
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using potassium bromide pellets and optical spectroscopy to assess the photodimerization of two trans-(trifluoromethyl)-cinnamic acid compounds†
Bayasgalan
Ulambayar
 a,
Khongorzul
Batchuluun
*a,
Chantsalnyam
Bariashir
ab,
Nergui
Uranbileg
ac,
Felix J.
Stammler
d,
Jav
Davaasambuu
ef and
Tobias E.
Schrader
*g
a,
Khongorzul
Batchuluun
*a,
Chantsalnyam
Bariashir
ab,
Nergui
Uranbileg
ac,
Felix J.
Stammler
d,
Jav
Davaasambuu
ef and
Tobias E.
Schrader
*g
aInstitute of Chemistry and Chemical Technology, Mongolian Academy of Sciences, 13330 Ulaanbaatar, Mongolia. E-mail: khongorzul.b@mas.ac.mn
bEnergy Science and Engineering, Daegu Gyeongbuk Institute of Science & Technology, Daegu, Republic of Korea
cDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea
dPhysical and Biophysical Chemistry, Bielefeld University, 33615 Bielefeld, Germany
eInstitute of Physics and Technology, Mongolian Academy of Sciences, 13330 Ulaanbaatar, Mongolia
fLaser Research Center, National University of Mongolia, 14201 Ulaanbaatar, Mongolia
gForschungszentrum Jülich, Jülich Centre for Neutron Science at Heinz Maier-Leibnitz Zentrum (MLZ), Lichtenbergstr. 1, 85747 Garching, Germany. E-mail: t.schrader@fz-juelich.de
First published on 18th July 2024
Abstract
The potential solid-state [2 + 2] photodimerization of two trans-cinnamic acid derivatives, trans-4-(trifluoromethyl)cinnamic acid (4-tfmca) and trans-3-(trifluoromethyl)cinnamic acid (3-tfmca), has been studied using potassium bromide (KBr) pellets and optical spectroscopy. As opposed to taking a powder or single crystal material as a sample, herein, we used polycrystalline samples incorporated in the KBr matrix (pellet) and were able to follow the course of the photochemical reaction via optical spectroscopy. The results show that the photodimerization of 4-tfmca within the KBr matrix yields a photodimer in roughly 100% conversion, while the changes observed for 3-tfmca within the KBr matrix suggest a subtle photochemical change which could not be observed when irradiating the neat powder. This suggests that the KBr matrix helps to couple light into the crystallites of the embedded sample and shows that the KBr matrix reveals photochemistry which might not have been found in the single crystal or powder state of the sample.
Introduction
After the discovery of photodimerization in 1867,1 extensive research efforts have been devoted to [2 + 2] photocycloaddition reactions in crystalline materials, especially in the latter half of the 1900s. [2 + 2] photocycloaddition reactions are essential in stereo synthesis2–5 as they afford stereospecific products that are difficult to obtain through solution chemistry in high or reasonable yields due to the constraints imposed on the molecular orientation by the crystal lattice.6 Such reactions have the advantage of producing only one product conformer. Additionally, these reactions do not require solvents and often do not need any purification steps, making them a greener approach compared to conventional organic synthesis methods.7,8One of the most common [2 + 2] photoreactions studied in crystalline materials is the [2 + 2] photodimerization of olefin containing compounds.9 Here, two nearby carbon–carbon double bonds turn into a four-membered ring of carbon atoms exhibiting a single-bond character with close to 90° bond angles. Schmidt and co-workers investigated the solid-state photodimerization properties of a series of trans-cinnamic acids and described the correlation between the packing arrangement and photoreactivity, which greatly aided the development of the topochemical principle that governs these types of solid-state reactions.10–12 Schmidt divided the trans-cinnamic acids into three types (α, β, γ) according to their crystal packing and [2 + 2] photodimerization properties.13 In the α type crystals, reactive molecules are arranged head-to-tail, while in the β type crystals, reactive molecules are arranged head-to-head. When exposed to UV radiation, both crystals undergo [2 + 2] photodimerization and produce a centrosymmetric α-truxillic acid dimer and a mirror-symmetric β-truxinic acid dimer, respectively (Fig. 1). On the other hand, the γ type crystals will remain unchanged under UV irradiation as they are photostable. From this structure–photoreactivity relationship, Schmidt reformulated the topochemical principle by emphasizing that the solid-state reactions are facilitated by minimum atomic movement when the reactive groups are in close vicinity and concluded the following rules: for photoactive molecules (1) the distance between the centroids of reactive groups should be less than 4.2 Å and (2) the reactive groups should be parallel to each other.13,14 However, many examples do not conform to these rules.15–17 Still, using these geometric criteria, one could evaluate and reason the photoreactivity of various molecules and design new photoresponsive materials.
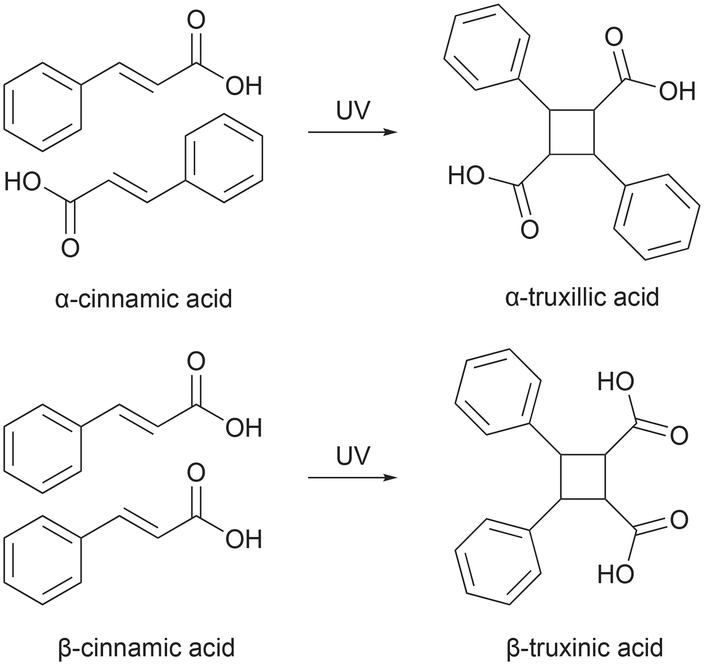 | ||
| Fig. 1 Reaction scheme of [2 + 2] photodimerization of α- and β-trans-cinnamic acid. | ||
Even though it is irreversible, photodimerization of trans-cinnamic acid remains a relevant subject in solid-state photoreaction studies, and it is often used as a model system for various studies in the field.18 A substantial amount of research has focused on the mechanism19 and kinetics20–22 of the reaction, utilizing different experimental techniques, most notably X-ray diffraction,23,24 vibrational spectroscopy,25–27 solid-state NMR,21 atomic force microscopy,28 and time-resolved spectroscopy.29–31 Pump–probe techniques, such as UV-pump and UV-visible probe spectroscopy, can elucidate the time scales of these reactions in the crystal phase.32 Combining UV-pump and infrared probe spectroscopy, mechanistic details along the reaction path can be elucidated.33 Probing with short X-ray pulses after initiating the photoreaction offers the possibility of following the reaction with the time-resolved single-crystal diffraction technique.34 However, despite these research efforts, certain aspects of photoreactions still lack complete understanding, and new model systems, as presented here, will help to shed more light on photodimerization reactions. In this work, we used the potassium bromide (KBr) pellet method, which is frequently used in infrared (IR) spectroscopy, to study the photodimerization process of two trans-cinnamic acid derivatives (Fig. 2). In the KBr pellet method, a mixture of the sample and KBr is pressed into a transparent pellet. As KBr exhibits low absorbance to UV radiation and shows inertness towards most organic compounds, it allows the sample within the pellet to absorb UV radiation effectively, making it possible to conduct a photoreaction inside the pellet. With direct recording of the absorption spectra in the pellet's IR and UV regions, the course of the photoreaction can be followed. Thus, herein, we report the spectroscopic investigations of the solid-state photochemical reactions of 4-tfmca and 3-tfmca by monitoring the absorption of the samples prepared in KBr pellets in the IR and UV regions along with the structure determination of 3-tfmca, 4-tfmca, and partially photodimerized 4-tfmca. It should be noted that among the derivatives studied, 4-tfmca has already been structurally characterized at low temperatures by Howard et al.35
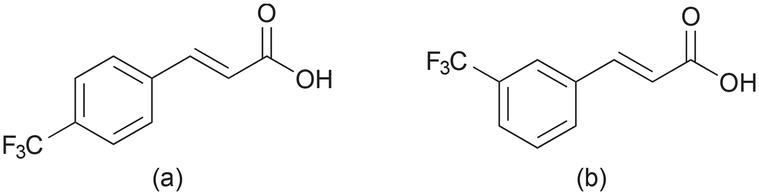 | ||
| Fig. 2 Molecular structures of trans-cinnamic acid derivatives used in this study: (a) 4-tfmca and (b) 3-tfmca. | ||
Experimental
Crystallization
4-tfmca (Aldrich, 99%) and 3-tfmca (Aldrich, 98%) were dissolved in ethanol at a typical concentration of 5 g l−1. Volumes of 17 ml solution were left in open vials to slowly evaporate at room temperature to yield plate-like crystals.UV irradiation
For the irradiation of the powders, a Petri dish filled with adequate amounts of the sample was exposed to UV radiation. The powder sample was mixed frequently to expose new surfaces to UV light. These samples were used for analysis in the case of PXRD and NMR. As for the irradiation of the pellet for FTIR and UV-vis analyses, powder samples were mixed with KBr and then pressed into 13 mm diameter pellets (the KBr to sample ratio is 2000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) before being exposed to UV radiation. The UV radiation was generated from a 500 W mercury–xenon lamp with a beam power of around 18–20 mW cm−2, for these irradiation experiments. As for the single crystals, the samples used for single-crystal X-ray diffraction analyses were exposed to monochromatic light of 334 nm.
:1) before being exposed to UV radiation. The UV radiation was generated from a 500 W mercury–xenon lamp with a beam power of around 18–20 mW cm−2, for these irradiation experiments. As for the single crystals, the samples used for single-crystal X-ray diffraction analyses were exposed to monochromatic light of 334 nm.
Single-crystal X-ray diffraction analyses
The single crystal X-ray data for the samples before and after UV irradiation were collected on a Rigaku Synergy-S diffractometer equipped with a microfocus Mo Kα X-ray source and a 150° HyPix-Arc detector. Data reduction and integration were carried out using CrysAlisPro software version 42.96a.36 Solution (direct methods, SHELXS and intrinsic phasing, SHELXT) and refinement (full matrix least squares minimization on F2) of all crystal structures were done using the SHELX program package37,38 connected to the OLEX2 graphical user interface.39 Anisotropic refinement was applied to non-hydrogen atoms. The hydrogen atoms attached to carbon atoms were placed geometrically and refined using a riding model with isotropic thermal parameters. For the hydrogen atoms of the carboxylic groups, positions were located from the difference density map, and distance restraints were put on the O–H bond lengths. In the case of the irradiated single crystal sample of 4-tfmca, an occupancy of 28.8% of the photoproduct could be modeled. One C-atom was modeled isotropically since it overlapped with the one from the photoproduct so much that the density could not be modeled unambiguously with two anisotropic ellipses. The disorder of the CF3 group was not easy to model, which explains the rather high R-factors of the deposited models.The crystallographic data for 4-tfmca, 3-tfmca, and partially photodimerized 4-tfmca can be found at the Cambridge Crystallographic Data Centre under the CCDC numbers 2331642, 2331099, and 2332753, respectively.
Powder X-ray diffraction
Powder X-ray diffraction measurements were recorded on both neat powder and powder samples embedded within the KBr pellet using a Shimadzu MAXima-X XRD 7000 in the case of 3-tfmca and on a Bruker Phaser D2 diffractometer in the case of 4-tfmca. Pellets used in the measurements were prepared by pressing the mixture with a sample-to-KBr ratio of 3:7. Measurements were recorded over the 2θ range between 10 and 40° at a scan speed of 1° min−1. The X-ray emission was generated from either a copper tube (λ = 1.5406 Å) in the case of 4-tfmca or a cobalt tube (λ = 1.7890 Å) in the case of 3-tfmca configured at 40 kV and 40 mA. Calculated PXRD and experimental PXRD data were compared to identify any additional phases or impurities if present. PXRD patterns were calculated from the single crystal X-ray diffraction data using Mercury software.40 Refinement of the unit cell sizes in the case of the powder samples embedded in the KBr matrix was performed using the software jana2020.41,42
Optical spectroscopy
The FTIR spectra of the pellets in the wavenumber range of 400–4000 cm−1 with a resolution of 1.0 cm−1 were recorded on a Perkin Elmer Spectrum Two spectrophotometer. The UV-vis spectra of the pellets were measured from 200 nm to 700 nm with a resolution of 1 nm on a Shimadzu UV-2550 spectrophotometer. For both FTIR and UV-vis measurements, the empty beam was taken as a blank reference for baseline correction. IR spectra were measured in situ while for the UV spectra measurements, the pellet had to be removed from the light exposure at regular intervals.Solution NMR
The solution 1H NMR spectra were measured at room temperature using a Bruker DMX 400 spectrometer at an operating frequency of 500 MHz. Samples were dissolved in nondeuterated dimethyl sulfoxide (DMSO), and chemical shift values were reported in parts per million (ppm) relative to the residual proton signal of the solvent (δ = 2.5 ppm).Results and discussion
X-ray diffraction analyses
To assess the photoreactivity of the derivatives, the crystal structure should be examined as the photodimerization in the solid state is influenced by crystal packing. Single crystal XRD analysis shows that 4-tfmca crystallizes in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) triclinic system and has two molecules in its asymmetric unit (Fig. 3a or ESI† Table S1). For each molecule in the asymmetric unit, the CF3 group shows disorder, where each fluorine atom is distributed across two different positions. The fundamental structural unit is a carboxylic acid dimer formed between pairs of molecules through hydrogen bonding (O(2)–H(2)⋯O(4), dO⋯O, 2.656(2) Å, O–H⋯O 172(2)°). Dimers in the same plane create a string of dimers through weak in-plane C–H⋯O and C–H⋯F interactions (Fig. 3b), which is a common feature observed among trans-cinnamic acids. Assembly of these strings in three dimensions leads to linear packing (Fig. 3c). Along the stack, every molecule in the carboxylic dimer has one neighboring molecule that it could react with, as the distance between the centroids of the C
triclinic system and has two molecules in its asymmetric unit (Fig. 3a or ESI† Table S1). For each molecule in the asymmetric unit, the CF3 group shows disorder, where each fluorine atom is distributed across two different positions. The fundamental structural unit is a carboxylic acid dimer formed between pairs of molecules through hydrogen bonding (O(2)–H(2)⋯O(4), dO⋯O, 2.656(2) Å, O–H⋯O 172(2)°). Dimers in the same plane create a string of dimers through weak in-plane C–H⋯O and C–H⋯F interactions (Fig. 3b), which is a common feature observed among trans-cinnamic acids. Assembly of these strings in three dimensions leads to linear packing (Fig. 3c). Along the stack, every molecule in the carboxylic dimer has one neighboring molecule that it could react with, as the distance between the centroids of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds is 3.78 Å. Since these molecules stack head-to-head, 4-tfmca is classified as a β type crystal. The crystal packing of 4-tfmca found at 293 K by our single-crystal X-ray diffraction analyses matches closely with the structure previously determined at 200 K.35
C bonds is 3.78 Å. Since these molecules stack head-to-head, 4-tfmca is classified as a β type crystal. The crystal packing of 4-tfmca found at 293 K by our single-crystal X-ray diffraction analyses matches closely with the structure previously determined at 200 K.35
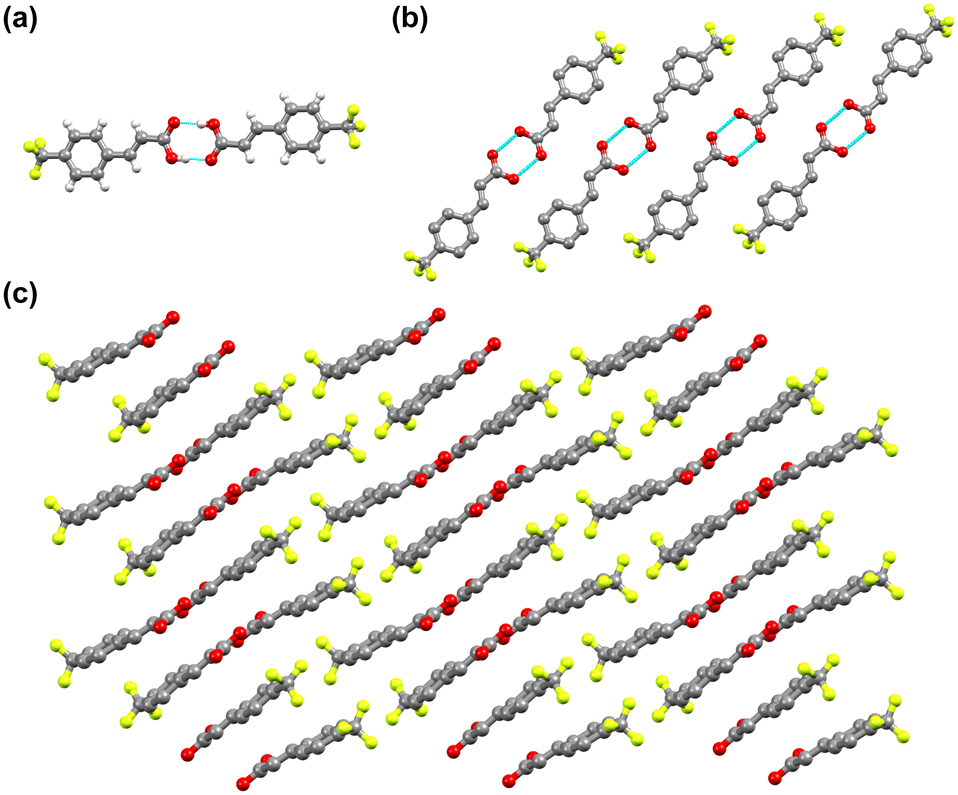 | ||
| Fig. 3 Crystal structure of 4-tfmca: (a) asymmetric unit, (b) one-dimensional string of dimers along the b axis, and (c) layered structure composed of the one-dimensional layers along the b axis. For b and c, hydrogen atoms are not displayed for clarity reasons. | ||
3-tfmca has a monoclinic crystal system and crystallizes in the P21/c space group. It has one molecule in its asymmetric unit (Fig. 4a, Table S1†), with the same disorder observed for the 4-tfmca molecule where the CF3 group exhibits two orientations. As in the case of 4-tfmca, the crystal of 3-tfmca is made up of carboxylic acid dimers (O(1)–H(1)⋯O(2), dO⋯O 2.632(3) Å, O–H⋯O 167(7)°) (Fig. 4b). However, the dimer units of 3-tfmca are stacked in two orientations almost perpendicular to each other. These dimers interact by weak out-of-plane C–H⋯O interactions, resulting in a zig-zag pattern (Fig. 4c). The closest distance between the centroids of the CC bonds of the neighboring molecules along the stack is 4.97 Å, which is well outside the topochemical limit for the photodimerization reaction. Thus, according to Schmidt's criteria, 3-tfmca is a γ type crystal.
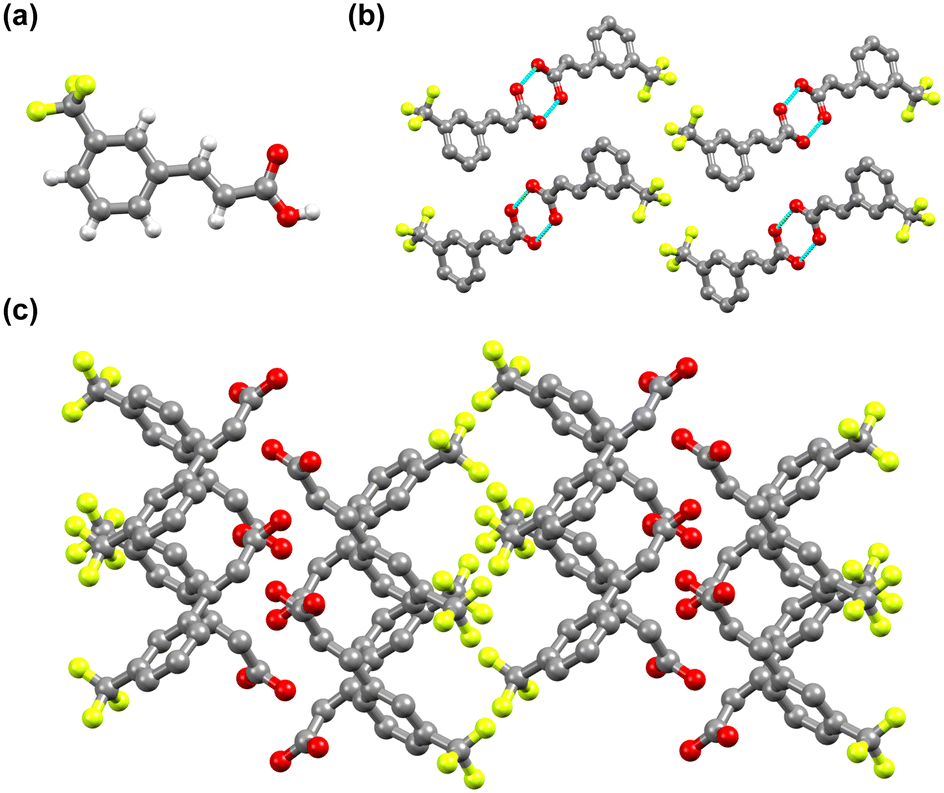 | ||
| Fig. 4 Crystal structure of 3-tfmca: (a) asymmetric unit, (b) two dimer orientations that are almost perpendicular to each other, and (c) zigzag structure. For b and c, the hydrogen atoms are not displayed for clarity reasons. | ||
The powder diffraction patterns of 3-tfmca and 4-tfmca are shown in Fig. S1.† Using the structure factors of the single crystal data, we calculated the powder diffraction patterns of the respective samples. Despite the slight mismatch in the peak positions, especially in the case of 4-tfmca, the calculated peak patterns agree well with the measured powder diffraction patterns showing that the powders of the respective samples exhibit the same crystal structures as the single crystals grown from the solution. The shifts in the peak positions can be attributed to minor variations in the unit cell dimensions.
It has been highlighted before that the KBr used to prepare FTIR pellets can potentially react with the embedded sample to form ionic co-crystals.43 To check whether this is the case for our samples, we prepared KBr pellets with a higher loading (around 30 wt%) and measured their powder patterns (Fig. 5). In the powder pattern, all the peaks of the KBr were visible, yet only a few low-intensity peaks can be seen from 4-tfmca and 3-tfmca. This is due to the lower concentration of 4-tfmca and 3-tfmca embedded in the pellet compared to their respective neat powders. A little effect of the applied pressure in the pellet leads to an upshift of all sample peaks in the pellet with respect to the neat powder. This can be explained by a slightly shrunk unit cell size in the potassium bromide pellet. Using jana2020, we could refine the unit cell size of the embedded 4-tfmca sample in the KBr matrix. We found a decrease in the a-axis by 0.06%, whereas the b-axis expanded by 0.17%. The largest change was observed in the c-axis, which shrunk by 0.28%. This may lead to a shrinking distance of the reactive molecules and may have an influence on the quantum yield of the photoreaction, which should be taken into consideration when using the KBr-pellet matrix to investigate the photoreaction of the embedded crystalline samples. Nevertheless, the peaks of 4-tfmca and 3-tfmca from the powder patterns indicate that incorporating samples within the KBr matrix does not involve profound structural rearrangement beyond that. This observation is supported by the close similarity between the FTIR spectra of 4-tfmca and 3-tfmca recorded in Nujol and KBr pellets over the CO stretching frequency region (Fig. S2†).
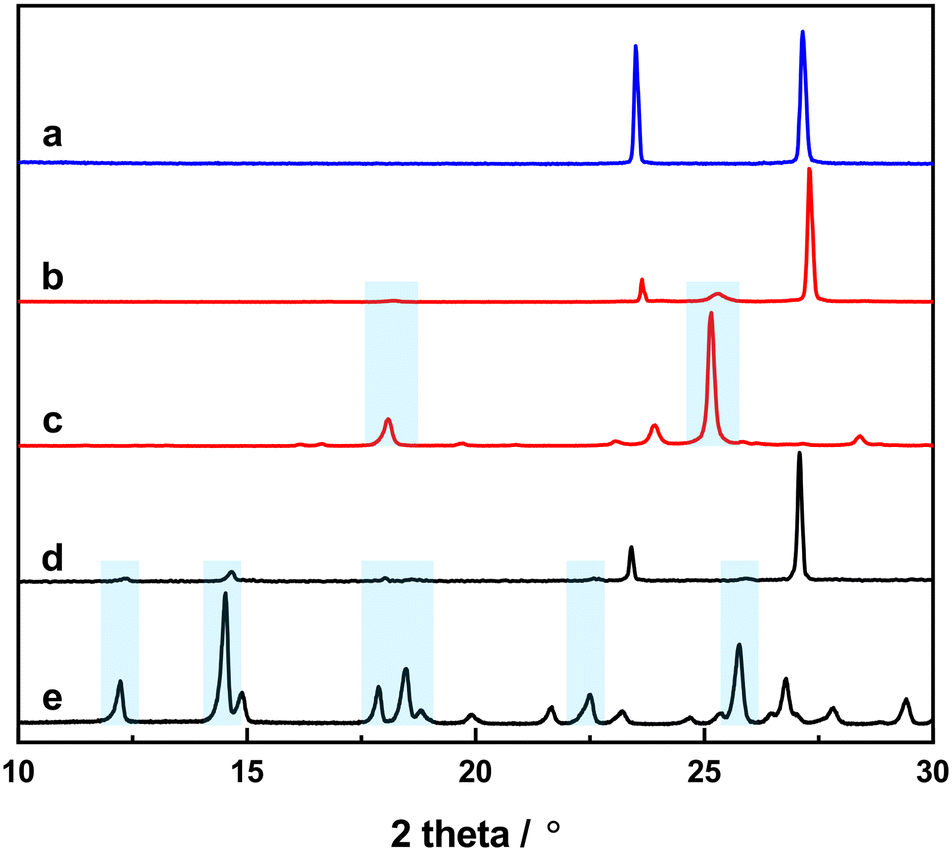 | ||
| Fig. 5 X-ray diffraction patterns of (a) an empty KBr pellet, (b) 4-tfmca embedded in a KBr pellet, (c) powder of 4-tfmca, (d) 3-tfmca embedded in a KBr pellet, and (e) powder of 3-tfmca. Cyan rectangles highlight the matching peaks between the powder samples and samples within the KBr pellet. | ||
Solid-state photoreaction
The crystal structure analysis revealed that 4-tfmca has the required geometry for the solid-state [2 + 2] photodimerization as the CC bonds of the neighboring molecules along the stacks are aligned parallel, with a center-to-center distance of 3.78 Å. On the other hand, for 3-tfmca the separation between the CC bonds of the neighboring molecules along the stacks is 4.97 Å, which is well beyond the topochemical limit. According to the topochemical postulate, only 4-tfmca should photodimerize to produce 4,4′-ditrifluoromethyl-β-truxinic acid. Nevertheless, the photoreactivity of the acids was examined by exposing their single crystals, previously used for structure determination, to UV radiation (8 hours for 4-tfmca and 4 hours for 3-tfmca). As expected, only 4-tfmca was able to undergo photodimerization, while 3-tfmca remained unchanged. The photodimer in the case of 4-tfmca was indeed 4,4′-ditrifluoromethyl-β-truxinic acid, and the occupancy was 28.8% (Fig. 6a). The UV irradiation already led to substantial turbidity of the crystal. A higher occupancy of the photoproduct could not be achieved since the crystal lattice was disrupted, leading to the destruction of the crystal. Crystallographic data of the partially photodimerized crystal are given in Table S1.† Each CF3 group of 4,4′-ditrifluoromethyl-β-truxinic acid is disordered between two orientations. Within the cyclobutane framework of 4,4′-ditrifluoromethyl-β-truxinic acid, the C–C–C angles slightly deviate from 90°, varying between 88(1)° and 91(1)° (Fig. 6b). Pairs of 4,4′-ditrifluoromethyl-β-truxinic acid molecules form carboxylic acid dimers through hydrogen bonding (Fig. 6c). Since hydrogen is a weak scatterer for X-ray, the hydrogen bonding pattern between the photoproduct molecules could not be refined unambiguously.
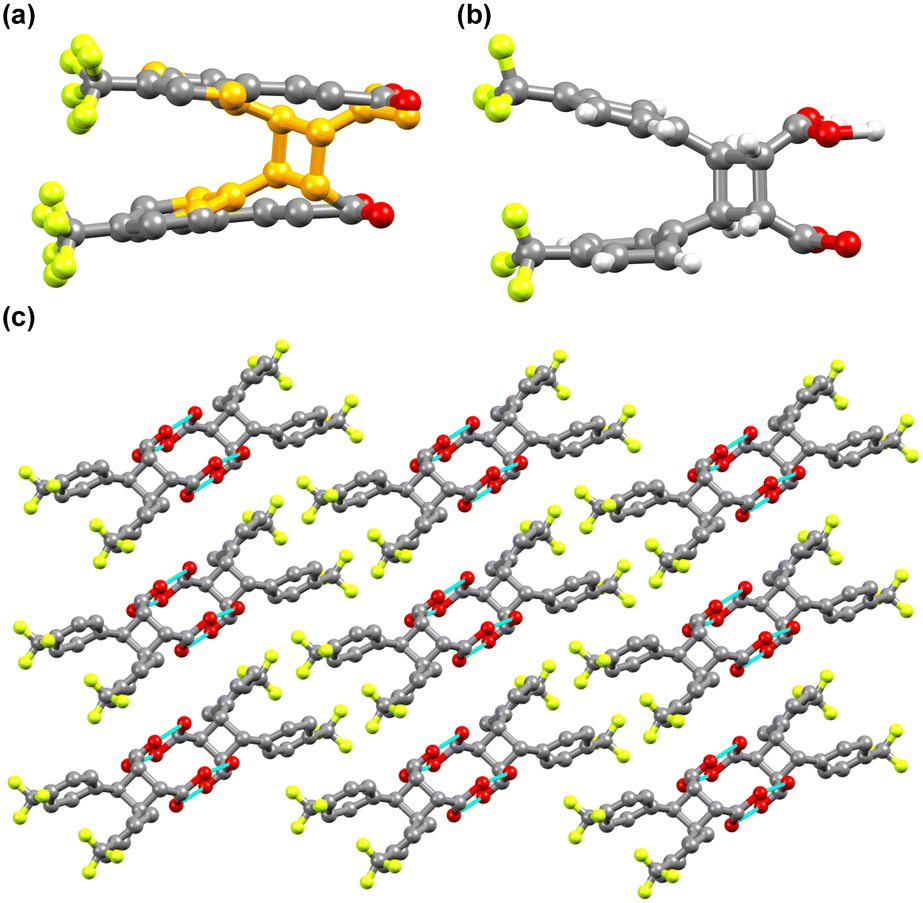 | ||
| Fig. 6 Crystal structure of the partially photodimerized 4-tfmca: (a) unit cell content after 8 hours of irradiation showing the formation of 4,4′-ditrifluoromethyl-β-truxinic acid (orange balls represent the carbon atoms of 4,4′-ditrifluoromethyl-β-truxinic acid); (b) molecular structure of 4,4′-ditrifluoromethyl-β-truxinic acid; (c) hydrogen bonded carboxylic acid dimer chain along the a axis of the unit cell. For a and c, the hydrogen atoms are not displayed for clarity reasons. | ||
We investigated the photodimerization of 4-tfmca with solution 1H NMR (Fig. 7 and S3†). Before the sample irradiation, the 1H NMR spectrum of the dissolved powder of the sample displayed the resonances of vinyl hydrogen atoms of the monomer at 7.76 ppm (1) and 6.68 ppm (2). After irradiating another aliquot of the sample with UV light and dissolving it in the same solvent, these vinyl hydrogen atom resonances disappeared, and new resonances that correspond to cyclobutane hydrogen atoms emerged at 4.38 ppm (3) and 3.92 ppm (4). This outcome indicates that, unlike the single crystal, in the powder case, the photodimer was obtained with a much higher conversion efficiency (nearly 100%).
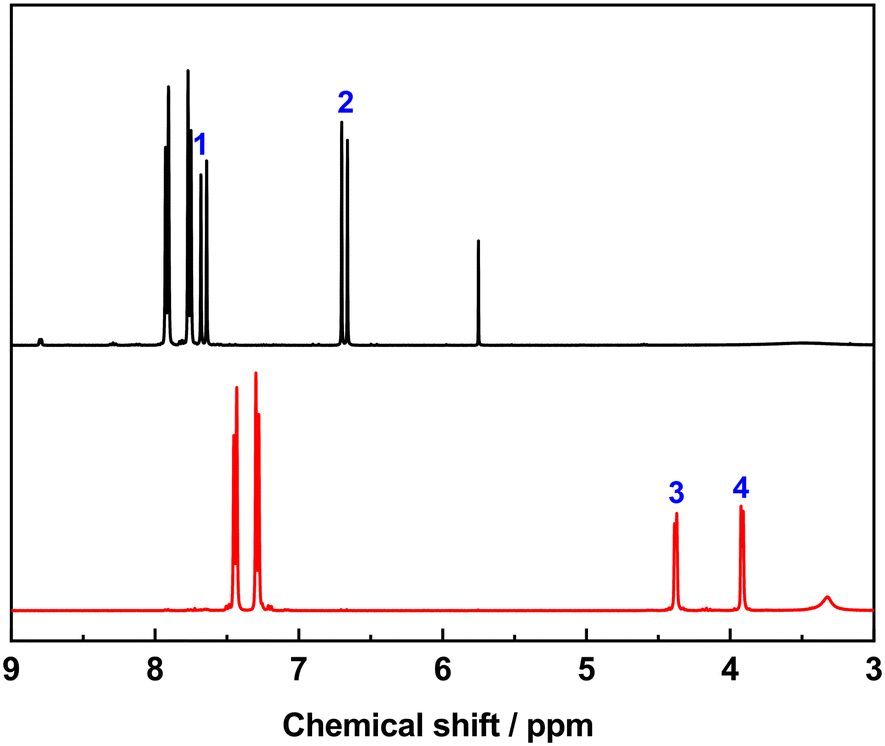 | ||
| Fig. 7 NMR spectra of 4-tfmca before (black line) and after (red line) irradiation. | ||
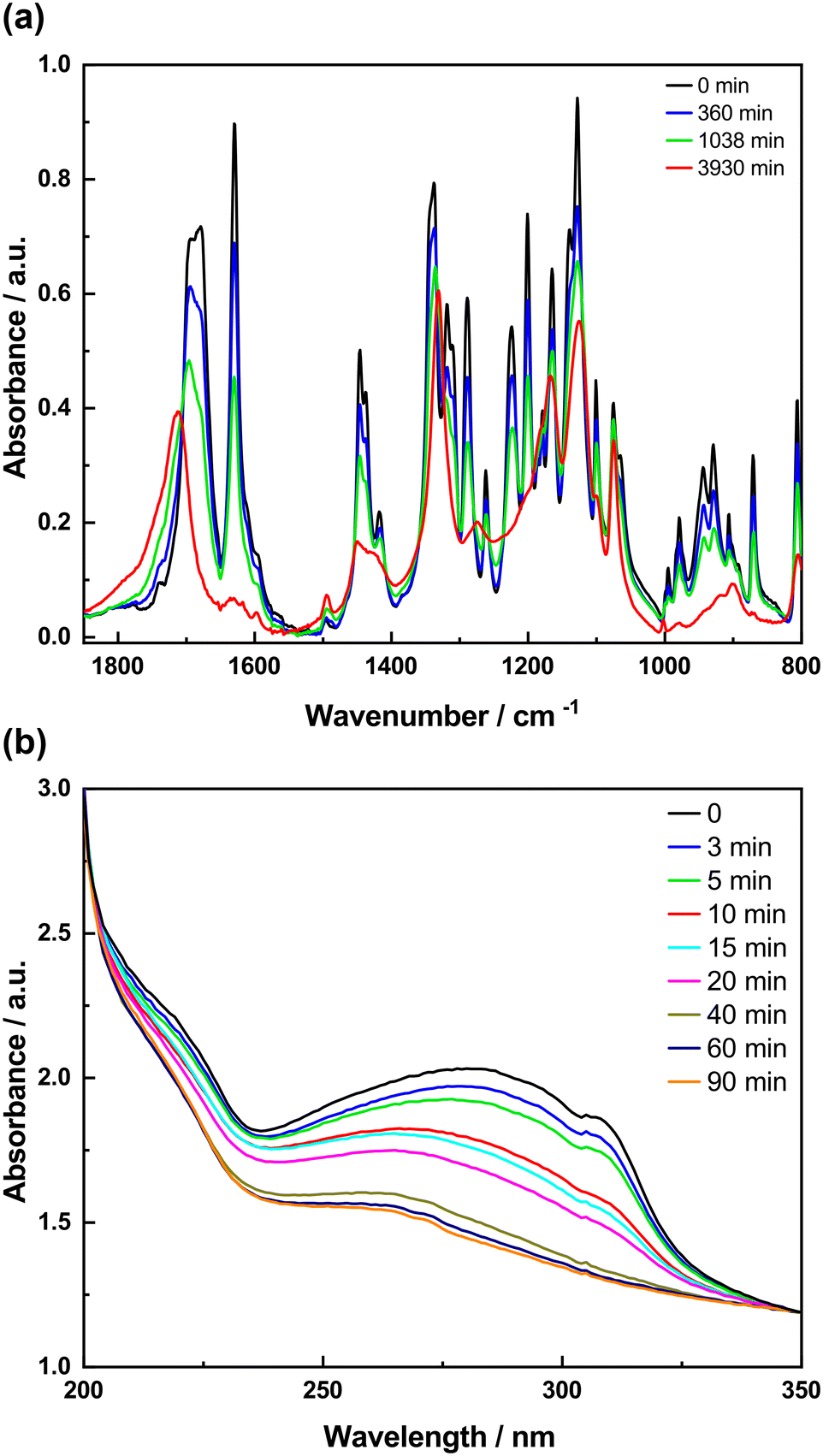
To study the photodimerization reaction in the KBr matrix, the absorption spectra in both the IR and UV regions of the samples prepared in KBr pellets were recorded as a function of irradiation time (Fig. 8 and 9). The FTIR spectra of 4-tfmca in the KBr pellet are presented in Fig. 8a, where two key changes confirm the photodimerization. First, the bands corresponding to νCC (1633 cm−1) and γC–H (990 cm−1) vibrations decay due to reaction-induced loss of the aliphatic CC bond. Second, the νCO vibration band shifts to a higher frequency from 1696 to 1734 cm−1. This change is attributed to the disruption of conjugation between the CO and CC bonds of the monomer as it transforms into the dimer.20,22,44–46 Furthermore, the UV analysis produces results (Fig. 8b) that agree with those obtained from the FTIR analysis. It is observed that the monomer absorption band centered at 272 nm decreases in intensity as irradiation time progresses and almost diminishes under prolonged irradiation. The occurrence of isosbestic points observed at around 1713 cm−1 in the FTIR spectra and around 228 nm in the UV spectra indicates a straightforward transformation from the monomer to the dimer without any by-products.
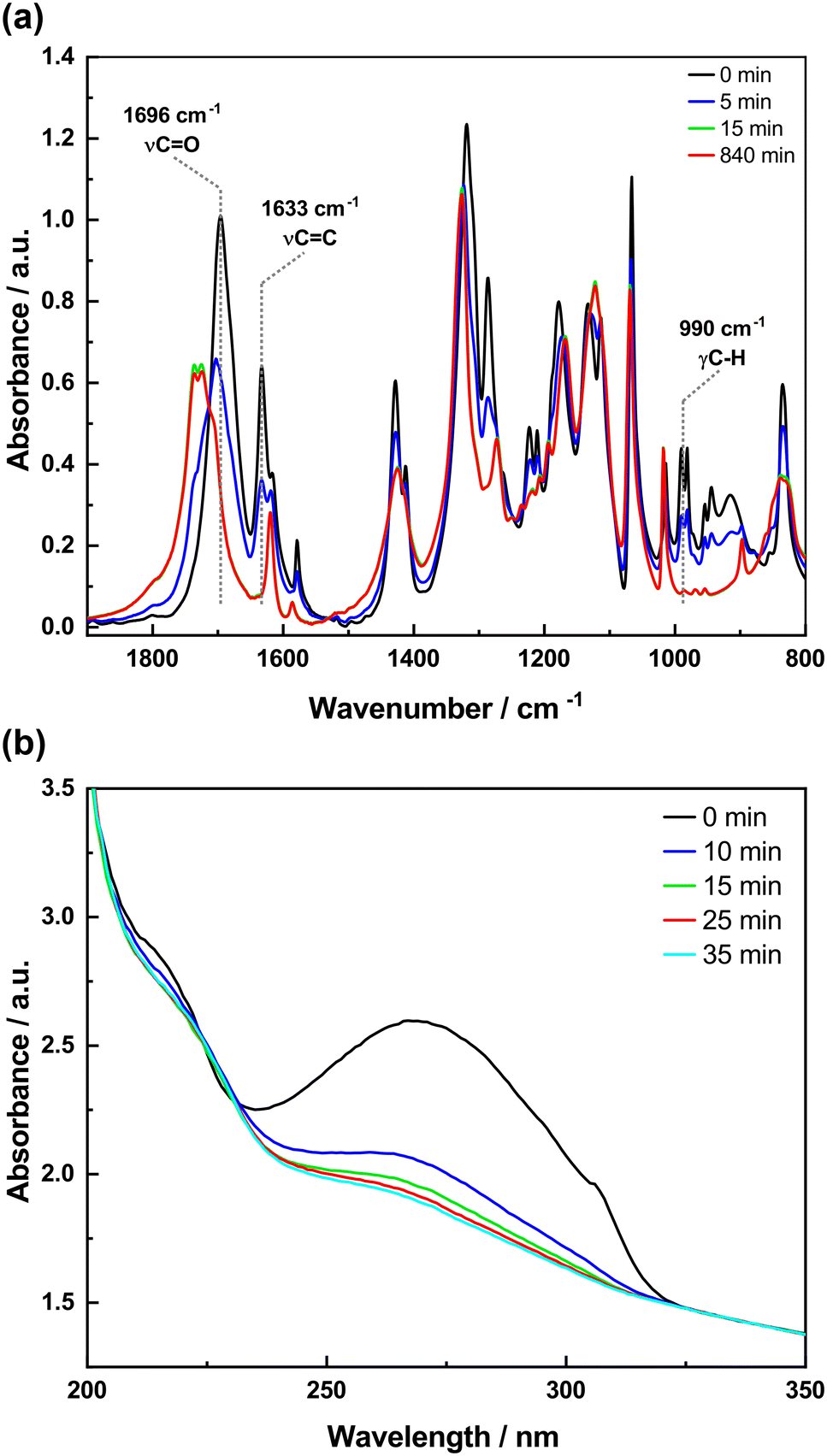 | ||
| Fig. 8 FTIR and UV spectra of 4-tfmca in KBr pellets as a function of irradiation time: (a) FTIR spectra and (b) UV spectra. | ||
 | ||
| Fig. 9 FTIR and UV spectra of 3-tfmca in KBr pellets as a function of irradiation time: (a) FTIR spectra and (b) UV spectra. | ||
The same spectroscopic investigations were conducted on 3-tfmca to investigate whether the photoreactivity observed for 4-tfmca is also evident in 3-tfmca within the KBr matrix. The FTIR spectra of 3-tfmca prepared in KBr pellets measured after certain irradiation times are shown in Fig. 9a. Surprisingly, despite the single crystal of 3-tfmca showing no signs of a photoreaction under UV radiation after 4 hours, significant changes were observed in the IR spectra indicating signs of a photoreaction. During the beginning of the irradiation (up to 1038 min), many vibrational bands in the IR spectra uniformly decay in intensity while only in view spectral regions intensity increases can be observed (above 1750 cm−1). This results in some isosbestic points suggesting a change between two species. Upon further irradiation, these isosbestic points disappear, while concurrently, the CO band shifts to a higher frequency indicating a change in the photoreaction dynamics which is possibly due to a more complex process such as photodecomposition, which could also be happening in parallel with the photoreaction. This explains the uniform decrease in intensity of the overall spectrum as the photodecomposition fragments might leave the pellet as a gas. The UV spectra of 3-tfmca embedded in KBr pellets recorded at different time intervals are summarized in Fig. 9b. The absorption band of the monomer centered at around 274 nm decreases in intensity as a function of irradiation time until complete consumption of the chromophores. It is worth mentioning that the difference in transformation rates between 4-tfmca and 3-tfmca is clear in both their IR and UV spectra.
Conclusion
In this communication, we have reported our findings on the structural and spectroscopic studies on the solid-state [2 + 2] photodimerization reaction of 4-tfmca and 3-tfmca. The crystal structure analysis revealed 4-tfmca to be a β type crystal, and 3-tfmca to be a γ type crystal. As expected, when exposed to UV radiation, the 4-tfmca crystal undergoes photodimerization to produce 4,4′-ditrifluoromethyl-β-truxinic acid, whereas the 3-tfmca crystal remains unchanged. During the photodimerization of 4-tfmca, the single crystal disintegrates into a polycrystalline material, which hinders the use of single-crystal X-ray diffraction when a conversion of 100% is to be achieved. But for a time-resolved investigation of the mechanism, one only wants to excite less than 10% of the molecules in the beam to avoid two-photon excitation effects. So, the fact that the crystal lattice decomposes in the single crystal case before a conversion efficiency of 100% is reached does not necessarily hinder its investigation with pump–probe techniques in the single crystal state. The hypothesis compatible with all our data so far would be that the photoproduct requires a different unit cell size, which puts strain on the crystal lattice when irradiating it with UV light. In the single crystal case, this results in decomposition of the crystal. In this context, it is noteworthy that we conclude from the solution NMR data that the conversion efficiency in the powder form of the 4-tfmca sample is nearly 100%, which is also supported by TGA measurements (Fig. S4†).The spectroscopic investigations on the crystals prepared in KBr pellets showed that the course of the photodimerization reaction can be conveniently followed using the KBr pellet method and optical spectroscopy. 4-tfmca could undergo photodimerization in the KBr matrix without any relevant fraction of side reactions, as evidenced by the isosbestic points observed in the IR and UV spectra. For comparison, 3-tfmca prepared in the KBr pellet was subjected to the same irradiation conditions. While showing no photoreaction in its single crystal, 3-tfmca displayed subtle photoreactivity under irradiation within the KBr matrix. The photoreaction of 3-tfmca is different from the faster photodimerization of 4-tfmca as it is slow and becomes much more complex with longer irradiation time. So, following the photodimerization reaction of 4-tfmca with time-resolved pump–probe techniques in the KBr matrix is also possible, but side reactions with the KBr matrix may have to be taken into account as has been reported by Braga et al.43 The improved index matching between the KBr matrix and the powder sample as compared to the irradiation of the neat powder sample allows the coupling of more light into the crystallites of the powder samples and helps to find this previously unknown photochemical reaction in the 3-tfmca compound.
Data availability
Crystallographic data for 4-tfmca, 3-tfmca, and partially photodimerized 4-tfmca have been deposited at the Cambridge Crystallographic Data Centre under the CCDC numbers 2331642, 2331099, and 2332753, respectively, and can be obtained from https://www.ccdc.cam.ac.uk/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Georg Stammler for helpful discussions and advice on the refinement of the single crystal structures. The authors thank Prof. Dr. Stephan Förster for funding a single crystal X-ray diffractometer for the outstation of the Forschungszentrum Jülich at the Heinz Meier-Leibnitz Zentrum in Garching. The authors would also like to thank Erdenedagva Amarzaya and Batbaatar Davaasuren for their help with the irradiation experiments and Sabreen Hammouda for her help with the powder diffraction measurements. This project was partially financially supported by the BMBF project 05K19PA3. The authors wish to thank the Mongolian Foundation for Science and Technology for supporting and partially funding project no 2020/38.Notes and references
- H. D. Roth, Angew. Chem., Int. Ed. Engl., 1989, 28, 1193–1207 CrossRef.
- B. B. Yagci, B. Munir, Y. Zorlu and Y. E. Türkmen, Synthesis, 2023, 55, 3777–3792 CrossRef CAS.
- B. B. Yagci, Y. Zorlu and Y. E. Tuürkmen, J. Org. Chem., 2021, 86, 13118–13128 CrossRef CAS PubMed.
- G. Ortega and A. Briceño, CrystEngComm, 2018, 20, 2932–2939 RSC.
- L. R. MacGillivray, J. L. Reid and J. A. Ripmeester, J. Am. Chem. Soc., 2000, 122, 7817–7818 CrossRef CAS.
- V. Ramamurthy and K. Venkatesan, Chem. Rev., 1987, 87, 433–481 CrossRef CAS.
- F. Toda, Acc. Chem. Res., 1995, 28, 480–486 CrossRef CAS.
- G. Kaupp, J. Phys. Org. Chem., 2008, 21, 630–643 CrossRef CAS.
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS PubMed.
- M. Cohen and G. Schmidt, J. Chem. Soc., 1964, 1996–2000 RSC.
- M. Cohen, G. Schmidt and F. Sonntag, J. Chem. Soc., 1964, 2000–2013 RSC.
- G. Schmidt, J. Chem. Soc., 1964, 2014–2021 RSC.
- G. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CrossRef CAS.
- M. D. Cohen, Angew. Chem., Int. Ed. Engl., 1975, 14, 386–393 CrossRef.
- M. Zahan, H. Sun, S. E. Hayes, H. Krautscheid, J. Haase and M. Bertmer, J. Phys. Chem. C, 2020, 124, 27614–27620 CrossRef CAS.
- S. d'Agostino, F. Spinelli, E. Boanini, D. Braga and F. Grepioni, Chem. Commun., 2016, 52, 1899–1902 RSC.
- V. Ramamurthy and J. Sivaguru, Chem. Rev., 2016, 116, 9914–9993 CrossRef CAS PubMed.
- T. Panda and P. Naumov, Cryst. Growth Des., 2018, 18, 2744–2749 CrossRef CAS.
- J. Davaasambuu, G. Busse and S. Techert, J. Phys. Chem. A, 2006, 110, 3261–3265 CrossRef CAS PubMed.
- S. L. Jenkins, M. J. Almond, S. D. Atkinson, M. G. Drew, P. Hollins, J. L. Mortimore and M. J. Tobin, J. Mol. Struct., 2006, 786, 220–226 CrossRef CAS.
- M. Bertmer, R. C. Nieuwendaal, A. B. Barnes and S. E. Hayes, J. Phys. Chem. B, 2006, 110, 6270–6273 CrossRef CAS PubMed.
- L. Pandolfi, A. Giunchi, T. Salzillo, A. Brillante, R. G. Della Valle, E. Venuti, F. Grepioni and S. D'Agostino, CrystEngComm, 2021, 23, 1352–1359 RSC.
- I. Abdelmoty, V. Buchholz, L. Di, C. Guo, K. Kowitz, V. Enkelmann, G. Wegner and B. M. Foxman, Cryst. Growth Des., 2005, 5, 2210–2217 CrossRef CAS.
- V. Enkelmann, G. Wegner, K. Novak and K. B. Wagener, J. Am. Chem. Soc., 1993, 115, 10390–10391 CrossRef CAS.
- S. D. Allen, M. J. Almond, J.-L. Bruneel, A. Gilbert, P. Hollins and J. Mascetti, Spectrochim. Acta, Part A, 2000, 56, 2423–2430 CrossRef PubMed.
- S. Chakrabarti, M. Gantait and T. Misra, Proc. - Indian Acad. Sci., Chem. Sci., 1990, 165–172 CrossRef CAS.
- M. Ghosh, S. Chakrabarti and T. Misra, J. Raman Spectrosc., 1998, 29, 263–267 CrossRef.
- G. Kaupp, Angew. Chem., Int. Ed. Engl., 1992, 31, 592–595 CrossRef.
- T. Hoyer, W. Tuszynski and C. Lienau, Chem. Phys. Lett., 2007, 443, 107–112 CrossRef CAS.
- G. Busse, T. Tschentscher, A. Plech, M. Wulff, B. Frederichs and S. Techert, Faraday Discuss., 2003, 122, 105–117 RSC.
- J. B. Benedict and P. Coppens, J. Phys. Chem. A, 2009, 113, 3116–3120 CrossRef CAS PubMed.
- C. Root, F. J. Lederer, T. E. Schrader, T. T. Herzog, T. Cordes, P. Gilch and M. Braun, Appl. Phys. A: Mater. Sci. Process., 2009, 96, 99–106 CrossRef CAS.
- W. J. Schreier, J. Kubon, N. Regner, K. Haiser, T. E. Schrader, W. Zinth, P. Clivio and P. Gilch, J. Am. Chem. Soc., 2009, 131, 5038–5039 CrossRef CAS PubMed.
- M. Braun, C. V. Korff Schmising, M. Kiel, N. Zhavoronkov, J. Dreyer, M. Bargheer, T. Elsaesser, C. Root, T. E. Schrader, P. Gilch, W. Zinth and M. Woerner, Phys. Rev. Lett., 2007, 98, 248301 CrossRef CAS PubMed.
- J. A. Howard and H. A. Sparkes, CrystEngComm, 2008, 10, 502–506 RSC.
- CrysAlisPRO, Rigaku Oxford Diffraction/Agilent Technologies, Yarnton, Oxfordshire, England, 2014, vol. 2014, pp. 1–2 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. Dolomanov, L. Bourhis, R. Gildea, J. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens and M. Towler, et al. , J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- V. Petíek, M. Duek and L. Palatinus, Z. Kristallogr. - Cryst. Mater., 2014, 229, 345–352 CrossRef.
- V. Petíek, L. Palatinus, J. Pláil and M. Duek, Z. Kristallogr. - Cryst. Mater., 2023, 238, 271–282 CrossRef.
- D. Braga, L. Maini, M. Polito and F. Grepioni, Chem. Commun., 2002, 2302–2303 RSC.
- Z. Wang, K. Randazzo, X. Hou, J. Simpson, J. Struppe, A. Ugrinov, B. Kastern, E. Wysocki and Q. R. Chu, Macromolecules, 2015, 48, 2894–2900 CrossRef CAS.
- S. D. Atkinson, M. J. Almond, G. A. Bowmaker, M. G. Drew, E. J. Feltham, P. Hollins, S. L. Jenkins and K. S. Wiltshire, J. Chem. Soc., Perkin Trans. 2, 2002, 1533–1537 RSC.
- S. D. Atkinson, M. J. Almond, S. J. Hibble, P. Hollins, S. L. Jenkins, M. J. Tobin and K. S. Wiltshire, Phys. Chem. Chem. Phys., 2004, 6, 4–6 RSC.
Footnote |
| † CCDC 2331099, 2331642 and 2332753. For crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00205a |
| This journal is © The Royal Society of Chemistry 2024 |