
Photon upconversion of Nd3+–Yb3+–Tb3+ doped core–shell–shell–shell nanoparticles†
Wenfei
Xun
a,
Zhipeng
Meng
*b,
Chuwei
Xie
c and
Suli
Wu
 *a
*a
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, 2# Linggong Road, Dalian 116024, P.R. China. E-mail: wusuli@dlut.edu.cn
bSchool of Chemistry and Chemical Engineering, Shandong University of Technology, Zibo 255049, P.R. China. E-mail: mengzhipeng@sdut.edu.cn
cSchool of Chemistry, Dalian University of Technology, 2# Linggong Road, Dalian 116024, P.R. China
First published on 30th May 2024
Abstract
808 nm excited Nd3+ doped upconversion nanoparticles (UCNPs) have become a new research hotspot due to their reduced thermal effect when applied in a water-rich system. Different from Tm3+-mediated energy migration upconversion of Tb3+, in this work, NaYF4:Yb/Tb@NaYF4:Yb@NaNdF4:Yb@NaYF4 core–shell–shell–shell nanoparticles were designed based on the Yb3+ sensitized cooperative energy-transfer process to realize photon upconversion. NaYF4:Yb served as the migration layer, separating the activator Tb3+ from the sensitizer Nd3+ and minimizing the deleterious cross-relaxation between them. At 40% Yb3+ doping concentration in the NaYF4:Yb layer, bright green luminescence was achieved under 808 nm excitation. Meanwhile, the presence of Yb3+ in the sensitization layer (NaNdF4:Yb) reduced Nd3+ cross-relaxation, leading to improved energy migration efficiency of Nd3+ → Yb3+. Finally, the incorporation of an inert NaYF4 shell resulted in a significant luminescence boost. These nanoparticles can be dispersed in water after surface modification and could be effectively excited after laser penetration of 3 cm of water, indicating the enormous potential of the designed UCNPs in the biological system.
Introduction
Lanthanide-doped UCNPs can absorb two or more photons and emit a single photon with a shorter wavelength.1 Anti-Stokes-shifted luminescent materials have been widely applied in displays, anti-counterfeiting, biological labelling, and other fields due to their sharp emission bands, high light stability, and low biological toxicity.2–7 Compared to typically 980 nm excited NaYF4:Yb/Er or NaYF4:Yb/Tm UCNPs, in which Er3+ or Tm3+ ions serve as activators and Yb3+ ions are used as sensitizers to absorb 980 nm NIR light,6,8–14 Nd3+ doped UCNPs can be excited by an 808 nm laser and then transfer energy to the activator (for example, Er3+) through Yb3+ as an energy transfer bridge (Nd3+ → Yb3+ → Er3+),15,16 greatly reducing the thermal effect caused by the strong absorption of water under 980 nm excitation. However, the inherently weak energy transfer efficiency limits their further applications. Researchers have tried to utilize different strategies such as changing the host lattice, energy transfer, surface passivation, surface plasmon coupling, and photonic crystals to modulate rare-earth luminescence.17–27 For example, Yan et al. built a NaGdF4:Yb/Er@NaGdF4:Nd/Yb core–shell structure, in which Er3+ and Nd3+ were doped separately in the core and shell, and the energy back transfer process can be inhibited through spatial separation.28 Furthermore, Yao et al. synthesized sandwich-structured nanoparticles (NPs) including a NaYF4:Yb quenching-shield layer between the activator core and Nd3+ doped shell.29 The quenching-shield layer eliminated the energy transfer between Nd3+ and activators at the core–shell interface. On the other hand, Yb3+ doped in the quenching-shield layer transferred the excitation energy from the outside shell to the core, reducing deleterious energy back transfer. These methods allow for the doping of high concentrations of Nd3+ in the shell layer to achieve higher absorption efficiency.29,30Although Tb3+ has typical green emission, compared to Yb–Er and Yb–Tm, Yb–Tb ion pairs are less used for achieving efficient upconversion luminescence (UCL) in rare earth fluorides due to the short lifetime of the intermediate level of Tb3+.31 There are two main strategies to realize UCL of Tb3+. One is Tm/Gd energy migration-mediated upconversion (EMU). For example, Liu et al. reported NaGdF4:Yb/Tm@NaGdF4:Tb core–shell nanoparticles. Utilizing high-energy excited states of Tm, the excitation energy is transferred through Yb3+ → Tm3+ → Gd3+ and finally transferred to the Tb3+ in the shell layer through Gd sublattice-mediated energy migration.32 A NaGdF4:Tb@NaGdF4:Yb/Tm@NaGdF4:Yb/Nd core–shell–shell structure was also prepared to realize UCL under 808 nm via spatial separation of Nd3+, Tm3+, and Tb3+ to suppress nonradiative decays.33 But the characteristic emission of Tm3+ inevitably appeared in the UCL spectrum of Tb3+. The other strategy is the Yb3+ sensitized cooperative energy-transfer (CET) process. Yan and Sun et al. prepared NaGdF4:Yb/A@NaGdF4:Nd/Yb (A = Tb, Eu), with Yb3+ having a higher virtual excited state, which can match the energy diagram of Tb3+ and Eu3+ to achieve green or red emission,34 showing the unique advantages in realizing pure Tb3+ luminescence.
In this work, a NaYF4:Yb/Tb (20%/2%)@NaYF4:Yb(40%)@NaNdF4:Yb(10%)@NaYF4 core–shell–shell–shell (CSSS) structure was designed to investigate the UCL of Tb3+ under 808 nm NIR excitation. As illustrated in Scheme 1, Tb3+ was doped in the inner core (Tb-core: NaYF4:Yb/Tb), and sensitizer Nd3+ was doped in the sensitization layer (Nd-layer: NaNdF4:Yb). Sensitizer Nd3+ and activator Tb3+ were separated by an energy migration layer (Yb-layer: NaYF4:Yb). On the one hand, the excited energy absorbed by Nd3+ could sensitize the co-doped Yb3+ in the Nd-layer by energy transfer (ET) and activated Tb3+ through energy migration (EM) of the Yb-layer, thereby achieving UCL of Tb3+. On the other hand, the negative impact of Tb3+ → Nd3+ energy back transfer occurring at the core–shell interface can be ignored compared to the core–shell structure without a migration layer, improving the brightness of UCL. Meanwhile, the doping concentration of Yb3+ ions in the Yb-layer was also investigated to optimize luminous intensity. Finally, an inert NaYF4-shell (Y-layer) was encapsulated on the structure to further enhance the luminescence by reducing the surface quenching effect. These particles can be effectively excited by an 808 nm laser and display bright green luminescence (5D4 → 7F5), demonstrating great potential for applications in the fields of displays and life sciences.
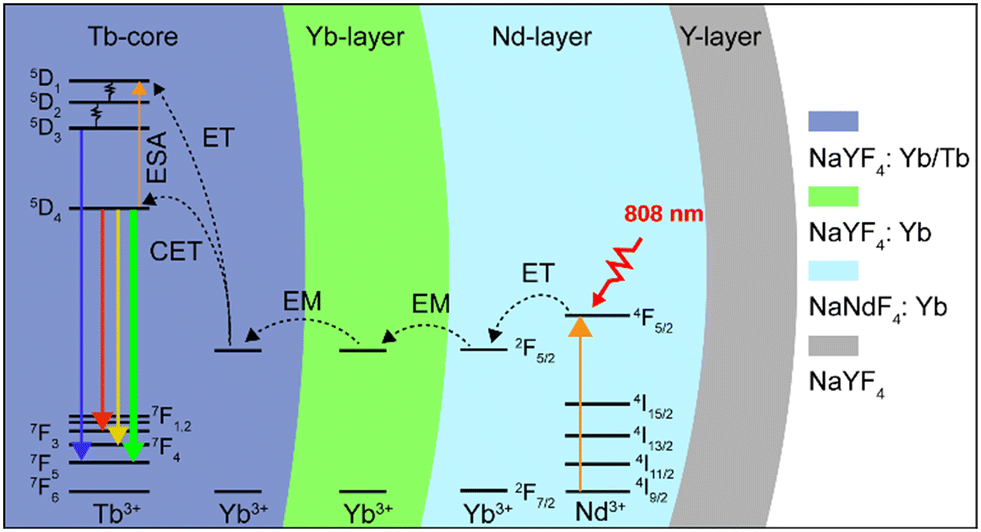 | ||
| Scheme 1 Energy transfer mechanisms in the CSSS nanoparticle under 808 nm laser excitation. | ||
Experimental
Materials
Yttrium(III) acetate hydrate (99.9%), ytterbium(III) acetate hydrate (99.9%), neodymium(III) acetate hydrate (99.9%), terbium(III) acetate hydrate (99.9%), oleic acid (AR) and 1-octadecene (90.0%) were purchased from Aladdin. Sodium hydroxide (NaOH; >96%) and polyethylene glycol (n = 6000) were purchased from TianDa Chemical Reagent Co., Ltd. and ammonium fluoride (NH4F; >96%) was purchased from Damao Chemical Reagent Factory. Cyclohexane was purchased from Tianjin Jindong Tianzheng Fine Chemical Reagent Factory. 2-Hydroxyethyl methacrylate (HEMA), acrylamide (AAm), ethylene glycol dimethacrylate (EGDMA), and 2′-hydroxy-1′-acetonaphthone (Darocur 1173) were purchased from Shanghai Macklin Biochemical Co., Ltd.Synthesis procedures
Characterization
High-resolution transmission electron microscopy and element mapping observations were performed on a JEM-F200 (200 kV). The upconversion luminescence spectra were recorded on an FLS-920 fluorescence spectrum instrument with an external 808 nm or 980 nm semiconductor laser (MDL-III-808 nm, MDL-III-980 nm, Changchun New Industries Optoelectronics Technology Co., Ltd., China). X-ray diffraction (XRD) spectra were recorded on a SmartLab 9 kW solid powder diffractometer.Results and discussion
To prepare the designed UCNPs, a layer-by-layer synthesis process was performed. NaYF4:Yb/Tb core NPs were first synthesized via the reported coprecipitation method.35 The TEM images in Fig. 1a and S1a† show that monodisperse core NPs with ∼20 nm size were obtained. Then, the Yb-layer (NaYF4:Yb) with about 5 nm thickness was coated on the core to form core–shell (CS) NPs (Fig. 1b and S1b†). Both the core and the CS NPs exhibit a uniform hexagonal prism morphology. Then the Nd-layer was grown, which only can selectively grow along the long axis of NaYF4 crystals due to the lattice mismatch between the NaYF4 and NaNdF4, leading to oblong core–shell–shell (CSS) nanostructures (Fig. 1c).36,37 Subsequently, the CSS NPs were coated with an inert NaYF4 layer to form the designed core–shell–shell–shell (CSSS) NPs (Fig. 1d) with a diameter of 70 nm (Fig. S1d†). The high-resolution TEM (HRTEM) image with a typical d spacing of about 0.52 nm (Fig. 1e) was consistent with the {10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0} crystal plane spacing of the hexagonal phase reported.38 Meanwhile, the fast Fourier transform (FFT) of the lattice (Fig. 1f) and the XRD pattern in Fig. S2† further revealed its hexagonal phase characteristics. The corresponding element mapping in Fig. 1g illustrates the distributions of different doped ions, where the higher Nd in the middle region is consistent with the design compositions for the CSSS particles.
0} crystal plane spacing of the hexagonal phase reported.38 Meanwhile, the fast Fourier transform (FFT) of the lattice (Fig. 1f) and the XRD pattern in Fig. S2† further revealed its hexagonal phase characteristics. The corresponding element mapping in Fig. 1g illustrates the distributions of different doped ions, where the higher Nd in the middle region is consistent with the design compositions for the CSSS particles.
 | ||
| Fig. 1 Scheme and TEM images of the as-prepared nanoparticles: (a) core NaYF4:Yb/Tb NPs, (b) NaYF4:Yb/Tb@NaYF4:Yb NPs, (c) NaYF4:Yb/Tb@NaYF4:Yb@NaNdF4:Yb NPs and (d) NaYF4:Yb/Tb@NaYF4:Yb@NaNdF4:Yb@NaYF4 NPs. (e and f) HRTEM images of CSSS NPs and their fast Fourier transform (FFT) pattern. (g) Element mappings of Y, Na, F, Yb, Tb, and Nd for CSSS NPs shown in (d). | ||
To verify the rationality of the structural design, we first investigated the importance of the spatial separation of the dopant ions Nd3+ and Tb3+. As shown in Fig. 2a, compared to NaYF4:Tb/Nd/Yb, the NaYF4:Yb/Tb@NaNdF4:Yb CS structure (Yb/Tb@Nd/Yb) produced visible green luminescence after excitation, demonstrating that the cross-relaxation process greatly quenched the luminescence when Nd3+ and Tb3+ were co-doped in the same layers. Furthermore, the CSS NPs (Yb/Tb@Yb@Nd/Yb) obtained an improved luminescence intensity by introducing an energy migration layer to prevent the deleterious cross-relaxation and energy back transfer of Tb3+ → Nd3+, allowing the energy of the excited Nd3+ ions to be transferred to the core layer to excite Tb3+ ions. Finally, an inert NaYF4 shell was coated on the CSS NPs, referred to as Yb/Tb@Yb@Nd/Yb@Y, to eliminate the surface quenching and greatly improve the brightness of the luminescence. The finally obtained UCNPs displayed typical 415 (5D3 → 7F5), 543 (5D4 → 7F5) and 584 (5D4 → 7F4) nm emission bands of Tb3+ under 808 nm excitation. The emissions at 525 and 658 nm should be assigned to impurity Er3+ ions mixed in the ytterbium acetate reagent. The emission at 477 nm was consistent with the previously reported cooperative luminescence from a pair of Yb3+ ions in the excited state.39 Meanwhile, the UCNPs can also be excited by a 980 nm laser. The photograph and luminescence spectrum in Fig. S3† show that similar luminescence was achieved under 808 and 980 nm laser excitation, respectively, illustrating the excellent energy transfer efficiency of the structure.
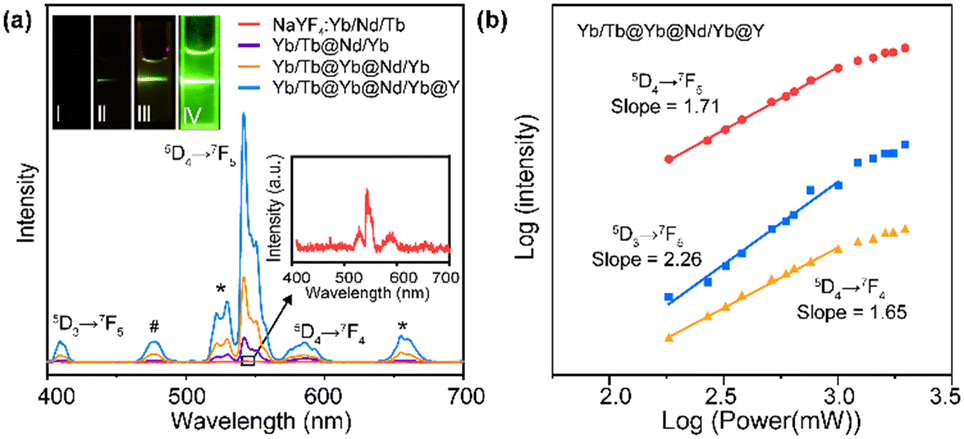 | ||
| Fig. 2 (a) UCL spectra of NaYF4:Yb/Nd/Tb, Yb/Tb@Nd/Yb, Yb/Tb@Yb@Nd/Yb and Yb/Tb@Yb@Nd/Yb@Y NPs. Symbol “*” represents the emission peaks assigned to Er3+ and symbol “#” represents the emission peak assigned to Yb3+. Insert: Digital photographs of NaYF4:Yb/Nd/Tb, Yb/Tb@Nd/Yb, Yb/Tb@Yb@Nd/Yb and Yb/Tb@Yb@Nd/Yb@Y NPs under 808 nm excitation. (b) Logarithmic fit of luminescence intensity–power of CSSS NPs excited by 808 nm NIR. | ||
To investigate the upconversion mechanism, the number of photons involved in the transition process was obtained using the logarithmic fit of luminescence intensity–power (I–P). As shown in Fig. 2b, the 5D4 → 7F5 and 5D4 → 7F4 transitions of Tb3+ followed two-photon processes, while the 5D3 → 7F5 transition followed three-photon processes, which is well correlated to the proposed energy transfer (Scheme 1) via the Yb3+-based CET process.40–42
According to the luminescence mechanism, a series of NPs doped with different concentrations of Yb3+ in the Yb-layer (from 10 to 100 mol%) were synthesized and their UCL spectra were recorded as shown in Fig. 3a. With the increase of doping concentration, the intensity of UCL was greatly enhanced first, and the strongest green luminescence was generated at the 40% Yb3+-doped structure. The result reveals that a high concentration of Yb3+ ions is required to activate the energy migration process.43 However, when the Yb3+ doping concentration was further increased, the UCL intensity decreased (Fig. 3a). The corresponding digital photos in Fig. 3b also demonstrated that the optimal luminous brightness occurs at 40% Yb3+ doping concentration in the Yb-layer. To detect the relaxation process during energy transfer, we synthesized NaYF4:m%Yb@NaNdF4:Yb@NaYF4 NPs and detected the emission lifetime at 980 nm (Yb3+ 2F5/2 level) under 808 nm excitation. When 40% Yb3+ was doped in the NaYF4:m%Yb core, the lifetime of Yb3+ was longer than that when 100% Yb3+ was doped (Fig. S5†), indicating the energy back transfer from Yb3+ to Nd3+ when 100% Yb3+ was doped. In addition, in CSSS UCNPs, the lifetime at 543 nm (Tb3+ 5D4 level) increased with increasing Yb3+ doping concentration in the Yb-layer (Fig. S6†), which was consistent with the energy cycle (EC) process between Yb3+ ions,44 indicating that the EC process was a pathway leading to energy loss.
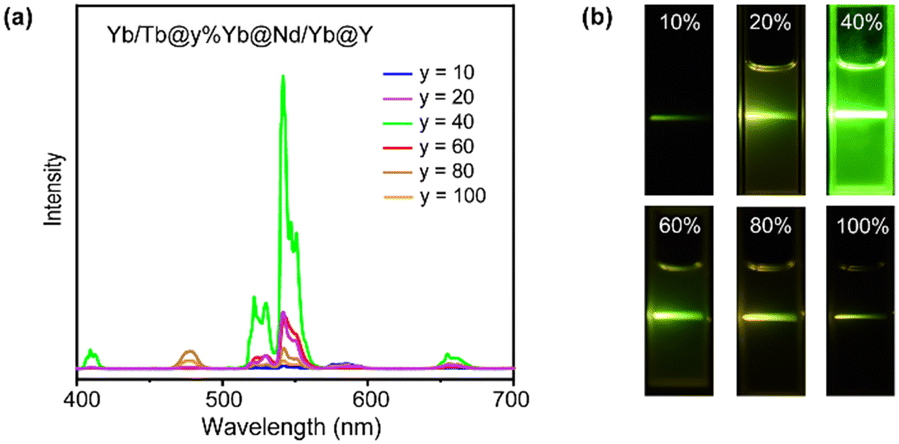 | ||
| Fig. 3 (a) Upconversion luminescence spectra of Tb/Yb@y%Yb@Nd/Yb@Y (y = 10–100) CSSS NPs. (b) Digital photos of Tb/Yb@y%Yb@Nd/Yb@Y (y = 10–100) CSSS NPs. | ||
To further understand the role of Yb sublattice energy migration, NaYF4:Yb/Tb@NaYF4:Yb@NaNdF4 (Yb/Tb@Yb@Nd) without Yb3+ in the Nd-layer was synthesized. The luminescence of Yb/Tb@Yb@Nd cannot be detected under 808 nm (Fig. 4a). In this case, direct Nd3+ → Yb3+ energy transfer only occurred at the core–shell interface (interfacial energy transfer, IET).45 Meanwhile, the multiple Nd3+ → Nd3+ EM processes with cross-relaxation in the sensitization layer caused non-radiative energy loss,46 thereby reducing the luminescence intensity greatly (Fig. 4a). For the Yb/Tb@Yb@Nd/Yb NPs, the Yb3+ ions doped in the sensitization shell can effectively collect the excitation energy of the nearby Nd3+ ions through the ET process of Nd3+ → Yb3+ and transfer to the Tb-core through the Yb sublattice EM procedure (Fig. 4b),47 and the energy loss was greatly reduced due to the simple excited state (2F5/2) of Yb3+ ions in this process.48 Hence, co-doped Yb3+ in the energy migration layer is essential to improve UCL.
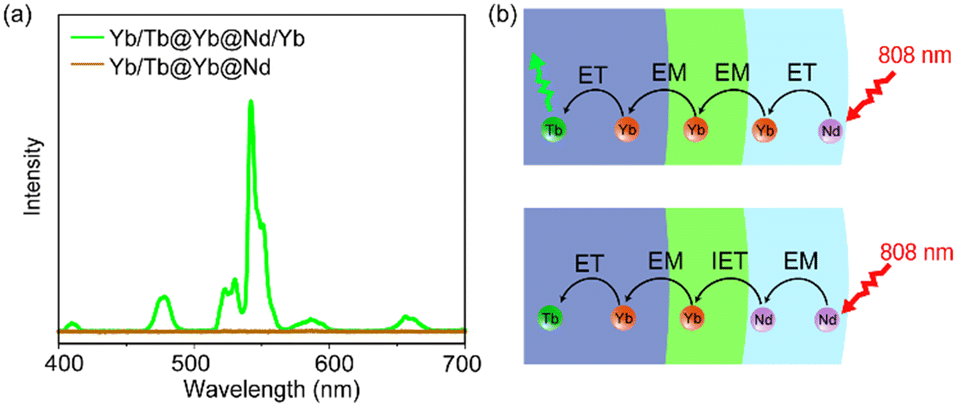 | ||
| Fig. 4 (a) Upconversion luminescence spectra of Tb/Yb@Yb@Nd/Yb and Tb/Yb@Yb@Nd CSS NPs. (b) Scheme of the CSS structure and energy transfer processes. | ||
We investigate the possibility of CSSS UCNPs for use in water-rich environments by inserting a cuvette containing deionized water between the UCNPs and the laser (Fig. 5a). UCNPs can still be activated when an 808 nm laser passed through 4 cm of water (Fig. 5b). Furthermore, surface modification of CSSS UCNPs was carried out using polyethylene glycol, and the surface modified UCNPs could be dispersed in deionized water. The digital photos in Fig. 5c showed UCL of surface modified UCNPs dispersed in deionized water after 980 nm and 808 nm laser penetration of 1–3 cm water. The surface modified UCNPs could still be excited after the 808 nm laser passed through 3 cm water and could not be excited after the 980 nm laser penetrated 1 cm of water. This finding highlights the enormous potential of Nd3+-doped materials in the application field of biology. The thermal effect of the laser in the water-rich environment was measured using P(AAm-HEMA) hydrogel as a biological model. The temperature of the hydrogel heated by the 808 nm laser was raised from 14.9 °C to 15.6 °C, while the hydrogel under the 980 nm laser was heated to 38.6 °C after 360 s exposure (Fig. S6a and b†).
 | ||
| Fig. 5 (a) Schematic diagram of excitation of surface modified UCNPs by an 808 nm laser penetrating water. (b) The digital photos of UCL of CSSS NPs excited by the 808 nm laser after passing through 1–4 cm water. (c) The digital photos of UCL of surface modified CSSS NPs dispersed in deionized water excited by 980 nm and 808 nm lasers after passing through 1–3 cm water. | ||
Conclusions
Based on the Yb3+ sensitized cooperative energy-transfer upconversion mechanism, we designed and synthesized NaYF4:Yb/Tb@NaYF4:Yb@NaNdF4:Yb@NaYF4 CSSS NPs that exhibit distinctive upconversion luminescence of Tb3+ when excited by an 808 nm laser. After comparing various Nd3+ and Yb3+ doped structures, we confirmed that effective UCL was achieved by introducing a migration layer between the Tb-core and the sensitizer Nd-layer. The migration layer can prevent deleterious cross-relaxation between Nd3+ and Tb3+, allowing the excited Nd3+ energy to be transferred to excite Tb3+ ions (Nd3+ → Yb3+ → Tb3+). Additionally, the ideal Yb3+ doping concentration in the Yb-layer was studied and the results indicated that the sample with 40% Yb3+ doping showed bright green fluorescence. Meanwhile, we found that co-doping the sensitization layer with Nd3+ and Yb3+ reduces cross-relaxation between Nd3+ ions and improves the energy transfer efficiency of Nd3+ → Yb3+. After surface modification, CSSS NPs could be dispersed in water and could be effectively excited after 808 nm laser penetration of 3 cm of water, demonstrating a promising future for 808 nm-stimulated UCNPs in water-rich settings, particularly in living tissues and cells.Author contributions
Wenfei Xun: conceptualization, investigation, methodology, writing – original draft. Zhipeng Meng: writing – original draft, validation. Chuwei Xie: characterization. Suli Wu: conceptualization, investigation, methodology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22178047, 22308200 and 21878042) and the Liaoning Province Science and Technology Plan Projects (2022JH2/101300233). The authors acknowledge the assistance of the DUT Instrumental Analysis Center.References
- O. Dukhno, S. Ghosh, V. Greiner, S. Bou, J. Godet, V. Muhr, M. Buchner, T. Hirsch, Y. Mély and F. Przybilla, ACS Appl. Mater. Interfaces, 2024, 16, 11217–11227 CrossRef CAS PubMed.
- J. Xu, P. Yang, M. Sun, H. Bi, B. Liu, D. Yang, S. Gai, F. He and J. Lin, ACS Nano, 2017, 11, 4133–4144 CrossRef CAS PubMed.
- Y. Li, S. Zeng and J. Hao, ACS Nano, 2019, 13, 248–259 CrossRef CAS PubMed.
- M. Wang, J. Song, F. Zhou, A. R. Hoover, C. Murray, B. Zhou, L. Wang, J. Qu and W. R. Chen, Adv. Sci., 2019, 6, 1802157 CrossRef PubMed.
- S. Hao, Y. Shang, Y. Hou, T. Chen, W. Lv, P. Hu and C. Yang, Sol. Energy, 2021, 224, 563–568 CrossRef CAS.
- Y. L. Xiong, J. N. Guo, L. Yang, S. R. Yao, P. Xiao and Y. H. Zhang, ACS Sustainable Chem. Eng., 2021, 9, 11171–11178 CrossRef CAS.
- S. Natalia, R. Marcin, W. Przemysław, L. Stefan and P. Du, NANO, 2022, 12, 799 Search PubMed.
- M. Gunaseelan, S. Yamini, G. A. Kumar and J. Senthilselvan, Opt. Mater., 2018, 75, 174–186 CrossRef CAS.
- F. Chen, Q. J. Lu, Y. Y. Zhang and S. Z. Yao, Sens. Actuators, B, 2019, 297, 126751 CrossRef CAS.
- J. J. Zhang, S. L. Zhao, Z. Xu, L. G. Zhang, P. F. Zuo and Q. X. Wu, RSC Adv., 2019, 9, 3688–3692 RSC.
- S. Z. Lv, K. Y. Zhang, L. Zhu and D. P. Tang, Anal. Chem., 2020, 92, 1470–1476 CrossRef CAS PubMed.
- G. Murali, S. Vattikuti, Y. K. Kshetri, H. Lee, J. Modigunta, C. S. Reddy, S. Park, S. Lee, B. Poornaprakash, H. Lee, Y. H. Park, J. Lee, S. Y. Park and I. In, Chem. Eng. J., 2021, 421, 129687 CrossRef CAS.
- E. M. Trifanova, M. E. Nikolaeva and V. K. Popov, Inorg. Mater. Appl. Res., 2022, 13, 426–433 CrossRef.
- M. Vukovic, I. Dinic, P. Jardim, S. Markovic, L. Veselinovic, M. Nikolic and L. Mancic, Adv. Powder Technol., 2022, 33, 103403 CrossRef CAS.
- D. Wang, B. Xue, X. G. Kong, L. P. Tu, X. M. Liu, Y. L. Zhang, Y. L. Chang, Y. S. Luo, H. Y. Zhao and H. Zhang, Nanoscale, 2015, 7, 190–197 RSC.
- D. Q. Chen, M. Xu, P. Huang, M. F. Ma, M. Y. Ding and L. Lei, J. Mater. Chem. C, 2017, 5, 5434–5443 RSC.
- B. Liu, Z. Meng, S. Wu, Y. Wu and S. Zhang, Nanoscale Horiz., 2018, 3, 616–623 RSC.
- Z. P. Meng, S. L. Wu and S. F. Zhang, J. Mater. Chem. C, 2018, 6, 13101–13107 RSC.
- X. S. Cui, Y. Cheng, H. Lin, Q. P. Wu, J. Xu and Y. S. Wang, J. Rare Earths, 2019, 37, 573–579 CrossRef CAS.
- S. Tek, B. A. Vincent, L. C. Mimun, A. Ponce and K. L. Nash, Cryst. Growth Des., 2020, 20, 2153–2163 CrossRef CAS.
- J. M. Bi, H. Y. Sun, X. X. Ke, L. Xu, R. D. Liu, L. L. Zhu and R. Qiao, J. Ind. Eng. Chem., 2023, 121, 452–461 CrossRef CAS.
- F. Liu, Y. Q. Wang, S. C. Zhang, F. Sun, D. Xu, W. L. Wang, X. Y. Li, W. S. Yu, H. Yu and X. T. Dong, Ceram. Int., 2023, 49, 26589–26603 CrossRef CAS.
- Y. Ma, T. Wen, K. Liu, D. Jiang, M. Zhao, C. Lin and Y. Wang, J. Mater. Chem. C, 2023, 11, 6588–6596 RSC.
- Z. Meng, W. Xun, S. Zhang and S. Wu, ACS Appl. Opt. Mater., 2023, 1, 421–429 CrossRef CAS.
- A. Skripka, M. Lee, X. Qi, J. A. Pan, H. R. Yang, C. Lee, P. J. Schuck, B. E. Cohen, D. Jaque and E. M. Chan, Nano Lett., 2023, 23, 7100–7106 CrossRef CAS PubMed.
- H. Wang, Z. He, K. Cai, L. M. Wei, Y. Xu, Y. Fu, M. M. Xing and Y. Tian, Chem. Eng. J., 2023, 468, 143558 CrossRef CAS.
- X. M. Yin, Q. Xiao, L. Lv, X. Y. Wu, X. Y. Dong, Y. Fan, N. Zhou and X. X. Luo, Spectrochim. Acta, Part A, 2023, 291, 122324 CrossRef CAS PubMed.
- Y. Wang, G. Liu, L. Sun, J. Xiao, J. Zhou and C. Yan, ACS Nano, 2013, 7, 7200–7206 CrossRef CAS PubMed.
- Y. Zhong, G. Tian, Z. Gu, Y. Yang, L. Gu, Y. Zhao, Y. Ma and J. Yao, Adv. Mater., 2014, 26, 2831–2837 CrossRef CAS PubMed.
- B. Liu, Y. Chen, C. Li, F. He, Z. Hou, S. Huang, H. Zhu, X. Chen and J. Lin, Adv. Funct. Mater., 2015, 25, 4717–4729 CrossRef CAS.
- B. Zhou, J. Huang, L. Yan, X. Liu, N. Song, L. Tao and Q. Zhang, Adv. Mater., 2019, 31, 1806308 CrossRef PubMed.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Nat. Mater., 2011, 10, 968–973 CrossRef CAS PubMed.
- T. Wang, H. F. Zhou, Z. C. Yu, G. J. Zhou, J. Zhou, D. P. Huang, L. L. Sun, P. Gao, Y. Z. Sun and J. F. Hu, J. Phys. Chem. C, 2018, 122, 10113–10124 CrossRef CAS.
- H. Dong, L. Sun, Y. Wang, J. Xiao, D. Tu, X. Chen and C. Yan, J. Mater. Chem. C, 2016, 4, 4186–4192 RSC.
- F. Wang, R. Deng and X. Liu, Nat. Protoc., 2014, 9, 1634–1644 CrossRef CAS PubMed.
- H. Peng, B. Ding, Y. Ma, S. Sun, W. Tao, Y. Guo, H. Guo, X. Yang and H. Qian, Appl. Surf. Sci., 2015, 357, 2408–2414 CrossRef CAS.
- Z. Lei, X. Ling, Q. Mei, S. Fu, J. Zhang and Y. Zhang, Adv. Mater., 2020, 32, 1906225 CrossRef CAS PubMed.
- H. Mai, Y. Zhang, L. Sun and C. Yan, J. Phys. Chem. C, 2007, 111, 13730–13739 CrossRef CAS.
- B. Lai, J. Wang and Q. Su, Appl. Phys. B: Lasers Opt., 2010, 98, 41–47 CrossRef CAS.
- J. Qiu, M. Shojiya, R. Kanno and Y. Kawamoto, Opt. Mater., 1999, 13, 319–325 CrossRef CAS.
- J. L. Adam, N. Duhamel-Henry and J. Y. Allain, J. Non-Cryst. Solids, 1997, 213–214, 245–250 CrossRef.
- R. C. Knighton, L. K. Soro, L. Francés-Soriano, A. Rodríguez-Rodríguez, G. Pilet, M. Lenertz, C. Platas-Iglesias, N. Hildebrandt and L. J. Charbonnière, Angew. Chem., Int. Ed., 2022, 61, e202113114 CrossRef CAS PubMed.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Nat. Mater., 2011, 10, 968–973 CrossRef CAS PubMed.
- R. Sun, J. Li, J. Chen, Y. Xie and L. Sun, Adv. Opt. Mater., 2024, 2302880 CrossRef CAS.
- B. Zhou, Q. Li, L. Yan and Q. Zhang, J. Rare Earths, 2020, 38, 474–482 CrossRef.
- J. Fan, L. Liang, Y. Gu and X. Liu, Opt. Mater.: X, 2021, 12, 100104 CAS.
- Y. Zhong, I. Rostami, Z. Wang, H. Dai and Z. Hu, Adv. Mater., 2015, 27, 6418 CrossRef CAS PubMed.
- X. Huang, Opt. Lett., 2015, 40, 3599–3602 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ce00373j |
| This journal is © The Royal Society of Chemistry 2024 |