP and CNP aggregates (see main text). | ||
The first noteworthy aspect of discussion is that although the sample changes continuously over time, DSC only reports one exothermic event. In fact, the instrument's resolution is insufficient to capture other structural modifications in the sample other than the formation of the α phase. Specifically, in the stearic sample (Fig. 7B), the α → β transition is not visible, as it occurs shortly after the α phase forms, and it takes a considerable amount of time to conclude, making it difficult to resolve in the DSC curve. In the case of the PS system, the polymorphic transition is visible at around 550 s (Fig. 7I).
Moreover, other structural rearrangements, such as the reduction of lamellar thickness, of CNP size or the changes in aggregation behavior of CNPs, do not exhibit sufficiently large thermal implications to be detected by DSC. This indicates that while DSC is an extremely useful technique to identify the biggest thermal events, such as the main crystallization peaks or the polymorphic form upon melt, whenever events are too closely spaced in time or structural modifications do not require big thermal energy, DSC fails to provide this information. In contrast, when sufficient time resolution is possible, XRS can capture these structural transitions.
A second important point is that not all length scales transform simultaneously. As seen in Fig. 7C for the 30% FHRO sample, at around 220 s the intensity at a q = 15.14 nm−1 (corresponding to the α phase) rapidly increases, indicating the rapid formation of the α polymorphic form. At around 300 seconds, the intensity of the α peak begins to decrease. This decline is coupled with a gradual increase in the intensity at q = 13.65 nm−1, indicative of the β phase. The further polymorphic transition into the triclinic β form occurs slowly and decelerates over time. Interestingly, although the α to β polymorphic form is clearly visible in the WAXS range between 200 s and 500 s (Fig. 7C and J), the long spacings do not transform simultaneously (Fig. 7D and K). The transition from 2L(α) to 2L(β) is notably slow for the stearic system, as the lamellar thickness remains at around 5 nm throughout the experiment. This result suggests that no variations in the angle of tilt are formed within the 15 minutes of the isothermal period. In the case of the palmitic system, the transition from a 2L(α) to a 2L(β) occurs at around 1000 s, well after the short spacings have transitioned.
To the best of our knowledge, this phenomenon has not yet been reported in the past. In fact, based on preliminary experiments (data not shown) and previous literature,14,18 we know that, in pure systems, WAXS and SAXS show simultaneous transformations. However, until now, time-resolved studies on microstructural transformations on TAGs have been mainly done on pure systems, rather than TAG ingredients, creating an important knowledge gap on the impact of impurities on the microstructural development of lipids. A very recent study performed by Seilert, et al.,16 reported a similar experiment for 2%, 10% and 20% dilutions of FHRO, however, their isothermal temperature was different than the one here used (25 °C), yielding to faster long spacing transitions.16 Due to the lack of secondary sources to validate the findings, the experiment was repeated on a lab-scale X-ray source (see Appendix 1). The experiment showed to be repeatable, and results suggest that the transformation from 2L(α) to 2L(β) can take up to 8 hours, as the system continuously evolves to smaller lamellar sizes with narrower FWHM, indicative of bigger CNPs. These findings confirm that commercial TAGs sources undergo structural modifications over longer timescales in comparison to their pure counterparts.
As lamellae assemble on top of each other, CNPs are formed. The volume-weighted average number of lamellae per CNP obtained following the BWA method is presented in Fig. 8. In both systems, the overall transformation is similar. Initially, in the α phase, the number of lamellae per CNP can be described as a unimodal distribution, skewed to the right, centered at around 7–8 lamellae. The distribution is broad, indicative of a high degree of polydispersity. As the transformation advances into the β phase, a secondary lamellae thickness distribution appears. The new distribution is skewed to the right and centered in a lower number of lamellae (at around 3 to 4). The CNPs formed in the β phase are less dispersed in the number of lamellae than those reported in the α phase. In the case of the stearic-based system, the transformation is still not completed after 15 minutes of isothermal time, as the last recorded distribution retains a bimodal character. In contrast, in the case of the PS, the broader α distribution seems to have fully transformed into the narrower β distribution.
 | ||
| Fig. 8 Crystallite thickness distributions for a 30% FHRO dilution in HOSO and a 30% PS dilution in HOSO. | ||
In addition to the distribution, the average CNP thickness values are presented in Fig. 7E and L, respectively. As described in the methodology section, three methods can be used to determine CNP thickness: the Scherrer equation, the BWA method and USAXS modeling. Regardless of the method used to calculate the CNP thickness, the conclusion is consistent: over time, there is a progressive reduction in CNP thickness, suggesting that CNPs of the β phase are thinner than those of the α phase. As previously mentioned, in the 30% PS sample, the CNP thickness decreases until it stabilizes upon completion of the 2L(β) transition. In the case of the stearic sample, the transition is not completed within 15 minutes, and therefore, only a progressive decrease in CNP thickness can be reported. For further discussion on the differences between the three methods, the reader is referred to Appendix 2.
Fig. 7F and M illustrate the changes in CNP width over time, while Fig. 7G and N show the changes in CNP aggregation behavior. Interestingly, new variations are observed at these length scales. In fact, until now, only reductions in thickness have been noted, transitioning from thicker to thinner domains, with only two distinct moments identified. In contrast, in this case, four distinct moments can be identified (indicated as I, II, III, and IV in Fig. 7). It is important to mention that aggregation behavior is here described via the power law P, where higher P values indicate increasingly dense aggregates, while lower P values indicate loser conformations (for more information on the interpretation of P, the reader is referred to the Appendix 3).
In the first phase (phase I), the α phase is formed. As seen in the WAXS region, the increase in the α peak is accompanied by an increase in the width of the CNPs. This indicates the formation of CNPs with increasing cross-sections, which conceptually aligns with the higher level of structural disorder characteristic of the α phase. These CNPs aggregate closer together as time develops, as indicated by an increase in P. During the second phase (phase II), the α-to-β polymorphic transition occurs, and the intensity of the α peak diminishes (Fig. 7C and K). As the α phase transitions, the average CNP size reduces, evidenced by a decrease in both the width and the thickness of the CNPs. Simultaneously, P decreases, suggesting that the reduction of the α phase leads to looser conformations and less aggregated CNPs. In the third phase (phase III), there is a rapid growth of the β phase. The width of the CNPs progressively increases until it reaches a maximum. Simultaneously, the value of P increases, indicating the formation of denser crystallites. Finally, in the fourth phase (phase IV), the β polymorphic form further develops. The CNP cross-sections decrease in size while maintaining the same level of aggregation. The equivalent microscopies of these four moments are presented in Fig. 9 (the equivalent supporting videos, where the transitions are clearer, are available in the ESI,† Videos S3 and S4).
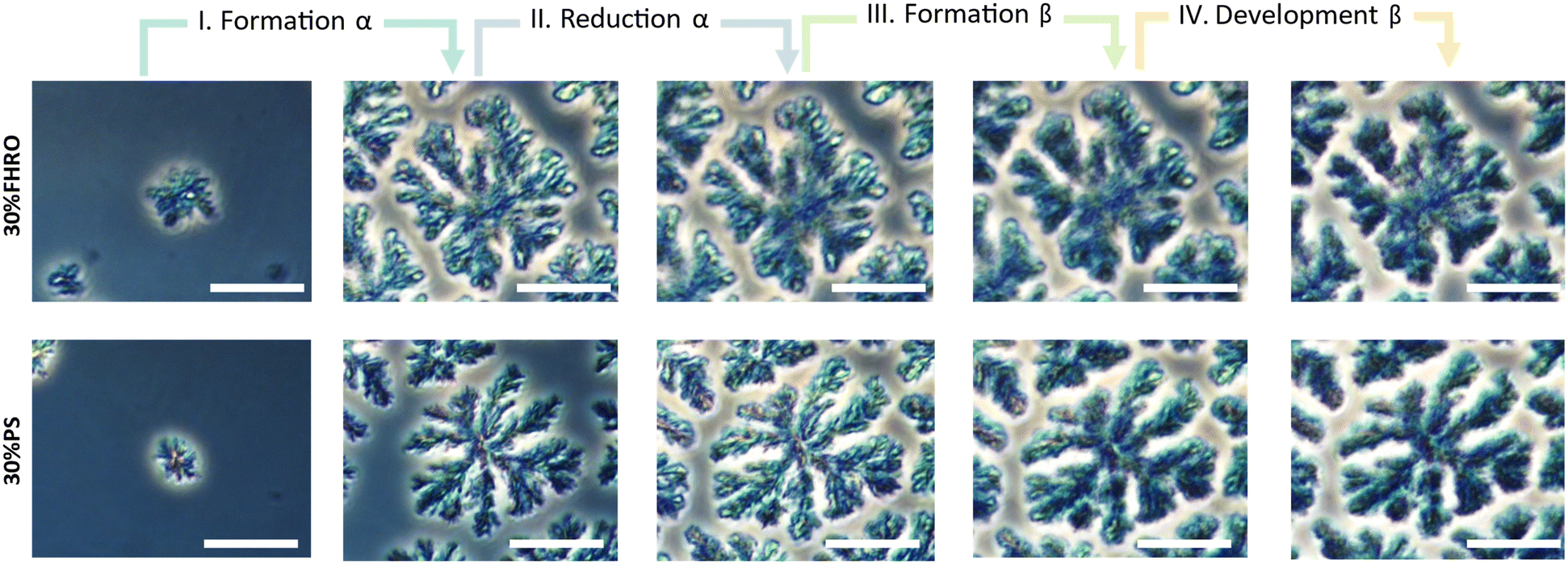 | ||
| Fig. 9 Phase contrast microscopy images illustrating the floc growth and development for a 30% FHRO dilution in HOSO and a 30% PS dilution in HOSO. Four distinct stages are identified. Corresponding videos are available in the ESI† (Video S3 and S4). The scale bar in the images represents 25 μm. | ||
Fig. 9 demonstrates that the β phase only emerges at the expense of the existing α phase. Consequently, the number of α nucleation points is a crucial factor in the formation of the final crystalline network (Fig. 2). In other words, upon polymorphic transition, the β phase emerges exclusively wherever there are pre-existing α crystalline structures, suggesting that the final architecture of the network is defined already by the nucleation of the α phase. This observed mechanism is similar to the one described by Stewart, et al.,41 where one polymorphic form expands at the expense of the other. The transformation mechanism involves the dissolution of the least stable polymorph followed by the crystallization of the more stable polymorph across a narrow liquid interface.
Conclusions
The crystallization of lipids is a complex multiscale process that requires the development of methods to better understand the self-assembly of each structural hierarchy level. In this article, we address how X-ray scattering techniques can help us understand triglyceride's multiple geometrical arrangements. From polymorphic identification (α, β′, β), to packing density and chain length arrangement (2L or 3L), to crystalline nanoplatelets (CNPs), X-ray scattering techniques allowed us to retrieve structural information of triglycerides at nano- and mesoscale non-accessible otherwise.In fact, while differential scanning calorimetry (DSC) serves as a valuable tool for identifying major thermal events such as primary crystallization peaks and polymorphic transitions upon melting, its ability to resolve closely spaced events or extract structural modifications without significant thermal energy is limited. In contrast, time-resolved X-ray scattering (XRS) techniques, provide tools for capturing these intricate structural transitions in triglycerides.
In the setup proposed, time-resolved synchrotron WAXS (1–10 Å), SAXS (10–100 Å), and USAXS (0.1–3.5 μm) were used to study the crystallization behavior of 30% wt palm stearin (PS) in high oleic sunflower oil (HOSO) and 30% wt fully hydrogenated rapeseed oil (FHRO) in HOSO. This comprehensive approach spans four orders of magnitude (from Å to μm), enabling us to capture dynamic transformations in triglyceride structures from the molecular to the mesoscale. For the first time, time-resolved USAXS results where provided for saturated monoacid triglycerides, allowing us to discerns important dynamics at the mesoscale, such as changes in CNP size and aggregation. The study was also complemented with microscopy visualizations. PCM allowed the visualization of time resolved transitions on the floc and network level, while simultaneously helping to identify differences in spherulite morphology between stearic and palmitic samples. In addition, Cryo-SEM facilitated the visualization of various aggregation units including lamellar structures, crystalline nanoplatelets (CNPs), CNP aggregates and crystal clusters.
The transition followed an α to β polymorphic pathway, with a 2L(α) to 2L(β) lamellar stacking configuration. Our results suggest that structural transformations in non-pure TAG systems progress over extended timescales post-crystallization, suggesting a gradual evolution across different length scales. This result highlights the distinctions between commercial systems and model systems, demonstrating that the presence of impurities or diversity in TAG composition, significantly influences the kinetics of crystallization. While the thickness of both lamellae and CNP only evolved from thicker to thinner, the overall mesoscale behavior was found to change in four different phases. In the initial phase, the α phase formation is characterized by the creation of larger CNPs that aggregate more closely over time. During the second phase, the α-to-β polymorphic transition occurs, leading to a decrease in the size of the CNPs as the α phase transitions, resulting in looser and less aggregated CNPs. In the third stage, a rapid growth in the β phase occurs, causing the CNPs to increase in size and become more densely aggregated. Finally, the β polymorphic form continues to develop. Altogether, the formation of the network is strongly determined by the nucleation of the α state. We believe that these findings showcase the capabilities of XRS in elucidating transitions in dynamic systems, and represent a starting point in the understanding of crystallization kinetics of triglycerides at several hierarchical levels.
Data availability
Following the ESRF data policy, the raw XRS data is made openly accessible to the public via the ESRF data portal (proposal numbers ME-1606, ME-1607 and ME-1642). Any additional data that substantiates the outcomes of this research are publicly are openly available in Zenodo at https://doi.org/10.5281/zenodo.13133005.Author contributions
Conceptualization: I. A. P., T. R., K. D., F. V. B.; data curation: I. A. P.; formal analysis: I. A. P., F. V. B.; funding acquisition: I. A. P., K. D., F. V. B.; investigation: I. A. P., F. D. W., T. R.; methodology: I. A. P., T. R., F. V. B.; project administration: K. D., F. V. B.; resources: K. D., F. V. B.; software: I. A. P., F. D. W.; supervision: T. R., K. D., F. V. B.; visualization: I. A. P.; writing – original draft: I. A. P.; writing – review & editing: F. D. W., T. R., K. D., F. V. B.Conflicts of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.Acknowledgements
The Research Foundation—Flanders (FWO) is acknowledged for providing the grant 1SA5321N in support of Ph.D. candidate Ivana A. Penagos and 1128923N in support of Ph.D. candidate Fien De Witte. The European Synchrotron Radiation Facility (ESRF) was recognized for providing financial support for the synchrotron-assisted XRS experiments in beamline ID-02 (proposal number ME-1606, ME-1607 and ME-1642). William Chevremont and Theyencheri Narayanan are thanked for their assistance during the experiments. The Hercules foundation is recognized for its financial support for the SEM JEOL JSM-7100F equipped with the cryogenic transfer system Quorum PP3000T (grant number FWO Hercules AUGE-09-029) and the XRS Xenocs Xeuss 3.0 (grant number FWO Hercules AUGE-17-029). Vandemoortele Lipids NV is acknowledged for financially supporting the UGent Vandemoortele Centre on ‘Lipid Science and Technology’. This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement No. 654000. Benny Lewille, Kato Rondou, Nathaniel J. Hendrik and Griet Spaepen, are thanked for their continuous support conducting the synchrotron experiments. This research reflects the views of the authors only, and hence, none of the above parties should be held responsible for the information contained here.References
- S. S. Narine and A. G. Marangoni, Food Res. Int., 1999, 32, 227–248 CrossRef CAS.
- A. G. Marangoni, N. Acevedo, F. Maleky, E. Co, F. Peyronel, G. Mazzanti, B. Quinn and D. Pink, Soft Matter, 2012, 8, 1275–1300 RSC.
- M. A. Rogers, in Fatty Acids, ed. M. U. Ahmad, Elsevier, 2017, pp. 541–559 Search PubMed.
- N. C. Acevedo and A. G. Marangoni, Annu. Rev. Food Sci. Technol., 2015, 6, 71–96 CrossRef CAS PubMed.
- K. Mishra, N. Kummer, J. Bergfreund, F. Kampf, P. Bertsch, R. Pauer, G. Nystrom, P. Fischer and E. J. Windhab, J. Colloid Interface Sci., 2023, 630, 731–741 CrossRef CAS PubMed.
- M. Kellens, W. Meeussen, C. Riekel and H. Reynaers, Chem. Phys. Lipids, 1990, 52, 79–98 CrossRef CAS.
- N. C. Acevedo and A. G. Marangoni, Cryst. Growth Des., 2010, 10, 3334–3339 CrossRef CAS.
- I. A. Penagos, F. De Witte, T. Rimaux, W. Chèvremont, I. Pintelon, K. Dewettinck and F. Van Bockstaele, Soft Matter, 2024, 5071–5085 RSC.
- D. Cholakova and N. Denkov, Adv. Colloid Interface Sci., 2024, 323, 103071 CrossRef CAS.
- S. Takeguchi, A. Sato, H. Hondoh, M. Aoki, H. Uehara and S. Ueno, Molecules, 2020, 25, 5086 CrossRef CAS.
- D. M. Small, J. Lipid Res., 1984, 25, 1490–1500 CrossRef CAS.
- C. E. Clarkson and T. Malkin, J. Chem. Soc., 1934, 666–671 RSC.
- D. Cholakova, S. Tcholakova and N. Denkov, Cryst. Growth Des., 2023, 23, 2075–2091 CrossRef CAS.
- K. Sato, Chem. Eng. Sci., 2001, 56, 2255–2265 CrossRef CAS.
- K. Sato, J. Phys. D: Appl. Phys., 1993, 26, B77 CrossRef CAS.
- J. Seilert, M. Rappolt, G. Dol and E. Flöter, Cryst. Growth Des., 2024, 24, 1146–1158 CrossRef CAS.
- L. Pellegrino, G. Tyagi, E. S. J. Robles and J. T. Cabral, Phys. Chem. Chem. Phys., 2022, 24, 29413–29422 RSC.
- M. Kellens, W. Meeussen, R. Gehrke and H. Reynaers, Chem. Phys. Lipids, 1991, 58, 131–144 CrossRef CAS.
- F. De Witte, I. A. Penagos, K. Rondou, K. Moens, B. Lewille, D. A. Tzompa-Sosa, D. Van de Walle, F. Van Bockstaele, A. G. Skirtach and K. Dewettinck, Crystals, 2024, 14, 142 CrossRef CAS.
- F. De Witte, I. A. Penagos, D. Van de Walle, A. G. Skirtach, K. Dewettinck and F. Van Bockstaele, Foods, 2024, 13, 1372 CrossRef CAS.
- N. C. Acevedo, in Structure-Function Analysis of Edible Fats, ed. A. G. Marangoni, 2018, pp. 1–19 Search PubMed.
- N. C. Acevedo, J. M. Block and A. G. Marangoni, Faraday Discuss., 2012, 158, 171–194 RSC.
- N. C. Acevedo and A. G. Marangoni, Cryst. Growth Des., 2010, 10, 3327–3333 CrossRef CAS.
- N. C. Acevedo, F. Peyronel and A. G. Marangoni, Curr. Opin. Colloid Interface Sci., 2011, 16, 374–383 CrossRef CAS.
- F. Maleky, N. C. Acevedo and A. G. Marangoni, Eur. J. Lipid Sci. Technol., 2012, 114, 748–759 CrossRef CAS.
- F. Peyronel, N. C. Acevedo, D. A. Pink and A. G. Marangoni, Crystallization of Lipids: Fundamentals and Applications in Food, Cosmetics, and Pharmaceuticals, 2018, pp. 143–181 Search PubMed.
- B. A. Macias-Rodriguez, F. Peyronel and A. G. Marangoni, J. Food Eng., 2017, 212, 87–96 CrossRef CAS.
- F. Peyronel, PhD, University of Guelph, 2014 Search PubMed.
- F. Peyronel, J. Ilavsky, G. Mazzanti, A. G. Marangoni and D. A. Pink, J. Appl. Phys., 2013, 114, 234902 CrossRef.
- F. Peyronel, B. Quinn, A. G. Marangoni and D. A. Pink, Food Biophys., 2014, 9, 304–313 CrossRef.
- F. Peyronel and D. A. Pink, in Structure-Function Analysis of Edible Fats, ed. A. G. Marangoni, 2018, pp. 267–285 Search PubMed.
- D. A. Pink, B. Quinn, F. Peyronel and A. G. Marangoni, J. Appl. Phys., 2013, 114, 304–313 CrossRef.
- P. R. R. Ramel Jr, F. Peyronel and A. G. Marangoni, Food Chem., 2016, 203, 224–230 CrossRef.
- P. Raimondi, C. Benabderrahmane, P. Berkvens, J. C. Biasci, P. Borowiec, J. F. Bouteille, T. Brochard, N. B. Brookes, N. Carmignani, L. R. Carver, J. M. Chaize, J. Chavanne, S. Checchia, Y. Chushkin, F. Cianciosi, M. Di Michiel, R. Dimper, A. D'Elia, D. Einfeld, F. Ewald, L. Farvacque, L. Goirand, L. Hardy, J. Jacob, L. Jolly, M. Krisch, G. Le Bec, I. Leconte, S. M. Liuzzo, C. Maccarrone, T. Marchial, D. Martin, M. Mezouar, C. Nevo, T. Perron, E. Plouviez, H. Reichert, P. Renaud, J. L. Revol, B. Roche, K. B. Scheidt, V. Serriere, F. Sette, J. Susini, L. Torino, R. Versteegen, S. White and F. Zontone, Commun. Phys., 2023, 6, 82 CrossRef CAS.
- T. Narayanan, M. Sztucki, T. Zinn, J. Kieffer, A. Homs-Puron, J. Gorini, P. Van Vaerenbergh and P. Boesecke, J. Appl. Crystallogr., 2022, 55, 98–111 CrossRef CAS.
- T. Narayanan, M. Sztucki, P. Van Vaerenbergh, J. Leonardon, J. Gorini, L. Claustre, F. Sever, J. Morse and P. Boesecke, J. Appl. Crystallogr., 2018, 51, 1511–1524 CrossRef CAS PubMed.
- S. Michael, SAXSutilities2: a graphical user interface for processing and analysis of Small-Angle X-ray Scattering data, Zenodo, 1.024 edn, 2011 Search PubMed.
- K. Rondou, F. De Witte, T. Rimaux, W. Dewinter, K. Dewettinck, J. Verwaeren and F. Van Bockstaele, J. Am. Oil Chem. Soc., 2022, 99, 1019–1031 CrossRef CAS.
- R. den Adel, K. van Malssen, J. van Duynhoven, O. O. Mykhaylyk and A. Voda, Eur. J. Lipid Sci. Technol., 2018, 120, 1800222 CrossRef.
- M. Doucet, J. H. Cho, G. Alina, Z. Attala, J. Bakker, W. Bouwman, P. Butler, K. Campbell, T. Cooper-Benun, C. Durniak, L. Forster, M. Gonzalez, R. Heenan, A. Jackson, S. King, P. Kienzle, J. Krzywon, R. Murphy, T. Nielsen, L. O'Driscoll, W. Potrzebowski, S. Prescott, R. Ferraz Leal, P. Rozyczko, T. Snow and A. Washington, SasView version 5.0.5, ed. W. Potrzebowski and A. Jackson, Zenodo, 5.0.4 edn, 2021 Search PubMed.
- D. I. Stewart, P. S. Chong and A. G. F. Stapley, Cryst. Growth Des., 2017, 17, 3005–3016 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ce00800f |
| This journal is © The Royal Society of Chemistry 2024 |