
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew solid forms of tetrahydrocurcumin with improved solubility†
Fanyu
Tian
ab,
Mengyuan
Xia
bc,
Hao
Wang
ab,
Bingrui
Zhang
bc,
Zhenfeng
Ding
bc,
Xiaoyi
Rong
b,
Bingqing
Zhu
*b and
Xuefeng
Mei
 *abc
*abc
aSchool of Chinese Materia Medica, Nanjing University of Chinese Medicine, 138 Xianlin Avenue, Nanjing 210023, China
bPharmaceutical Analytical & Solid-State Chemistry Research Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China. E-mail: xuefengmei@simm.ac.cn
cUniversity of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing 100049, China
First published on 25th April 2024
Abstract
Tetrahydrocurcumin (THC), one of the active metabolites of curcumin, possesses pharmacological activities similar to those of curcumin, such as hypoglycemic, hypolipidemic and antioxidant effects. However, poor water solubility of THC leads to unfavorable absorption, which limits its application. The aim of this study was to improve the solubility of THC through cocrystallization. Five cocrystals of THC with L-proline (THC–L-PRO), D-proline (THC–D-PRO), DL-proline (THC–DL-PRO), 4,4′-bipyridine (THC–BPY), and betaine (THC–BTN) were discovered for the first time using liquid-assisted grinding. Single crystal structures of all THC co-crystals were analyzed and comprehensive characterization was performed through PXRD, thermal analysis, DVS and FT-IR. Dissolution studies were further conducted on two edible cocrystals THC–L-PRO and THC–BTN, and THC–L-PRO exhibits the highest apparent solubility compared with THC–BTN and the raw material. Moreover, THC-form II was found in THC–L-PRO dissolution experiments, and subsequently, the melt crystallization method was used to obtain the pure form of THC-form II. THC-form II was characterized for its physicochemical properties and the result of the dissolution experiment showed that it has better solubility compared to THC-form I.
1. Introduction
Curcumin is a natural active ingredient extracted from the rhizome of Curcuma longa, a plant of the family Zingiberaceae, and is widely used as a yellow coloring agent and spice in food. It has a variety of pharmacological activities such as anti-inflammatory,1 antioxidant,2 antidiabetic,3 anti-bacterial,4 and anti-viral5 effects, and its safety has been widely demonstrated.6–8 However, curcumin's powerful colouring ability also causes problems in practical use, such as the appearance of the product turns yellow after adding curcumin to cosmetics, and consumers' skin also turns yellow after applying it. In addition, water insolubility,9 chemical instability under physiological conditions10,11 and serious metabolism of curcumin12 limit its absorption. In particular, in a buffer system at 37 °C and pH 7.2, curcumin decomposed by about 90% within 30 minutes.13 Tetrahydrocurcumin (THC) is an active metabolite of curcumin and appears as a white crystalline powder with excellent chemical stability; it was not decomposed when incubated in 0.1 M phosphate buffer for 8 h regardless of pH.14 It not only exhibits better pharmacological activities than curcumin in anti-glycemic,3 anti-hyperlipidemic3 and antioxidant aspects,15 but also solves the coloring problem of curcumin skin care products. THC is now considered “generally recognised as safe” (GRAS) and has been evaluated for safety by the EFSA,16 and might be a promising substitute for curcumin. However, poor water solubility17 and rapid metabolism18 challenge the absorption of THC. Self-nanoemulsifying drug delivery systems (SNEDDSs),19 THC–hyaluronic acid (THC–HA) conjugate20 and THC–diglutaric acid (TDG) prodrug,21 have been reported to improve THC solubility, but no bioavailability is available to validate these. It is still necessary to explore more process-friendly methods that can effectively improve THC solubility and its bioavailability.As an emerging crystal engineering strategy, cocrystallization provides a new way to improve the physicochemical properties of active ingredients.22–26 Cocrystals are defined as “solids that are crystalline single phase materials composed of two or more different molecular and/or ionic compounds generally in a stoichiometric ratio which are neither solvates nor simple salts”.27 The active pharmaceutical ingredient (API) and coformers are generally bonded together in a crystalline lattice by noncovalent interactions such as hydrogen bonding, π–π interactions and halogen bonding. Hydrogen bonding is currently the most widely studied in cocrystal design due to its saturation and directivity.28,29 Dozens of literature studies have reported the ability of cocrystallization to improve the solubility and bioavailability of APIs.30–34 For compounds lacking ionizable functional groups, cocrystallization provides valuable opportunities to modulate physicochemical properties. More than 20 cocrystals of curcumin have been reported (Table S1†), but no reports on THC cocrystals are available so far. Cocrystal design of THC was conducted in virtue of its competitive hydrogen bonding sites such as phenolic hydroxyl groups and carbonyl groups.
In the present study, based on the molecular structure of THC, coformers containing carboxylic acid groups and nitrogen heterocycles that may form intermolecular hydrogen bonds with THC were selected for cocrystallization (Table S2†). Finally, five cocrystals, i.e., THC–L-proline (THC–L-PRO), THC–D-proline (THC–D-PRO), THC–DL-proline (THC–DL-PRO), THC–4,4′-bipyridine (THC–BPY), and THC–betaine (THC–BTN) were obtained. Single crystal structures and physicochemical properties were thoroughly analyzed. Notably, THC–L-PRO significantly enhanced the solubility of THC. And THC-form II was found in the dissolution experiments of THC–L-PRO and its physicochemical properties were also analyzed. Dissolution study showed that it increased the solubility of THC (Scheme 1).
 | ||
| Scheme 1 Chemical structures of THC and coformers. | ||
2. Experimental section
2.1. Materials
THC used in the present work was purchased from Shanghai DEMO Medical Technology Co., Ltd. (Shanghai, China), with a purity greater than 99%. Coformers were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). All analytical-grade solvents were purchased from Sinopharm Chemical Reagent Company. HPLC-graded methanol was purchased from Merck Company. All solvents were used without further purification.2.2. Synthesis of cocrystals via liquid-assisted grinding (LAG)
Bulk powders of the cocrystals (THC–L/D/DL-PRO, THC–BPY and THC–BTN) were prepared by a liquid-assisted grinding method using an XM-2017S oscillatory ball mill (Shanghai Jingxin Industrial Development Co., Ltd., Shanghai, China) with stainless steel grinding jars (25 mL). 372 mg of THC (1 mmol) and an equal molar amount of coformer were mixed along with 100 μL of methanol in a 25 mL grinding jar with stainless steel balls. Samples were ground in a ball mill for 30 min at a rate of 40 Hz. Products were dried overnight under ambient conditions. The cocrystals were identified by means of powder X-ray diffraction (PXRD) (Fig. S1†).2.3. Preparation of single crystals
All single crystals of THC cocrystals were obtained through slow evaporation. About 20 mg of pure cocrystal sample was dissolved into 1 mL of methanol solutions. After slow evaporation at room temperature, the single crystals (THC–L/D/DL-PRO and THC–BPY) were obtained. The single crystal of THC–BTN was obtained by slow evaporation of 1 mL ethanol solutions containing about 20 mg of pure THC–BTN at 4 °C.2.4. Synthesis of THC-form II via melt crystallization
The raw material (THC-form I) was heated at 120 °C to melt and stirred continuously at room temperature until a solid sample was completely generated, which was then pulverized with a pestle and the form purity was confirmed by PXRD.2.5. Powder X-ray diffraction (PXRD)
All PXRD measurements were performed at ambient temperature using a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation. The voltage and current of the generator were set to 40 kV and 40 mA, respectively. The scanning interval was 3–40° 2θ with time (0.1 s) per step. Data were imaged and integrated with RINT Rapid and peak-analyzed with Jade 6.0 from Rigaku.2.6. Single-crystal X-ray diffraction (SCXRD)
Single crystals suitable for X-ray crystallography studies were selected by polarized light microscopy. Single crystal X-ray diffraction data were collected on a Bruker Apex II CCD diffractometer using Mo Kα radiation (λ = 0.71073 Å) at 100 K or 170 K. Integration and scaling of intensity data were accomplished using the Bruker SAINT program, and corrections of data for the effects of absorption were performed using a multiscan method (SADABS). The crystal structures were solved by direct methods and refined with the full-matrix least-squares technique using the SHELX-2017 program package in Olex 2 software. Non-hydrogen atoms were refined with anisotropic displacement parameters, and hydrogen atoms were placed in calculated positions and refined with a riding model. Crystallographic data in cif format have been deposited in the Cambridge Crystallographic Data Center and the CCDC No. 2334117–2334121 are for THC–L-PRO, THC–D-PRO, THC–DL-PRO, THC–BPY, and THC–BTN, respectively. CCDC-Mercury and POV-ray were used to create molecular graphics. Crystallographic data and refinement details are summarized in Table 1, and hydrogen bond parameters are provided and listed in Table S3.†| THC–L-PRO | THC–D-PRO | THC–DL-PRO | THC–BPY | THC–BTN | |
|---|---|---|---|---|---|
| Formula | C26H33NO8 | C26H33NO8 | C26H33NO8 | C31H32N2O6 | C26H35NO8 |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21212 | P21212 | P2/n | C2 | P21/c |
| Temperature (K) | 170 | 100 | 170 | 170 | 170 |
| a (Å) | 18.4944(8) | 18.370(4) | 20.106(4) | 13.421(5) | 15.254(3) |
| b (Å) | 23.2557(11) | 23.190(8) | 5.4996(10) | 6.843(3) | 7.9952(14) |
| c (Å) | 5.4935(2) | 5.4660(16) | 23.399(5) | 14.762(4) | 20.500(3) |
| α (°) | 90 | 90 | 90 | 90 | 90 |
| β (°) | 90 | 90 | 114.231(5) | 101.980(12) | 96.512(5) |
| γ (°) | 90 | 90 | 90 | 90 | 90 |
| V (Å3) | 2362.76(17) | 2328.5(12) | 2359.4(8) | 1326.2(8) | 2484.0(7) |
| D Cal (g cm−3) | 1.371 | 1.391 | 1.373 | 1.324 | 1.309 |
| Z | 4 | 4 | 4 | 2 | 4 |
| λ (Mo-Kα) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Independent reflns. | 4792 | 4722 | 4448 | 2378 | 5068 |
| GooF | 1.081 | 1.060 | 1.090 | 1.063 | 1.039 |
| R int | 0.0692 | 0.0881 | 0 | 0.0545 | 0.0955 |
| R 1 | 0.0516 | 0.0657 | 0.0993 | 0.0719 | 0.0871 |
| wR2 | 0.1215 | 0.1592 | 0.2087 | 0.1985 | 0.2048 |
| CCDC | 2334117 | 2334118 | 2334119 | 2334120 | 2334121 |
2.7. Thermogravimetric analysis (TGA)
Thermogravimetric analysis (TGA) experiments were carried out on TGA 55 equipment (TA Instruments, New Castle, DE, USA). Samples weighing 5–10 mg were placed in open aluminum oxide pans and heated from 25 °C to 410 °C at a rate of 10 °C min−1. Nitrogen was used as the purge gas at 60 mL min−1.2.8. Differential scanning calorimetry (DSC)
Differential scanning calorimetry (DSC) measurements were carried out on a DSC Q2000 instrument (PerkinElmer) under a nitrogen gas flow of 50 mL min−1 purge. Accurately weighed samples were sealed in nonhermetic aluminum pans and heated at 10 °C min−1. Two-point calibration with indium and tin was used to check the temperature axis and heat flow of the instrument.2.9. Dynamic vapor sorption (DVS)
The hygroscopicity behaviors were investigated on an Intrinsic DVS instrument from Surface Measurement Systems, Ltd. Samples were placed on the balance, and the water sorption and desorption were measured from one cycle over a range of 0–95–0% relative humidity (RH) at 25 °C. The next RH step with an interval of 5% was performed if the mass change was less than 0.02% within 10 min.2.10. Fourier-transform infrared (FTIR) spectroscopy
Fourier-transform infrared (FT-IR) spectra of samples were recorded using a ThermoFisher Nicolet iS50 FT-IR spectrometer in the range from 4000 to 400 cm−1, with an average of 32 scans and a resolution of 4 cm−1 under ambient conditions.2.11. Equilibrium solubility and powder dissolution
To avoid the particle size effect on dissolution results, two edible cocrystals (THC–-PRO and THC–BTN) were sieved through 100-mesh sieves. The equilibrium solubility of THC–L-PRO and THC–BTN was measured in pH 2.0 (hydrochloric acid–glycine), pH 4.5 (citric acid–disodium hydrogen phosphate), and pH 6.8 (sodium dihydrogen phosphate–disodium hydrogen phosphate) buffers. About 15 mg of sample was added to 2 mL of buffers and slurried at a stirring rate of 200 rpm for 24 h at ambient temperature. The suspensions were centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min, and the supernatants were subjected to HPLC analysis. Each test was triplicated. After equilibrium solubility experiments, the remaining solids were dried in a vacuum and analyzed using PXRD to confirm the composition.
000 rpm for 5 min, and the supernatants were subjected to HPLC analysis. Each test was triplicated. After equilibrium solubility experiments, the remaining solids were dried in a vacuum and analyzed using PXRD to confirm the composition.
Powder dissolution experiments were conducted in pH 2.0, pH 4.5, and pH 6.8 buffers at 37 °C and 50 rpm. Excess amounts of 30 mg of THC (or corresponding cocrystals and THC-form II) were added to 15 mL of the buffer pre-equilibrated at 37 °C. Without supplementing the fresh medium, about 0.5 mL of solutions were withdrawn at 5, 10, 15, 30, 45, 60, 90 and 120 min and centrifuged at 14000 rpm for 5 min. Then the supernatant was diluted immediately with pure methanol to avoid recrystallization prior to HPLC analysis. Each test was triplicated. The precipitates after centrifugation were dried in a vacuum for PXRD analysis to confirm the composition.
2.12. High-performance liquid chromatography (HPLC)
The accurate concentration of THC was analyzed on an Agilent 1260 series HPLC (Agilent Technologies Co., Ltd., Santa Clara, CA, USA) equipped with a quaternary pump (G1311C), a diode-array detector (G1315D) and a 4.6 × 150 mm, 5 μm Agilent ZORBAX Extend C18 column. The injected volume was set to 20 μL, and the UV-vis wavelength was set to 290 nm. The column temperature was 35 °C. The mobile phase consisting of 0.05% ammonia liquor aqueous solution (eluent A) and methanol (eluent B) was run at 1.0 mL min−1. The gradient elution procedure started at 90% A, and then linearly decreased to 10% A over 6 min and held for 2 min. Finally, it returned to the initial mobile phase within 0.1 min and held for 4 min.3. Results and discussion
3.1. Cocrystal design and PXRD analysis
On the basis of the hydrogen bonding possibilities of THC, coformers containing carboxylic acid groups and nitrogen heterocycles were selected for cocrystallization (Table S2†). Various methods for screening the cocrystals of THC were used, including evaporation, cooling, slurry, and grinding. As a result, THC–L/D/DL-PRO, THC–BPY and THC–BTN cocrystals with high crystallinity were successfully synthesized by liquid-assisted grinding.PXRD analysis was performed to verify the cocrystal formation and identify the phase purity of the products. The powder diffraction peaks of the resulting materials after grinding are clearly different from those of THC and the corresponding coformer, suggesting the generation of new phases. Compared with the raw material, THC–BPY presents unique characteristic peaks at 2θ = 14.4, 15.1, 16.2, 17.9, 20.0, 22.0 and 25.0°, and THC–BTN at 2θ = 13.3, 16.2, 18.2, 18.6, 21.2, 21.6 and 24.3°. Notably, PXRD patterns of THC–L-PRO, THC–D-PRO and THC–DL-PRO are of high similarity, sharing peaks at 2θ = 9.4, 15.9, 18.9, and 24.3°. And all the patterns of powder samples experimentally obtained closely match with the simulated patterns calculated from the single crystal diffraction data, proving that the new phases are of high purity (Fig. 1). PXRD patterns of the THC cocrystals and corresponding coformers are provided in Fig. S1.†
 | ||
| Fig. 1 PXRD patterns of (a) THC–L-PRO, THC–D-PRO and THC–DL-PRO cocrystals, and (b) THC–BPY and THC–BTN cocrystals. | ||
3.2. Single crystal structure
 | ||
| Fig. 2 Molecular overlay of THC in raw materials (red), THC–L-PRO (yellow), THC–BPY (light green) and THC–BTN (deep green). | ||
 | ||
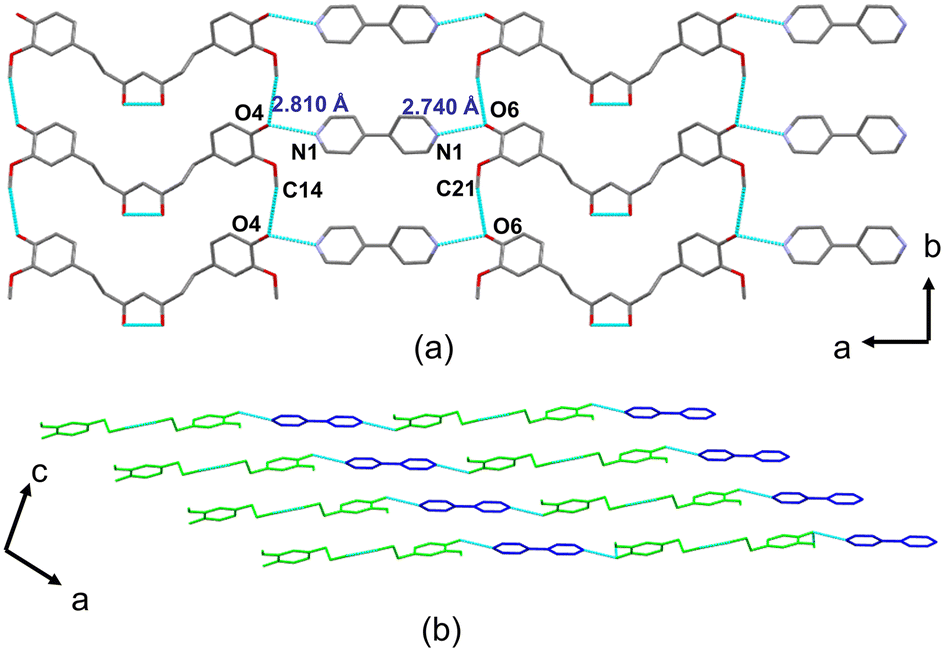
| Fig. 3 Crystal structure of THC–L-PRO: (a) hydrogen bonding mapping and 2D layer parallel to the bOc plane, and (b) 2D layers stacked via van der Waals forces to give a 3D architecture. | ||
 | ||
| Fig. 4 Crystal structure of THC–BPY: (a) hydrogen bonding mapping and 2D layer parallel to the aOb plane and (b) 2D layers stacked via van der Waals forces to give a 3D architecture. BPY is marked in blue, and THC is marked in light green. Hydrogen atoms have been omitted for clarity. | ||
 | ||
| Fig. 5 Crystal structure of THC–BTN: (a) hydrogen bonding profiles and 2D layer along the b-axis direction, (b) crystal centers of symmetry in the unit cell, indicated by orange balls (c) 2D layers stacked via van der Waals forces to give a 3D architecture. Hydrogen atoms have been omitted from the (b) and (c) diagrams for clarity. Each molecule is coloured according to the symmetry relationship it has with the asymmetric unit. | ||
3.3. Thermal analysis
DSC profiles of the THC cocrystals and corresponding coformers are superimposed in Fig. S4.† Overlays of TGA and DSC curves are shown in Fig. S5.† The TGA curves show that the cocrystals have no significant mass loss before melting, indicating that no water or organic solvents are contained in their solid form. DSC profiles show that all the cocrystals have a single sharp endothermic peak, indicating that there is no phase transition prior to their melting and high purity of the new phases.The onset temperature of THC, the THC cocrystals and the corresponding coformers is summarized in Table 2. The melting enthalpy (ΔHm) of THC and its cocrystals is summarized in Table S4.† All of the cocrystals substantially increased the melting point of THC. The melting point of the cocrystal correlates with the coformers.35,36 The melting point of the THC cocrystals increase with that of coformers. Notably, the three proline related cocrystals exhibit a similar onset temperature, probably due to the similarity of the coformers and the spatial arrangement.
| Form | T onset (°C) | ||
|---|---|---|---|
| THC | Cocrystal | Coformer | |
| THC–L-PRO | 96.5 ± 0.0 | 138.3 ± 0.7 | 229 |
| THC–D-PRO | 138.6 ± 0.2 | 229 | |
| THC–DL-PRO | 140.1 ± 0.2 | 210 | |
| THC–BPY | 114.6 ± 0.7 | 110 | |
| THC–BTN | 156.3 ± 0.3 | 305 | |
3.4. FT-IR analysis
FT-IR can recognize shifts in functional group eigenfrequencies of a compound, providing support for the identification of cocrystals. FT-IR spectra of THC, coformers, and THC cocrystals are shown in Fig. S6.† THC has two phenolic hydroxyl groups, whose characteristic stretching frequency is at 3401 cm−1 (O–H stretch). In the THC cocrystals, the signal corresponding to the O–H stretching vibration in THC is red-shifted to 3291 and 3177 cm−1 for THC–L-PRO, 3286 and 3175 cm−1 for THC–D-PRO, 3304 and 3176 cm−1 for THC–DL-PRO, 3255 cm−1 for THC–BTN and 3046 cm−1 for THC–BPY, suggesting that the hydroxy groups of THC engaged in the hydrogen bonding interactions in these cocrystals. Typical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration appears at 1614 cm−1 for L-PRO, 1613 cm−1 for D-PRO, 1596 cm−1 for DL-PRO, and 1614 cm−1 for BTN. In the THC cocrystals, the carbonyl stretching band is blue-shifted to 1626 cm−1 for THC–L-PRO, 1625 cm−1 for THC–D-PRO, 1629 cm−1 for THC–DL-PRO, and 1640 cm−1 for THC–BTN since it acts as a hydrogen bond acceptor. The signal corresponding to the CN stretching vibration in BPY (1587 cm−1) is blue-shifted to 1595 cm−1 in THC–BPY.
O stretching vibration appears at 1614 cm−1 for L-PRO, 1613 cm−1 for D-PRO, 1596 cm−1 for DL-PRO, and 1614 cm−1 for BTN. In the THC cocrystals, the carbonyl stretching band is blue-shifted to 1626 cm−1 for THC–L-PRO, 1625 cm−1 for THC–D-PRO, 1629 cm−1 for THC–DL-PRO, and 1640 cm−1 for THC–BTN since it acts as a hydrogen bond acceptor. The signal corresponding to the CN stretching vibration in BPY (1587 cm−1) is blue-shifted to 1595 cm−1 in THC–BPY.
3.5. Hygroscopicity
Fig. S7† illustrates the moisture sorption and desorption curves of THC and the five cocrystals, whereby mass absorption was monitored as a function of relative humidity (RH) at room temperature. Among the five cocrystals, THC–BPY has the lowest hygroscopicity and almost no moisture increase. The remaining cocrystals absorb no more than 1.5% moisture at 95% RH. All cocrystals provide excellent physical stability in that no significant changes were observed in the PXRD patterns of the samples after DVS experiments (Fig. S8†).3.6. Dissolution studies (equilibrium solubility and powder dissolution)
Solid drugs need to be dissolved in order to be absorbed by the body, and the low solubility of drugs often limits their bioavailability. Cocrystal technology can be used to improve the solubility of drugs. THC–L-PRO and THC–BTN were selected for dissolution studies with regard to safety and edibility of L-PRO and BTN. The equilibrium solubility and powder dissolution experiments for pure THC, THC–L-PRO and THC–BTN were performed in three different pH buffers (pH 2.0, pH 4.5 and pH 6.8 buffers).The results of the equilibrium solubility experiments showed that both THC–L-PRO and THC–BTN were converted to THC, with solubility similar to that of the THC raw material (Fig. 6 and S9†). In the dissolution experiment, THC–BTN exhibited a dissolution profile similar to that of THC due to its rapid conversion to THC. Notably, THC–L-PRO significantly increased the dissolution rate and apparent solubility of THC (Fig. 6). The solution concentrations of THC–L-PRO at 30 min were 2.9, 2.0 and 1.8 times higher than those of THC at pH 2.0, 4.5 and 6.8, respectively (Table S5†). The supersaturation advantage of THC–L-PRO could be sustained for a long period of time of at least 2 hours, which might be favorable for in vivo absorption.18
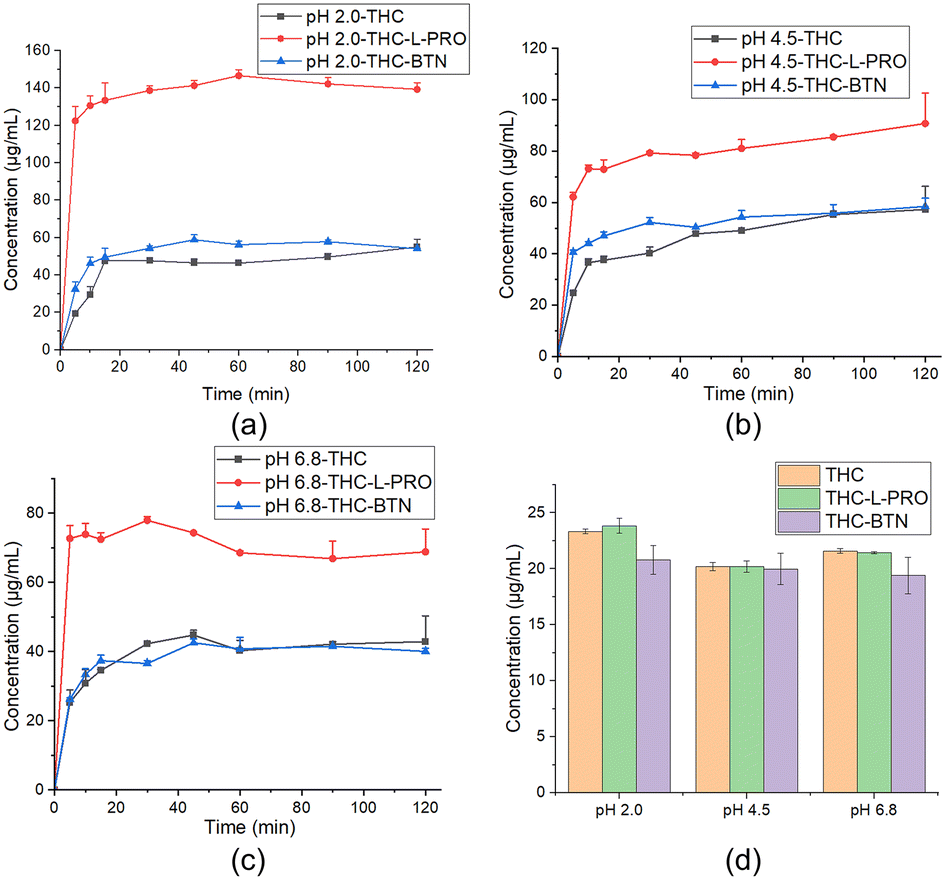 | ||
| Fig. 6 Powder dissolution profiles and equilibrium solubility of THC and its two cocrystals: powder dissolution profiles at 37 °C in (a) pH 2.0, (b) pH 4.5, and (c) pH 6.8 buffers. (d) Equilibrium solubility after 24 h at ambient temperature; data are expressed as mean ± SD (n = 3). | ||
PXPD characterization showed that the precipitates of THC–L-PRO from the dissolution experiments exhibited a mixture of THC and a new phase (Fig. S10†). The new phase was observed in the precipitates under all three pH conditions. The DSC curve of the mixture shows the presence of an endothermic peak at 68 °C before the melting of THC, suggesting that a new crystal form of THC (form II) appeared during the dissolution process (Fig. S10†). The pure phase of THC-form II was further obtained by melting crystallization (Fig. S12†). The PXPD, DSC, TGA, DVS and FT-IR patterns of THC form II are shown in Fig. S10 and S11.† The onset temperature of THC-form II is 68.1 ± 0.8 °C, and the melting enthalpy is 23.4 ± 1.6 kJ mol−1.
PXRD patterns of the mixtures of the two crystal forms illustrate that a high degree of supersaturation prompted the recrystallization of THC into the metastable crystal form II, which still has high solubility. And afterwards, form II was continuously transformed into the stable crystal form I.37 So the higher apparent solubility can be maintained for more than 2 h.
As shown in Fig. 7, powder dissolution experiments were further carried out for THC-form II in three different pH buffers (pH 2.0, pH 4.5 and pH 6.8 buffers). At pH 2.0, 4.5, and 6.8, the solution concentration of THC-form II was 2.3, 2.1, and 1.6 times the concentration of THC at 30 min, but at 120 min the solution concentration of THC-form II was 1.2, 1.1, and 1.3 times the concentration of THC, respectively (Table S6†). The residue powder after the dissolution experiments showed that the undissolved solid of THC-form II exhibited a mixture of two crystal forms dominated by THC-form I (Fig. S13†). The PXRD patterns showed that the metastable THC-form II slowly recrystallized to the stable THC-form I after it was dissolved, leading to a decrease in solubility.37 So the dissolution advantage cannot be maintained for 2 hours.
 | ||
| Fig. 7 Powder dissolution profiles of THC-form I and THC-form II: powder dissolution profiles at 37 °C in (a) pH 2.0, (b) pH 4.5, and (c) pH 6.8 buffers. Data are expressed as mean ± SD (n = 3). | ||
4. Conclusions
THC, which has similar biological activity to curcumin and better physicochemical properties than curcumin, faces low solubility and bioavailability issues in its application. Five THC cocrystals were synthesized for the first time by choosing appropriate coformers. The crystal structures of all the cocrystals were analyzed. The physicochemical properties of the cocrystals were comprehensively evaluated, among which the THC–L-PRO cocrystal presented higher solubility compared with THC. And a new polymorph of THC was found during the dissolution of THC–L-PRO. The physicochemical properties of THC-form II were characterised and dissolution experiments showed an increase in solubility compared to THC-form I. The availability of new solid forms that enhance the solubility of THC provides more options for the application of THC.Conflicts of interest
There are no conflicts to declare.References
- Z. Liu and Y. Ying, Front. cell dev. biol., 2020, 8, 479 CrossRef.
- Q. Ma, Y. Ren and L. Wang, Food Hydrocolloids, 2017, 70, 286–292 CrossRef CAS.
- P. Murugan and L. Pari, J. Ethnopharmacol., 2007, 113, 479–486 CrossRef CAS.
- A. Adamczak, M. Ożarowski and T. M. Karpiński, Pharmaceuticals, 2020, 13, 153 CrossRef CAS PubMed.
- R. K. Thimmulappa, K. K. Mudnakudu-Nagaraju, C. Shivamallu, K. J. T. Subramaniam, A. Radhakrishnan, S. Bhojraj and G. Kuppusamy, Heliyon, 2021, 7, e06350 CrossRef CAS PubMed.
- N. Chainani-Wu, J. Altern. Complementary Med., 2003, 9, 161–168 CrossRef PubMed.
- V. Soleimani, A. Sahebkar and H. Hosseinzadeh, Phytother. Res., 2018, 32, 985–995 CrossRef CAS.
- C. D. Lao, M. T. Ruffin, D. Normolle, D. D. Heath, S. I. Murray, J. M. Bailey, M. E. Boggs, J. Crowell, C. L. Rock and D. E. Brenner, BMC Complementary Altern. Med., 2006, 6, 10 CrossRef PubMed.
- B. T. Kurien, A. Singh, H. Matsumoto and R. H. Scofield, Assay Drug Dev. Technol., 2007, 5, 567–576 CrossRef CAS.
- Y. Nimiya, W. Wang, Z. Du, E. Sukamtoh, J. Zhu, E. Decker and G. Zhang, Mol. Nutr. Food Res., 2015, 60, 487–494 CrossRef PubMed.
- M. Kharat, Z. Du, G. Zhang and D. J. McClements, J. Agric. Food Chem., 2017, 65, 1525–1532 CrossRef CAS PubMed.
- B. Wahlström and G. Blennow, Basic Clin. Pharmacol. Toxicol., 2009, 43, 86–92 Search PubMed.
- Y. J. Wang, M. H. Pan, A. L. Cheng, L. I. Lin, Y. S. Ho, C. Y. Hsieh and J. K. Lin, J. Pharm. Biomed. Anal., 1997, 15, 1867–1876 CrossRef CAS PubMed.
- M. H. Pan, T. M. Huang and J. K. Lin, Drug Metab. Dispos., 1999, 27, 486–494 CAS.
- N. P. Morales, S. Sirijaroonwong, P. Yamanont and C. Phisalaphong, Biol. Pharm. Bull., 2015, 38, 1478–1483 CrossRef CAS PubMed.
- D. Turck, T. Bohn, J. Castenmiller, S. De Henauw, K. I. Hirsch-Ernst, A. Maciuk, I. Mangelsdorf, H. J. McArdle, A. Naska, C. Pelaez, K. Pentieva, A. Siani, F. Thies, S. Tsabouri, M. Vinceti, F. Cubadda, T. Frenzel, M. Heinonen, R. Marchelli, M. Neuhäuser-Berthold, M. Poulsen, M. Prieto Maradona, J. R. Schlatter, H. van Loveren, R. Ackerl, E. Kouloura and H. K. Knutsen, EFSA J., 2021, 19, e06936 CAS.
- A. Kanshide, M. R. Peram, N. Chandrasekhar, A. Jamadar, V. Kumbar and M. Kugaji, J. Pharm. Innov., 2022, 18, 980–998 CrossRef.
- J. Novaes, R. Lillico, C. Sayre, K. Nagabushanam, M. Majeed, Y. Chen, E. Ho, A. Oliveira, S. Martinez, S. Alrushaid, N. Davies and T. Lakowski, Pharmaceutics, 2017, 9, 45 CrossRef PubMed.
- I. K. A. Astuti, T. R. I. Suliatin and R. Wahyuningrum, Int. J. Appl. Pharm., 2019, 11, 97–102 CrossRef CAS.
- X. Tang, M. Zhang, H. Zhang, Y. Pan, Q. Dong, Y. Xin, C.-T. Ho and Q. Huang, Int. J. Biol. Macromol., 2021, 182, 1322–1330 CrossRef CAS PubMed.
- N. Jongjitphisut, R. Phumsuay, W. Thitikornpong, P. Rashatasakhon, C. Muangnoi, O. Vajragupta and P. Rojsitthisak, J. Pharm. Invest., 2022, 52, 477–487 CrossRef CAS.
- G. Bolla and A. Nangia, Chem. Commun., 2016, 52, 8342–8360 RSC.
- G. Bolla, B. Sarma and A. K. Nangia, Chem. Rev., 2022, 122, 11514–11603 CrossRef CAS PubMed.
- M. Karimi-Jafari, L. Padrela, G. M. Walker and D. M. Croker, Cryst. Growth Des., 2018, 18, 6370–6387 CrossRef CAS.
- S. N. Wong, Y. C. S. Chen, B. Xuan, C. C. Sun and S. F. Chow, CrystEngComm, 2021, 23, 7005–7038 RSC.
- P. S. Panzade and G. R. Shendarkar, Drug Dev. Ind. Pharm., 2020, 46, 1559–1568 CrossRef CAS PubMed.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. Rodriguez-Hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. Kumar Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147–2152 CrossRef CAS.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed.
- A. J. Smith, P. Kavuru, L. Wojtas, M. J. Zaworotko and R. D. Shytle, Mol. Pharmaceutics, 2011, 8, 1867–1876 CrossRef CAS PubMed.
- L. Zhang, D. Kong, H. Wang, L. Jiao, X. Zhao, J. Song, D. Yang, H. Yang, S. Yang, G. Du and Y. Lu, Molecules, 2021, 26, 2677 CrossRef CAS PubMed.
- D. Yang, J. Cao, L. Jiao, S. Yang, L. Zhang, Y. Lu and G. Du, ACS Omega, 2020, 5, 8283–8292 CrossRef CAS PubMed.
- P. Li, T. Ramaiah, M. Zhang, Y. Zhang, Y. Huang and B. Lou, Cryst. Growth Des., 2019, 20, 157–166 Search PubMed.
- S. J. Bethune, N. Schultheiss and J.-O. Henck, Cryst. Growth Des., 2011, 11, 2817–2823 CrossRef CAS.
- M. K. Stanton and A. Bak, Cryst. Growth Des., 2008, 8, 3856–3862 CrossRef CAS.
- S. Kumar, Indian J. Pharm. Sci., 2017, 79, 858–871 Search PubMed.
- D. D. Bavishi and C. H. Borkhataria, Prog. Cryst. Growth Charact. Mater., 2016, 62, 1–8 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2334117–2334121. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00259h |
| This journal is © The Royal Society of Chemistry 2024 |