
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural properties and lattice phonons evolution in phenothiazine/iminostilbene solid solutions†
Andrea
Giunchi‡
 a,
Lorenzo
Pandolfi§
a,
Raffaele G.
Della Valle
a,
Tommaso
Salzillo
a,
Elisabetta
Venuti
*a,
Nicola
Demitri
b,
Hans
Riegler
c,
Christina
Petschacher
c,
Jie
Liu
d and
Oliver
Werzer
e
a,
Lorenzo
Pandolfi§
a,
Raffaele G.
Della Valle
a,
Tommaso
Salzillo
a,
Elisabetta
Venuti
*a,
Nicola
Demitri
b,
Hans
Riegler
c,
Christina
Petschacher
c,
Jie
Liu
d and
Oliver
Werzer
e
aDipartimento di Chimica Industriale “Toso Montanari”, Università di Bologna, viale del Risorgimento, 4, 40136, Bologna, Italy. E-mail: elisabetta.venuti@unibo.it
bElettra – Sincrotrone Trieste, S.S. 14 Km 163.5 in Area Science Park, 34149 Basovizza (Trieste), Italy
cDepartment for Pharmaceutical Technology and Biopharmacy, Institute of Pharmaceutical Sciences, Graz University, Universitätsplatz 1, 8010 Graz, Austria
dDepartment of Physics, University of Warwick, CV4 7AL, Coventry, UK
eJoanneum Research Forschungsgesellschaft mbH, Franz-Pichler Str. Nr. 30, 8160 Weiz, Austria
First published on 5th November 2024
Abstract
Together with co-crystals, solid solutions of molecular systems are vital in the design of multicomponent solids that exhibit improved physical and chemical properties compared to those of pure substances. In this work, both the bulk and thin film phases of the molecular solid solutions of the active pharmaceutical ingredients (APIs) phenothiazine (PTZ) and iminostilbene (ISB) are characterized structurally, while low frequency Raman spectroscopy coupled with DFT simulations is employed to understand the impact of the loss of perfect periodicity of the mixed system on its lattice dynamics. X-ray diffraction methods show the statistical distribution of the two molecules in the structure, and the steady variation of the structural parameters with solution composition, confirming that we are dealing with monophasic mixtures. The spectroscopic properties are demonstrated to be different depending on the nature of the vibrational mode. While the vibrational spectra of molecules can always be decomposed into a superposition of the spectra of the two pure compounds, the lattice phonons exhibit a continuous evolution throughout the solution series.
Introduction
When molecules are mixed in the solid state,1 the resulting material may have physical and chemical properties significantly different from the individual components in crystal structures,2 phase diagrams and electronic transport characteristics.3 This is especially relevant in applications where functionality operates on the molecular scale, with many examples in fields such as organic electronics and pharmaceutical science. The mixed systems are usually labelled according to the relative stoichiometric ratios of the components.1,4–7 Co-crystal are structurally homogeneous crystalline materials that contain two or more neutral building blocks that are present in definite stoichiometric amounts6 in the crystal unit cell and can be made of molecules very different both structurally and chemically, interacting via hydrogen and halogen bonds or π–π stacking in supramolecular synthons. A special situation arises in the solid solutions, where one specie “dissolves” in the lattice of another one with a random occupancy of the crystallographic sites.4,8–10 Consequently, these systems may display a continuous variation of one component in the unit cell of the others (or vice versa) and the phase separation eventually occurs only in miscibility gaps of the constituents. With few exceptions,8 the formation of molecular solid solutions over the entire range of relative compositions requires components with very similar molecular geometries and intermolecular interaction patterns, and results in isomorphic structures with matching unit cell parameters, so that the crystal packing of the mixtures is very similar to that of the pure components. When considering the system from a thermodynamic point of view, the maximum entropy condition caused by mixing is the leading factor, indicating that a phase separation yielding either saturated solutions or co-crystals would not result in a favourable balance of the total free energy.11 Despite the similarity, the presence of more molecular species still alters a number of important solid's properties, such as solubility, melting, long-time stability and thermal coefficients.Various techniques are available for investigating the characteristics of both amorphous and crystalline states, applicable in both industrial and academic settings. Among these, methods utilizing X-ray diffraction are preferred for analysing periodic structures. Nonetheless, in the last years, Raman spectroscopy has proven to be able to provide useful information on the material's chemical footprint, with the added bonus of not requiring the sample preparation prior to measurements, a feature that makes the technique applicable, for instance, in processes of in-line quality control. The Raman investigation of the molecular solid state reaches its maximum effectiveness when it encompasses the low-wavenumber region of lattice-phonon modes, which represent the collective vibrations that probe the weak intermolecular interactions of the lattice structure and therefore the characteristics of the lattice itself. Exploring the lattice phonon modes yields valuable information on the molecular solid state, such as structural properties, phase identification, and transformations.
In this study, we investigate the properties of solid solutions composed of two prototypical molecules: phenothiazine (PTZ, 10H-dibenzo-[b,e]-1,4-thiazine) and iminostilbene (ISB, 5H-dibenz[b,f]azepine). These compounds belong to the bisarylimine class and are known for their potential therapeutic effects in treating cancer, Parkinson's disease, epilepsy, Plasmodium falciparum, and other in vitro applications.12–15 While PTZ and ISB exhibit high potency, their poor selectivity hinders their broader medicinal use. In drug therapy these molecules are modified by side groups making them more selective for medical targets, e.g. chlorpormazine is used to treat psychotic disorders while carbamazepine treats epilepsy and neuropathic pain. The additional side groups result in more complex polymorphism.16 Anyway, their chemical affinity and structural similarities hints to similar intermolecular interaction patterns in their structures, potentially yielding monophasic mixed systems characterized by distinct physical–chemical and therapeutic properties that could make them lead materials.
The results obtained in this work from X-ray single crystal and grazing incidence techniques, applied to both single crystals and thin films of the mixed PTZ/ISB systems, demonstrate the near-ideal monophasic nature of the mixtures, which are found to exhibit continuous property changes across various compositions. Unlike co-crystals with stoichiometric ratios, these structures lack the necessary periodicity for lattice vibrations. Nevertheless, the persistence of low-frequency spectral features observed in previous studies involving solid solutions17 prompted us to further investigation into the lattice dynamics of the PTZ/ISB mixtures. Thus, alongside structural characterization, the micro-Raman technique was employed in both high and low-frequency energy intervals, with the support of solid-state DFT simulations, with the aim of detecting the lattice fingerprints which allow for the safe identification of the solid solution formation.
Methods
Materials and crystallization
PTZ and ISB were purchased from Sigma Aldrich company and used without further modification. All the samples were prepared starting from tetrahydrofuran (THF) solutions with concentration of 10 mg mL−1. Solid solutions with varying compositions were obtained by mixing THF solutions of the parent systems in precise ratios, yielding the desired molar fractions of PTZ and ISB in both bulk materials and thin films. As substrates, silicon wafers hosting a native oxide layer (Siegfert Wafers, Germany) were cut into 2 × 2 cm2 pieces and cleaned in an acetone sonication bath, rinsed with ethanol and isopropanol and finally dried in a dry nitrogen stream. Thin film samples were obtained by spin coating 100 μL of solution and rotation at a speed of 1000 rpm for 60 s. For the preparation of drop casting samples and single crystals, the same batch of THF solutions were taken and 25 to 100 μl drops of were placed onto 1 × 1 cm2 silicon wafers which were covered by Petri dishes to slow solvent evaporation.Grazing incidence X-ray diffraction (GIXD) measurements
The SAXS beamline at the synchrotron ELETTRA used a wavelength of 0.154 nm. The hexapot aligned the sample with respect to the incoming beam. An underlying azimuth rotation increased the statistics of the sample. After collecting the intensity with a Pilatus 1 M detector, the experimental data was processed with the software GIDVis.18 LaB6 was the calibrant for the setup. All GIXD images were collected under an incidence angle of 1.2°. While this is above the critical angle of silicon oxide (about 0.2°) this high angle reduces the footprint and thus limited smearing at the detector side which is inherent to a setup without parallel beam collimation.19Single crystal experiments
Data collections at ELETTRA were done at the beamline XRD1 (ref. 20) on a standard Kappa – geometry (HUBER Diffraktionstechnik GmbH & Co. KG). Samples were obtained by removing individual crystals from the silicon wafers with small fragments of adhesive tape, consequently, single crystals appear as very thin elongated plates with limited diffracting power (0.9–1.0 Å). Then crystals were mounted on the goniometer head using the tape as a support. Complete datasets were collected at room temperature through rotating crystal method using monochromatic X-ray of a wavelength of 00.700 Å on a Pilatus 2 M hybrid-pixel area detector (DECTRIS Ltd., Baden-Daettwil, Switzerland). The diffraction data were indexed and integrated using XDS.21 Scaling were done using CCP4-Aimless.22,23 The structures were solved by the dual space algorithm implemented in the SHELXT.24 Fourier analysis and refinement were performed by the full-matrix least-squares methods based on F2 implemented in SHELXL-2014 (ref. 25) and the modelling was performed using the Coot program.26 Data have been collected at room temperature to allow direct comparison with grazing incidence data. To overcome model refinement problems associated with poor resolutions limits, crystals have been also collected at 100 K, using a cold nitrogen stream (Oxford Cryosystems Ltd.). This approach gave datasets with significantly better statistics and led to structural models where occupancies of the PTZ:IBS mixture could also be reliably refined. Data obtained by pure PTZ and ISB crystals, produced in the same conditions, were also useful and has been used as references for geometrical restrains (needed in the refinement of disorder in the mixtures).Hirshfeld surface (HS) analysis
Hirshfeld surface is the density weight function of the specific interested molecule (i.e. the promolecule) over the same sum of density of its nearest neighbor, supplying information about intermolecular interactions.27 Different surface properties such as i.e. dnorm, electrostatic potential, shape-index and curvedness can be calculated and mapped on HS using the software CrystalExplorer17.28,29 Distance from the surface to the nearest atom interior to the surface is represented by di; and de reflects distance from the surface to the nearest atom exterior to the surface. Curvedness, C, is given by:where, k1 and k2 is principal curvatures. The two-dimensional fingerprint plot derived from a Hirshfeld surface.30,31 The color on the plot with a range from blue (relatively few points) through green (moderate fraction) to red (highest fraction) reflects the contribution from different interatomic contacts.

Raman experiments
The spectra were collected on a Horiba Jobin Yvon T64000 spectrometer equipped with three monochromators in double subtractive + additive configuration. The spectrometer was coupled to an Olympus BX40 confocal microscope equipped with various objectives; a lateral resolution of 1 μm can be obtained with the 100× objective. Spectra were recorded with the excitation wavelength of the 647.1 nm line of a Kr+ gas laser.For the polarized Raman measurements, a half-wave plate was used to rotate the polarization of the incident light, while that of the scattered light was selected by means of a wire grid polarizer. The crystal faces were identified based on the results of X-ray indexing and the morphology of the samples. Using a polarizing microscope, the samples were aligned under crossed polarizers along the extinction directions, which, in the orthorhombic systems analyzed here, corresponded to alignment along one of the principal crystallographic axes.
DFT simulations
DFT simulations on both PTZ and ISB pristine structures32,33 were performed using the code VASP (Vienna Ab initio Simulation Package)34–39 including the effects of dispersive interactions using the D3-BJ pair-wise corrections.40 Energy convergence was achieved with identical settings for both cases: a plane wave cutoff of 800 eV proved to be adequate in combination with (2 × 3 × 1) Monkhorst–Pack k-point sampling, for the short, middle and long axis, respectively, and selected to obtain the same density of points in each direction. Firstly, Atomic coordinates and cell axes were fully relaxed, halting when residual forces fell below 1 meV A−1, and for this the GADGET package41 was employed. For both structures of PTZ and ISB, energy differences converge is faster than absolute energies within 0.006 meV per atom. Then, the vibrational modes and the polarizability tensors were computed at k = 0 based on the experimental unit cell volumes using the software Phonopy.42 Polarizability tensors were calculated for each Phonopy displacement, and the system symmetry operations were applied to complete the tensors. Finally, the numerical derivatives were performed. Unpolarized and polarized Raman spectra of powders and main crystallographic faces (ab, bc, ac) were simulated by selecting the proper combinations of the components of the symmetric 3 × 3 polarizability tensors. Raman intensities were finally adjusted by accounting for the excitation wavelength and temperature dependence.43Results and discussion
PTZ and ISB crystallize in an orthorhombic crystal system with space group Pnma,32,33 with small variations in the unit cell volume and axes (Fig. 1a and Table 1). Their structural differences are attributable to the chemical nature of the central rings, two benzene rings fusing to an azepine group in the case of ISB and to a thiazine for PTZ. Each molecule structure shows a characteristic bend around the hinge which is defined by the chemical composition of the central rings; i.e. ISB bends approximately 27° compared with 21° in PTZ (Fig. 1b), resulting from the substitution of two carbon atoms in ISB molecule with a sulfur atom in PTZ molecule (Fig. 1c). | ||
| Fig. 1 a) Packing of PTZ and ISB molecules in their lattices and their mixed 1/2 structure PTZ/ISB. The cell orientation has been upright, as it is on the substrate surface. b) The projection of PTZ and ISB molecular structures to illustrate the similarity of the molecular geometry and the bending angles. PTZ is displayed in blue and ISB is in red. c) Molecular structure of PTZ and ISB. | ||
| Compounds | PTZ | PTZ/ISB (1/2) | ISB | |
|---|---|---|---|---|
| Temperature/K | 298 | 298 | 298 | |
| Unit cell/Å | a | 7.923(2) | 8.169(2) | 8.239(2) |
| b | 20.978(4) | 20.915(4) | 20.450(4) | |
| c | 5.897(1) | 6.053(1) | 6.036(1) | |
Single crystal diffraction
The single crystal structures of PTZ, ISB and a mixed PTZ/ISB crystal prepared with a molar ratio 1/2 of the two components were determined at room temperature by carefully removing the crystals from the substrates of drop casting samples. The lattice constants are summarized in Table 1. Structure refinement of the room temperature results of the pristine materials PTZ and ISB match those deposited in the Cambridge Structural Database (refcodes: BZAZPO and PHESAZ01). This finding confirms that the quality of single crystals obtained from the preparation route allows for structure determination by single crystal diffraction experiments.Structure characterization of the mixed 1/2 PTZ/ISB crystal (Table 1) indicates that it also belongs to an orthorhombic crystal system with the same space group Pnma as PTZ and ISB. Inspection of the unit cell dimensions of ISB and mixed structure reveals that for the mixed system PTZ/ISB the axial length increases along the a-axis, decreases along the b-axis, and remains nearly identical along the c-axis (Table 1) on increasing the ISB fraction. The structure of the mixed system (Fig. 1) reveals a non-stoichiometric packing of the PTZ and ISB molecules, as the crystallographic position in this particular case was occupied by PTZ and ISB molecules with a ratio 34%/66% which reflects the statistical distribution of the molecules in the unit cell sites and also agrees well with the relative amounts of PTZ and ISB used for preparation of the system.
Since packing mode similarities and subtle differences necessarily affect the physiochemistry properties (Fig. 1), it is crucial to visually investigate the feature for PTZ, ISB and the mixed PTZ/ISB molecules through the Hirshfeld Surface (HS) analysis (Fig. 2). The shape of the curvedness surfaces of the three molecules is subjected to the division of patches associated with contacts between adjacent molecules, varying from uncovered rectangles to triangles (marked by red rectangles), resulting directly in an increase in the bending angle from PTZ (21°) to ISB (27°). Inspection of the fingerprint plots in Fig. 2 clearly depicts the major packing differences amongst the crystal structures. The fingerprints in Fig. 2a show two separated and sharp peaks in PTZ resulting from the S⋯H and C⋯H short contacts respectively, which then converge into broad shoulders on the side of the plot with the characteristic superposition S⋯H and C⋯H in the mixed structure PTZ/ISB (Fig. 2b), whereas the interaction S⋯H is absent in ISB (Fig. 2c), where the thiazine group is replaced by the azepine ring. A careful investigation of the spike between the wings along the diagonal of the streak on the plot clearly identifies the H⋯H contacts (which appear where de ≈ di, or shorter than the H-atom van der Waals radius of 1.20 Å) representing the molecular interaction between two adjacent sheets. The peak is sharper and the distances of H⋯H contacts are closer in PTZ (de ≈ di = 1.1 Å) than in ISB (de ≈ di > 1.2 Å), which are shown with green rectangles in the packing diagram of PTZ while absent in ISB (Fig. S1†). However, these H⋯H contacts, shorter than the sum of H-atom van der Waals radii of 1.20 Å, usually are thought to be repulsive in nature.44,45 Note that the H⋯H contacts in ISB span a high concentration in a narrow distance range (de = 1.3–1.6 Å, marked with a yellow circle in Fig. 2c), and are shorter compared to the PTZ's (de > 1.8 Å). As shown in Fig. S2,† H⋯H is the dominant contribution to the surfaces area of molecules in PTZ, PTZ-ISB and ISB, whilst it is higher in ISB (49.8%) than that in PTZ (43.8%). So, the presence of H⋯H contacts necessarily plays an important effect on the bonding and stabilizing interactions in within crystal structure. Moreover, The C–H⋯π contacts also contribute more towards the total surface ISB (45.6%) than that in PTZ (38.7%). Based on the above investigation, this implies that the crystal packs more closely in ISB structure than that in PTZ.
 | ||
| Fig. 2 The curvedness surfaces and 2D fingerprint plots for (a) PTZ, (b) PTZ-ISB and (c) ISB. The centre section of the curvedness are marked with empty rectangles in red. The characteristic short contacts are pointed to with red arrows. The area circled in yellow illustrates the intense concentration range of H⋯H contacts span. | ||
Grazing incidence diffraction (GIXD)
Grazing incidence diffraction (GIXD) measurements are ideally performed on sample prepared in flat surfaces. Since the spin cast samples provide the best statistical in-plane randomness, only these were examined over the entire range of the relative compositions of the two components.19 Some GIXD results are shown in Fig. S3† with clear diffraction peaks corresponding to a 2D-powder or fibre texture.19 The indexing of the peaks, i.e. assigning the hkl value to each peak following the literature data, indicates that the ac plane is the contact plane and the crystals grow with the molecules standing upright on the substrate (Fig. 1). This finding is further supported by specular diffraction results, which only exhibit peaks corresponding to the 020 face and its higher order reflections (Fig. S4†). This structural finding is typical for rod like molecules which often form fibre textures with the long molecular axis perpendicular to SiOx substrate surfaces, e.g. molecules such as acenes46 or alkyl substitutes thiophenes47 grow upright standing.When the two chemical species are both present in the same lattice, the unit cell changes result in a smooth changing in peak patterns (Fig. S4†). The colour coded space map in Fig. 3 summarises the shift of the peaks on the change of composition. As the amount of ISB increases the peaks shift upwards along the qz direction. In this direction the information on the unit cell extension along the b-axis is represented, and corresponds to the unit cell of PTZ shortening along this axis to host the ISB guest molecule.
 | ||
| Fig. 3 Summary of grazing incidence X-ray diffraction peak positions of various fractions of ISB:PTZ measured on spin coated samples. The extracted cell dimensions of the various samples hosting different amounts of PTZ and ISB. | ||
The unit cell parameters of the solid solutions are obtained from the peak positions of the specular X-ray diffraction scans and the GIXD (Fig. 3a and S3†). The extracted parameters show that the length of a and c axes increase as the ISB fraction increases while the b-axis length decreases. The volume increases steadily as the larger ISB molecules need to be accommodated in the unit cell. This also agrees very well with the single crystal data (see Table 1 and Fig. 1).
Raman spectroscopy
A series of Raman spectra of single crystals and films made by drop casting or spin coating of PTZ, ISB and their mixtures are given in Fig. 4 for a selected intramolecular energy range (Fig. 4a) and the low energy range (Fig. 4b). All spectra display similar features, regardless of the preparation procedure of the solid solution. In the high wavenumber range, the strong marker bands at 1034 cm−1 for PTZ and at 1040 cm−1 for ISB can be identified, corresponding to an in-plane bending ring vibration coupled a C–S–C stretch mode (ref. 48 and this work) and an in-plane bending ring vibration coupled a C–N–C stretch mode (ref. 49 and this work), respectively. In the mixed crystals, each spectrum is the overlap of the spectra of the two components, with the amplitude ratio between the PTZ and ISB marker bands proportional to the mixture molar fraction. Also, no measurable frequency shifts are detected with respect to the bands of the pure compound crystals. Such findings are expected, as in the limiting case of negligible coupling molecules can be vibrationally excited in the crystal independently of each other, but also indicates that the interaction patterns in the mixed and in pure compound systems are alike.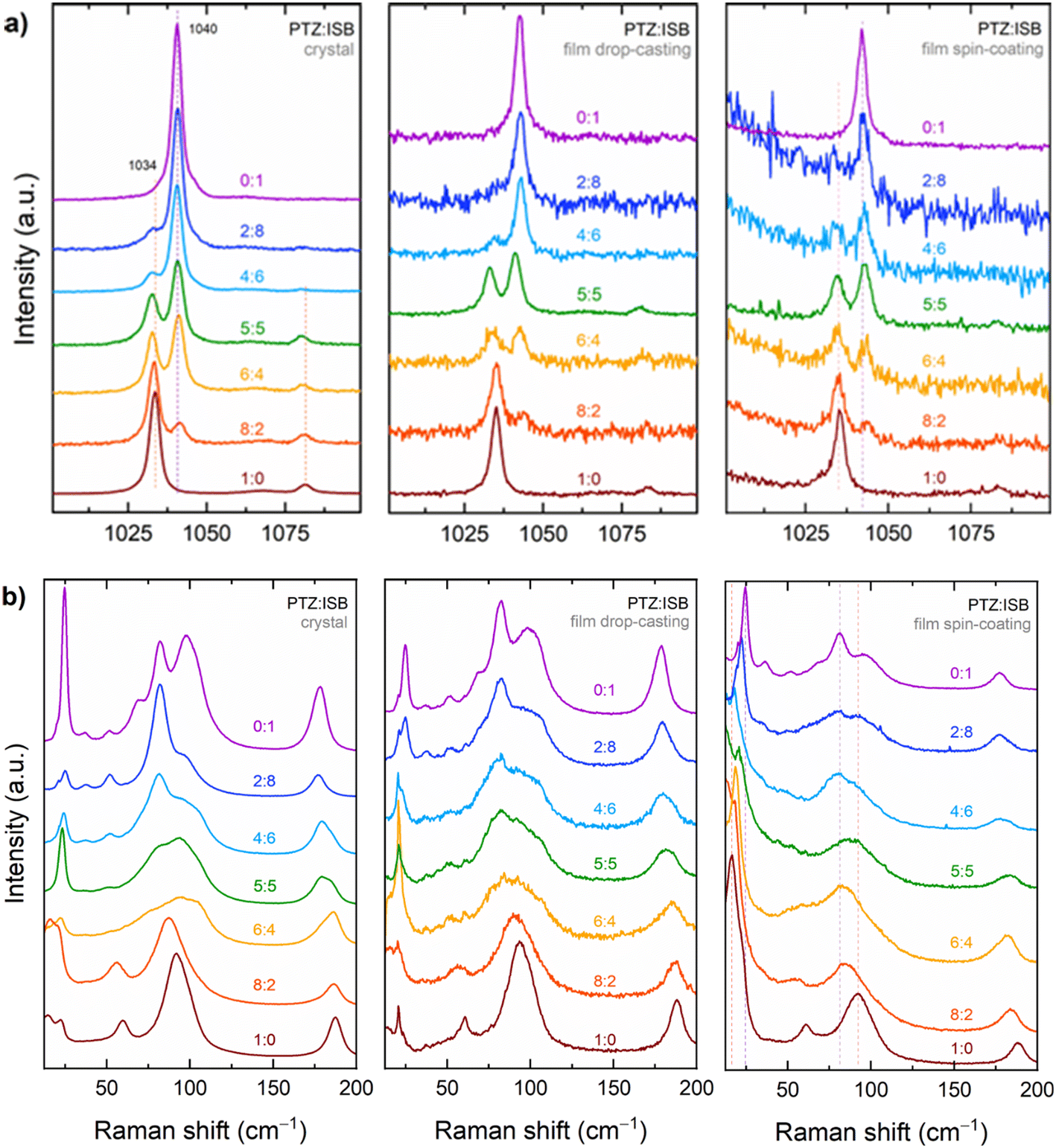 | ||
| Fig. 4 Raman spectra of single crystals, and polycrystalline drop cast and spin coated samples at different mixing ratios of phenothiazine and imminostilbene. The spectra are recorded in (a) a selected energy range of the intramolecular vibrations and (b) in the lattice phonon vibration range. | ||
Fig. 4b shows the evolution of the lattice-phonon Raman patterns in the various samples. Peaks for PTZ are detected at 17, 23, 61, 90 and 188 cm−1 and for ISB at 24, 37, 82, 97 and 178 cm−1. Both single crystal and thin films of the pure compounds display similar spectral features, albeit with different relative intensities, which are likely to arise from different orientations of the microcrystalline structure of the films. In particular, the spin coated films features most resemble those of the crystalline powder (Fig. 6, where the powder experimental spectra are reported), indicating that the crystallites are randomly oriented in-plane, i.e. along the surface. Such a behaviour agrees with the findings of the X-ray measurements, which found that while the film samples are highly textured with ac being the contact plane as in the single crystals, the crystallite in-plane orientation around the surface normal can take any value. With the only exception of the bands found in the pure PTZ and ISB at 188 cm−1 and 178 cm−1, which can be assigned to molecular ring deformations, in the mixed systems the lattice phonons frequencies change continuously from one pristine spectrum to the other as a function of the PTZ to ISB ratio, through energy shifts and band broadenings. As a result, each spectral pattern is representative of a specific structure. This is the important difference with respect to the spectra of the intramolecular vibrations of Fig. 4a, which can always be described as the superposition of the spectra of the two pure compounds. As an example, in the spectra of the films the band peaked at 17 cm−1 of pure PTZ shifts to higher frequencies as the fraction of ISB increases: the broad band moves continuously up to 24 cm−1, where it merges with the 24 cm−1 pure ISB band. A similar behaviour is detected around 75–100 cm−1, where the broad band of the pure PTZ shifts gradually shifts in the mixtures.
The general trend of the solid solution lattice phonons with composition and cell changes is not easy to describe or rationalize. For a more accurate analysis, polarized Raman measurements of single crystals can be employed, which display the same behaviour as the films, but with the advantage of having known and easily oriented crystal faces. Besides, measurements provide a significant spectral simplification, due to the lower numbers of peaks arising from spectroscopic symmetry selection rules for these crystals. Polarization properties of the spectra of the systems can be deduced from the vibrational analysis of the crystals of the two pure compounds. The X-ray measurements of this work demonstrate that for both of them the crystal ac face is the one most often in contact with the substrate in the film, and this applies also to the single crystals removed from the surface. In the orthorhombic Pnma cell with with Z = 4 molecules, out of the 12 lattice phonon modes of symmetries 3Ag + 3B1g + 3B2g + 3B3g, predicted by the rigid molecule approximation in the orthorhombic Pnma cell with with Z = 4 molecules, only the six 3Ag + 3B2g can be detected on the ac face of the pure compounds. In the SI a more detailed description of the characteristics of polarized spectra in these systems can be found, with Fig. S6† illustrating the experimental geometry and highlighting the chosen orientation of the sample with respect to the laser beam excitation and scattering polarizations.
The spectra reported in Fig. 5 display separately the evolution of the lattice-phonon modes of Ag symmetry (Fig. 5a and b) and of B2g symmetry (Fig. 5c). The discrimination based on symmetry allows for an overall simplification and a clearer view of the spectral trend, with each polarization configuration characterized by its own unique trend, throughout the entire set of relative compositions. The blue shift of the totally symmetric frequency mode of lowest frequency of PTZ (Fig. 5a and b), with its intensity gain and final merge into the 24 cm−1 band of ISB is, in this respect, exemplary. More complex to disentangle is the behaviour for the broad band comprising the B2g modes in PTZ grouped around 100 cm−1, which transform into the modes of the same symmetry in ISB, as shown in the spectrum of Fig. 5c, with positive and negative shifts and large intensity variations. Again, we need to stress the difference between the intramolecular portion of the vibrational spectrum in the solid solution, which shows no frequency shifts but just spectral overlap, and the low-frequency region, which instead probes the change of the pattern of intermolecular interactions with the presence of a guest in a host lattice. The intrinsic anisotropic nature of such interactions in the organic system, even when the species involved are quite geometrically and chemically similar as PTZ and ISB, implies that the heuristic Vegard's law does not apply to the structural parameters. Analogously, the lattice vibrations do not, and are not expected to, follow the simple rule of shifting to lower frequencies on expansion of the unit cell, as predicted by the application of the Grüneisen's rule, which describes the effect that changing the volume of a crystal has on its vibration properties as a function of anharmonicity. In fact, we have observed that both kind of positive or negative shifts accompany the transformation from PTZ to ISB.
 | ||
| Fig. 5 Polarized lattice-phonon Raman spectra of PTZ:ISB solid solutions at given molar ratios in crystal samples: labels Y(XX)Y and Y(ZZ)Y identify an experimental configuration in which modes of Ag symmetry are detected; label Y(XZ)Y identifies an experimental configuration in which modes of B2g symmetry are detected. | ||
Calculations
Raman spectra of the pure PTZ and ISB crystals
The DFT simulations of the Raman spectrum of the intramolecular modes of the powders of ISB and PTZ are shown in Fig. S7† over the energy range 200 to 1700 cm−1, along with the experimental results. PTZ and ISB have alike chemical structures but display distinct spectroscopic fingerprints which are correctly reproduced by the simulations and allow for their identifications also in the mixtures. The comparison of the literature DFT calculations performed on the isolated ISB and PTZ molecules48–50 with the results of this work for crystals shows that they agree quite well with the experimental vibrational pattern both in frequencies and intensities, demonstrating that molecular geometries and intramolecular vibrational modes are very little affected by the solid state packing and interactions.The experimental lattice-phonon spectra of the powders of pure PTZ and ISB between 10 and 200 cm−1 are reported in Fig. 6 along with the results of the DFT simulations over the same energy range. For the simulations, a vertical line marks frequency and intensity of the calculated mode, while a colour code is used to identifying its symmetry. Single bands have been drawn with a Lorentzian shape and a FWHM's of 5 cm−1 chosen to match the experimental features. As reported above, bands of PTZ are detected at 17, 23, 61, 90 and 188 cm−1 and of ISB at 24, 37, 82, 97 and 178 cm−1 and DFT calculations reproduce very well the experimental Raman spectra of both ISB and PTZ powders and help disentangle the many bands lying under the broad features. Table S1† lists the 18 Raman active modes computed over the analyzed energy range for both ISB and PTZ.
 | ||
| Fig. 6 Lattice-phonon Raman spectra of pure PTZ (brown bottom line) and ISB (violet top line) commercial powders compared to the corresponding simulated spectra. The colour code which identifies the symmetry of the simulated modes is reported in the legend. | ||
The eigenvector analysis for each crystal shows that 12 modes can be described as librations/translations of the entire molecule as a rigid body, and are therefore lattice phonons, while the remaining 6 correspond to molecular vibrations with negligible mixing with the lattice phonons. Such a distinction is important as the former ones are the most sensitive to cell and chemical environment changes, and are diagnostic of the changes of the lattice dynamics in the solid solution formation. The eigenvector analysis of the low-frequency modes of the PTZ and ISB crystals also show that such vibrations correspond to very similar motions in the two crystals. For instance, the modes calculated at 27.4 cm−1 or 23.6 cm−1; 87.0 or 76.9 cm−1; and 88.7 or 81.9 cm−1 for PTZ or ISB respectively, all correspond to the same kind of rigid-body librations/translations around the L, M, N inertia axes of the molecules. The X-ray diffraction finding that the film samples have the crystal ac face as the most often in contact with the substrate is further confirmed by comparison of the spectra in polarized light of both PTZ and ISB single crystals with the simulations for the various crystal faces as reported in Fig. S8 and S9.† Experiments and simulations on pristine materials can thus be brought to a very satisfactory agreement, laying the groundwork for the subsequent analysis of solid solutions.
Low-frequency Raman spectra of PTZ/ISB solid solutions
The computational description of the solid solutions lattice phonons is not trivial. To simulate the properties of any crystalline system, periodic boundary conditions must be adopted. For perfect crystals, DFT calculations can be performed using standard procedures. The computational cost is low for the process of energy optimization and easily sustainable for the vibrational frequencies, as only the atoms present in the unit cell require consideration. Dealing with structures without a perfect periodicity like defective structures, containing impurities or with no fixed stoichiometry of the unit cell is instead quite troublesome. This arises from the fact that in principle a number of supercells should be used51 to account for all statistic deviations of a pure crystal, and this would increase greatly the total number of atoms required in the DFT calculations. To the best of our knowledge, very few computations have been reported in the literature for the description of mixed organic crystalline systems.51–53 So, for the case studied here, it is relatively easy to compute the properties of the pristine structures but quite unfeasible to follow the Raman experiment, simulating the behaviour of the various solid solutions of different PTZ/ISB ratios using a standard approach. To overcome these limitations, and to better understand how the vibrational modes of pure component crystals are modified in a statistical mixture, we used the same computational strategy adopted to describe the solid solutions formed during the photodimerization of cinnamic acids,17 which allowed us to understand the properties of solid solutions in relation to those of the pure components. In this approach, the vibrational properties of one component are computed in the lattice of the other. Specifically, the PTZ molecule was projected into the larger ISB cell, and the vibrational modes for this system, called PTZ@ISBcell, were thus computed after relaxing the PTZ atomic positions, while maintaining fixed the ISB cell axes and angles. The structure obtained from this “experiment” is compared with the that of pristine PTZ in Fig. 7a. On assuming that lattice phonons are on a first order approximation depending on the volume changes, such an operation should return a computation of the vibrational modes in which the volume effects are enucleated. In Fig. 7b the result of the dot product between the vibrational eigenvectors of the PTZ and PTZ@ISBcell structures is reported graphically. Notwithstanding the shifts in frequencies, that will be discussed below, the PTZ normal modes can be seen largely to maintain their nature and description in the ISB cell. This is expected for modes lying at wavenumbers higher than 200–230 cm−1, with full or prevailing intramolecular character. Applied to the lattice phonons, it shows that the “host” ISB lattice can take in the PTZ molecules as guests and that the different chemical nature of the guest is less important than the changes in the unit cell dimensions. | ||
| Fig. 7 a) Unit cell of the pristine PTZ (black) superimposed to the PTZ@ISBcell (orange); (b) graphic representation of the result of the dot product between the vibrational eigenvectors of the two structures (purple = 0, yellow = 1). c) Frequency shifts for the modes computed in the 14–240 cm−1 spectral window, where the mode eigenvectors are decomposed by projection along the crystal axes of the PTZ@ISB system. | ||
To account for the frequency shifts, we assume that, as a result of the anisotropic cell volume changes which connect the host and guest lattices, the lattice vibrations will undergo shifts and modifications with magnitudes which are dependent on the characteristics of their motion, i.e. on how their eigenvectors are projected along the changing crystal axes. To evaluate such an effect, the weighted averaged frequency shifts for each i-th mode is computed as the sum of the differences between the i-th eigenvalue in the PTZ structure and all the j-th eigenvalues in the PTZ@ISBcell structure, each one multiplied by a weight given by the dot product of the corresponding eigenvectors:

Conclusions
In this study we have presented by X-ray diffraction and Raman spectroscopy how the structural isomorphism of the pure phases of the APIs PTZ and ISB results in the formation of solid solutions over the entire span of relative compositions, with no miscibility gap. In addition, regardless of the crystal growth method, only one polymorph was found under the operating conditions. This was also observed for the mixtures, suggesting a deep free energy minimum for this particular system, which may result in long-term stability. The formation of solid solutions likely allows for continuous and predictable modification of structures and properties, directly impacting their potential as active pharmaceutical ingredients.The Raman investigation was focused on the lattice phonon behaviour, and aimed to detect how the vibrational degrees of freedom characteristic of a periodic system get modified when such a periodicity is continuously albeit slightly broken by the statistical replacement of a molecular entity with another one. By Raman spectroscopy the changes of the chemical composition as well as the structural changes accompanying the formation of the solid solution lattices were monitored. Whereas the spectra of the molecular vibration can always be analysed as the superposition of the spectra of the two pure compounds, the lattice phonons evolve seamlessly throughout the solution series. The experiments performed in polarized light help providing a clearer picture of the vibrational patterns of the mixed systems. Calculating the vibrations in the PTZ@ISB system has allowed us to analyse the volume effects on the lattice-phonons, mimicking the physical condition of a component which replaces the other, whose packing order is preserved. The mapping of the PTZ modes in its cell on those in the ISB cell shows that they do not basically undergo a change of description in the host lattice, and the bulk of the spectral modifications in the solution can be accounted for by the different molecular volumes of the two species. Most importantly, the stability of the PTZ@ISB phase, for which it has been possible to compute a minimum energy structure, demonstrates that the formation of the solid solution takes place because of the full compatibility between the lattices of the two compounds. In fact, the experimental evidence of the persistence of low-frequency features in these systems and the nearly bi-univocal correspondence between the normal modes of the parent lattices of the two components found by the DFT simulations suggest that the packing of different molecules in the unit cells can be viewed as a perturbation of perfect periodicity and symmetry, which alters but does not disrupt the dynamical characteristics of the lattice.
Data availability
Data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to thank the synchrotron ELETTRA and especially Luisa Barba and Heinz Amenitsch for great support at their respective beamlines, i.e. XRD1 and SAXS. Project funded under the National Recovery and Resilience Plan (NRRP), Mission 04 Component 2 Investment 1.5 – NextGenerationEU, call for tender no. 3277 dated 30/12/2021 (award number: 0001052 dated 23/06/2022). This work has also been supported by the project “POLYPHON” funded by the MUR Progetti di Ricerca di Rilevante Interesse Nazionale (PRIN) Bando 2022 – grant 2022XZ2ZM8. T. S. thanks the Programma per Giovani Ricercatori “Rita Levi Montalcini” year 2020 (grant PGR20QN52R) of the Italian Ministry of University and Research (MUR) for the financial support.References
- Molecular Mixed Crystals, ed. M. À. Cuevas-Diarte and H. A. J. Oonk, Physical Chemistry in Action, Springer Cham, 2021, DOI:10.1007/978-3-030-68727-4.
- L. Vegard and H. Schjelderup, Die Konstitution Der Mischkristalle, Phys. Z, 1917, 18, 93–96 Search PubMed.
- E. M. Engler, B. A. Scott, S. Etemad, T. Penney and V. V. Patel, Organic Alloys: Synthesis and Properties of Solid Solutions of Tetraselenafulvalene-Tetracyano-p-Quinodimethane (TSeF-TCNQ) and Tetrathiafulvalene-Tetracyano-p-Quinodimethane (TTF-TCNQ), J. Am. Chem. Soc., 1977, 99(18), 5909–5916, DOI:10.1021/ja00460a011.
- A. I. Kitaigorodsky, Mixed Crystals, Springer Science & Business Media, 2012, vol. 33 Search PubMed.
- G. R. Desiraju, Crystal and Co-Crystal, CrystEngComm, 2003, 5(82), 466–467, 10.1039/B313552G.
- C. B. Aakeröy and D. J. Salmon, Building Co-Crystals with Molecular Sense and Supramolecular Sensibility, CrystEngComm, 2005, 7(72), 439–448, 10.1039/B505883J.
- T. Salzillo, M. Masino, G. Kociok-Köhn, D. Di Nuzzo, E. Venuti, R. G. Della Valle, D. Vanossi, C. Fontanesi, A. Girlando, A. Brillante and E. Da Como, Structure, Stoichiometry, and Charge Transfer in Cocrystals of Perylene with TCNQ-Fx, Cryst. Growth Des., 2016, 16(5), 3028–3036, DOI:10.1021/acs.cgd.5b01663.
- E. Schur, E. Nauha, M. Lusi and J. Bernstein, Kitaigorodsky Revisited: Polymorphism and Mixed Crystals of Acridine/Phenazine, Chem. – Eur. J., 2015, 21(4), 1735–1742, DOI:10.1002/chem.201404321.
- R. Tsunashima, Molecular Solid Solutions for Advanced Materials – Homeomorphic or Heteromorphic, CrystEngComm, 2022, 24(7), 1309–1318, 10.1039/D1CE01632F.
- M. Lusi, Engineering Crystal Properties through Solid Solutions, Cryst. Growth Des., 2018, 18(6), 3704–3712, DOI:10.1021/acs.cgd.7b01643.
- A. Hinderhofer and F. Schreiber, Organic–Organic Heterostructures: Concepts and Applications, ChemPhysChem, 2012, 13(3), 628–643, DOI:10.1002/cphc.201100737.
- P. Hajieva, J. B. Mocko, B. Moosmann and C. Behl, Novel Imine Antioxidants at Low Nanomolar Concentrations Protect Dopaminergic Cells from Oxidative Neurotoxicity, J. Neurochem., 2009, 110(1), 118–132, DOI:10.1111/j.1471-4159.2009.06114.x.
- C. Behl and B. Moosmann, Oxidative Nerve Cell Death in Alzheimers Disease and Stroke: Antioxidants as Neuroprotective Compounds, Biol. Chem., 2002, 383(3–4), 521–536, DOI:10.1515/BC.2002.053.
- A. D. Mosnaim, V. V. Ranade, M. E. Wolf, J. Puente and M. Antonieta Valenzuela, Phenothiazine Molecule Provides the Basic Chemical Structure for Various Classes of Pharmacotherapeutic Agents, Am. J. Ther., 2006, 13(3), 261–273, DOI:10.1097/01.mjt.0000212897.20458.63.
- H. M. Lee and A. Graudins, Iminostilbene Anticonvulsant: Carbamazepine/Oxcarbazine, in Critical Care Toxicology, ed. J. Brent, K. Burkhart, P. Dargan, B. Hatten, B. Megarbane, R. Palmer and J. White, Springer Cham, 2017 Search PubMed.
- B. Schrode, B. Bodak, H. Riegler, A. Zimmer, P. Christian and O. Werzer, Solvent Vapor Annealing of Amorphous Carbamazepine Films for Fast Polymorph Screening and Dissolution Alteration, ACS Omega, 2017, 2(9), 5582–5590, DOI:10.1021/acsomega.7b00783.
- A. Giunchi, L. Pandolfi, T. Salzillo, A. Brillante, R. G. Della Valle, S. d'Agostino and E. Venuti, Visualizing a Single-Crystal-to-Single-Crystal [2+2] Photodimerization through Its Lattice Dynamics: An Experimental and Theoretical Investigation, ChemPhysChem, 2022, 23(12), e202200168, DOI:10.1002/cphc.202200168.
- B. Schrode, S. Pachmajer, M. Dohr, C. Röthel, J. Domke, T. Fritz, R. Resel and O. Werzer, It GIDVis: A Comprehensive Software Tool for Geometry-Independent Grazing-Incidence X-Ray Diffraction Data Analysis and Pole-Figure Calculations, J. Appl. Crystallogr., 2019, 52(3), 683–689, DOI:10.1107/S1600576719004485.
- O. Werzer, S. Kowarik, F. Gasser, Z. Jiang, J. Strzalka, C. Nicklin and R. Resel, X-Ray Diffraction under Grazing Incidence Conditions, Nat. Rev. Methods Primers, 2024, 4(1), 15, DOI:10.1038/s43586-024-00293-8.
- A. Lausi, M. Polentarutti, S. Onesti, J. R. Plaisier, E. Busetto, G. Bais, L. Barba, A. Cassetta, G. Campi, D. Lamba, A. Pifferi, S. C. Mande, D. D. Sarma, S. M. Sharma and G. Paolucci, Status of the Crystallography Beamlines at Elettra, Eur. Phys. J. Plus, 2015, 130(3), 43, DOI:10.1140/epjp/i2015-15043-3.
- W. Kabsch, XDS, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(2), 125–132 CrossRef PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Overview of the CCP4 Suite and Current Developments, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67(4), 235–242, DOI:10.1107/S0907444910045749.
- P. R. Evans and G. N. Murshudov, How Good Are My Data and What Is the Resolution?, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69(7), 1204–1214 CrossRef PubMed.
- G. M. Sheldrick, SHELXT – Integrated Space-Group and Crystal-Structure Determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71(1), 3–8, DOI:10.1107/S2053273314026370.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71(1), 3–8, DOI:10.1107/S2053229614024218.
- P. Emsley and K. Cowtan, Coot: Model-Building Tools for Molecular Graphics, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60(12), 2126–2132, DOI:10.1107/S0907444904019158.
- M. A. Spackman and D. Jayatilaka, Hirshfeld Surface Analysis, CrystEngComm, 2009, 11(1), 19–32, 10.1039/B818330A.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, It CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals, J. Appl. Crystallogr., 2021, 54(3), 1006–1011, DOI:10.1107/S1600576721002910.
- M. J. Turner, J. J. Mckinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, Perth, 2017 Search PubMed.
- M. A. Spackman and J. J. McKinnon, Fingerprinting Intermolecular Interactions in Molecular Crystals, CrystEngComm, 2002, 4(66), 378–392, 10.1039/B203191B.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Novel Tools for Visualizing and Exploring Intermol\-Ecular Interactions in Molecular Crystals, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60(6), 627–668, DOI:10.1107/S0108768104020300.
- J. J. H. McDowell, The Crystal and Molecular Structure of Phenothiazine, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32(1), 5–10, DOI:10.1107/S0567740876002215.
- J. P. Reboul, B. Cristau, J. C. Soyfer and J. Estienne, Structure de La 5H-Dibenzo[b,f]Azépine (Iminostilbène), Support Tricyclique d'analogues Structuraux Des Antidépresseurs Imipraminiques, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36(11), 2683–2688, DOI:10.1107/S0567740880009685.
- G. Kresse and J. Hafner, Ab Initio Molecular Dynamics for Liquid Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47(1), 558–561, DOI:10.1103/PhysRevB.47.558.
- G. Kresse and J. Hafner, Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal–Amorphous-Semiconductor Transition in Germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49(20), 14251–14269, DOI:10.1103/PhysRevB.49.14251.
- G. Kresse and J. Furthmüller, Efficiency of Ab Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6(1), 15–50, DOI:10.1016/0927-0256(96)00008-0.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186, DOI:10.1103/PhysRevB.54.11169.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979, DOI:10.1103/PhysRevB.50.17953.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775, DOI:10.1103/PhysRevB.59.1758.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32(7), 1456–1465, DOI:10.1002/jcc.21759.
- T. Bučko, J. Hafner and J. G. Ángyán, Geometry Optimization of Periodic Systems Using Internal Coordinates, J. Chem. Phys., 2005, 122(12), 124508, DOI:10.1063/1.1864932.
- A. Togo and I. Tanaka, First Principles Phonon Calculations in Materials Science, Scr. Mater., 2015, 108, 1–5, DOI:10.1016/j.scriptamat.2015.07.021.
- D. Michalska and R. Wysokiński, The Prediction of Raman Spectra of Platinum(II) Anticancer Drugs by Density Functional Theory, Chem. Phys. Lett., 2005, 403(1–3), 211–217, DOI:10.1016/j.cplett.2004.12.096.
- C. F. Matta, J. Hernández-Trujillo, T.-H. Tang and R. F. W. Bader, Hydrogen-Hydrogen Bonding: A Stabilizing Interaction in Molecules and Crystals, Chemistry, 2003, 9(9), 1940–1951, DOI:10.1002/chem.200204626.
- J. J. McKinnon, F. P. A. Fabbiani and M. A. Spackman, Comparison of Polymorphic Molecular Crystal Structures through Hirshfeld Surface Analysis, Cryst. Growth Des., 2007, 7(4), 755–769, DOI:10.1021/cg060773k.
- R. Ruiz, B. Nickel, N. Koch, L. C. Feldman, R. F. Haglund, A. Kahn and G. Scoles, pentacene Ultrathin Film Formation on Reduced and Oxidized Si Surfaces, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67(12), 125406, DOI:10.1103/PhysRevB.67.125406.
- M. Defaux, F. Gholamrezaie, J. Wang, A. Kreyes, U. Ziener, D. V. Anokhin, D. A. Ivanov, A. Moser, A. Neuhold, I. Salzmann, R. Resel, D. M. de Leeuw, S. C. J. Meskers, M. Moeller and A. Mourran, Solution-Processable Septithiophene Monolayer Transistor, Adv. Mater., 2012, 24(7), 973–978, DOI:10.1002/adma.201103522.
- M. Alcolea Palafox, M. Gil, J. L. Núñez and G. Tardajos, Study of Phenothiazine and N-Methyl Phenothiazine by Infrared, Raman, 1H-, and 13C-NMR Spectroscopies, Int. J. Quantum Chem., 2002, 89(3), 147–171, DOI:10.1002/qua.10314.
- G. M. Kuramshina, T. Mogi and H. Takahashi, Structures and Vibrational Spectra of 5H-Dibenz [b,f]Azepine and 5H-Dibenzo[a,d]Cycloheptene-5-Ol on the Basis of Quantum Mechanical Calculations, J. Mol. Struct., 2003, 661–662, 121–139, DOI:10.1016/j.molstruc.2003.07.016.
- M. Bolboaca, T. Iliescu, Cs Paizs, F. D. Irimie and W. Kiefer, Raman, Infrared, and Surface-Enhanced Raman Spectroscopy in Combination with Ab Initio and Density Functional Theory Calculations on 10-Isopropyl-10H-Phenothiazine-5-Oxide, J. Phys. Chem. A, 2003, 107(25), 5144–5144, DOI:10.1021/jp030615t.
- M. Habgood, R. Grau-Crespo and S. L. Price, Substitutional and Orientational Disorder in Organic Crystals: A Symmetry-Adapted Ensemble Model, Phys. Chem. Chem. Phys., 2011, 13(20), 9590–9600 RSC.
- N. M. Villeneuve, J. Dickman, T. Maris, G. M. Day and J. D. Wuest, Seeking Rules Governing Mixed Molecular Crystallization, Cryst. Growth Des., 2023, 23(1), 273–288, DOI:10.1021/acs.cgd.2c00992.
- A. Hill, W. Kras, F. Theodosiou, M. Wanat, D. Lee and A. J. Cruz-Cabeza, Polymorphic Solid Solutions in Molecular Crystals: Tips, Tricks, and Switches, J. Am. Chem. Soc., 2023, 145(37), 20562–20577, DOI:10.1021/jacs.3c07105.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1562931, 1562932, 1562929, 1562930, 1562933, and 1562934 contain the supplementary crystallographic data for pristine PTZ at 298 K and 100 K, ISB at 298 K and 100 K and the PTZ/ISB mixture at 298 K and 100 K, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00605d |
| ‡ Present address: CINECA, via Magnanelli 6/3, 40033 Casalecchio di Reno (Bologna), Italy. |
| § Present address: CNR-INO, via Branze 45, 25123 Brescia, Italy. |
| This journal is © The Royal Society of Chemistry 2024 |