
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBilirubin and its crystal forms†
Matthew L.
Bracken
 ,
Manuel A.
Fernandes
* and
Sadhna
Mathura
*
,
Manuel A.
Fernandes
* and
Sadhna
Mathura
*
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, PO Wits 2050, Johannesburg, South Africa. E-mail: Manuel.Fernandes@wits.ac.za; Sadhna.Mathura@wits.ac.za; Fax: +27 11 7176749; Tel: +27 11 7176723/81
First published on 26th March 2024
Abstract
Bilirubin IXα is the major product of the degradation of heme from red blood cells and is of biological and medical relevance. It is insoluble in aqueous media leading to crystals in the gall bladder and urine indicating liver dysfunction. The degradation process leads to enantiomers – the M- and P-helical conformers – which are mirror versions of each other. The molecule is rigid because of strong intramolecular hydrogen bonding. We have grown two forms of bilirubin and analyzed another that was published in 1980. All forms crystallize in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Form I was first published in 1978, but the original authors mentioned that the molecules showed unresolved disorder. We have regrown this form and resolved the disorder. There are two molecular sites in the asymmetric unit in Form I, and both sites are a disordered mixture of M- and P-helical conformers, with no voids in the structure. Form II is a solvate (void space: 5% of the unit cell volume) and is a new form. It contains two molecular sites in the asymmetric unit with the same helical conformer in both sites, with an ordered molecule in one site and rotationally disordered molecules in the other site. Form III is also a solvate (void space: 25% of the unit cell volume) and contains methanol and chloroform. There is no strong intermolecular hydrogen bonding in any of the three forms, and the molecules aggregate through weak interactions. Despite this, CLP-PIXEL calculations showed that the three most stable molecule⋯molecule arrangements between the three forms are geometrically consistent.
. Form I was first published in 1978, but the original authors mentioned that the molecules showed unresolved disorder. We have regrown this form and resolved the disorder. There are two molecular sites in the asymmetric unit in Form I, and both sites are a disordered mixture of M- and P-helical conformers, with no voids in the structure. Form II is a solvate (void space: 5% of the unit cell volume) and is a new form. It contains two molecular sites in the asymmetric unit with the same helical conformer in both sites, with an ordered molecule in one site and rotationally disordered molecules in the other site. Form III is also a solvate (void space: 25% of the unit cell volume) and contains methanol and chloroform. There is no strong intermolecular hydrogen bonding in any of the three forms, and the molecules aggregate through weak interactions. Despite this, CLP-PIXEL calculations showed that the three most stable molecule⋯molecule arrangements between the three forms are geometrically consistent.
1. Introduction
Bilirubin IXα (henceforth simply “bilirubin”) is a structurally complex and functionally enigmatic biological chromophore, generated by the systematic catabolism of heme in senescent erythrocytes.1 The heme moiety, an iron-containing macrocyclic tetrapyrrole, arguably evolved to harness the oxidative properties of iron to support life in an oxidizing environment.2 Coupled to the appropriate apoprotein, heme executes a variety of life-essential physiological functions from respiration to electron transport.3 Paradoxically, free heme is cytotoxic at high concentrations, generating reactive oxygen species that lead to irreversible cell damage.4 To mitigate against this cytotoxicity, natural systems carefully regulate the concentrations of free heme. In mammals for example, this is achieved by systematically disassembling the macrocycle in ageing red blood cells and eliminating the metabolites.A typical mammalian heme catabolic pathway is a multistep process. The general process begins with opening of the macrocyclic tetrapyrrole by cleavage at one of the four methene bridges (α, β, γ or δ) initiated by heme oxygenase (EC 1.14.99.3) as shown in Scheme 1a. This step releases carbon monoxide, liberates the metal ion and, leaves behind the corresponding hydrophilic green-yellow linear tetrapyrrolic biliverdin isomers.5 In the case of cleavage at the α-position, biliverdin IXα is rapidly and enzymatically reduced by biliverdin reductase-A (EC 1.3.1.24) in the liver, forming the hydrophobic orange-yellow tetrapyrrole, bilirubin IXα (Scheme 1b). This biosynthetic form of bilirubin, which may be toxic at elevated concentrations, is subsequently conjugated to glucuronic acid by uridine 5′-diphosphate glucuronosyl transferase (EC 2.4.1.17) resulting in the hydrophilic bilirubin diglucuronide which can then be eliminated from the body. The concentration of free heme is thus regulated, and cytotoxicity is mitigated through the elimination of the heme metabolites.6,7
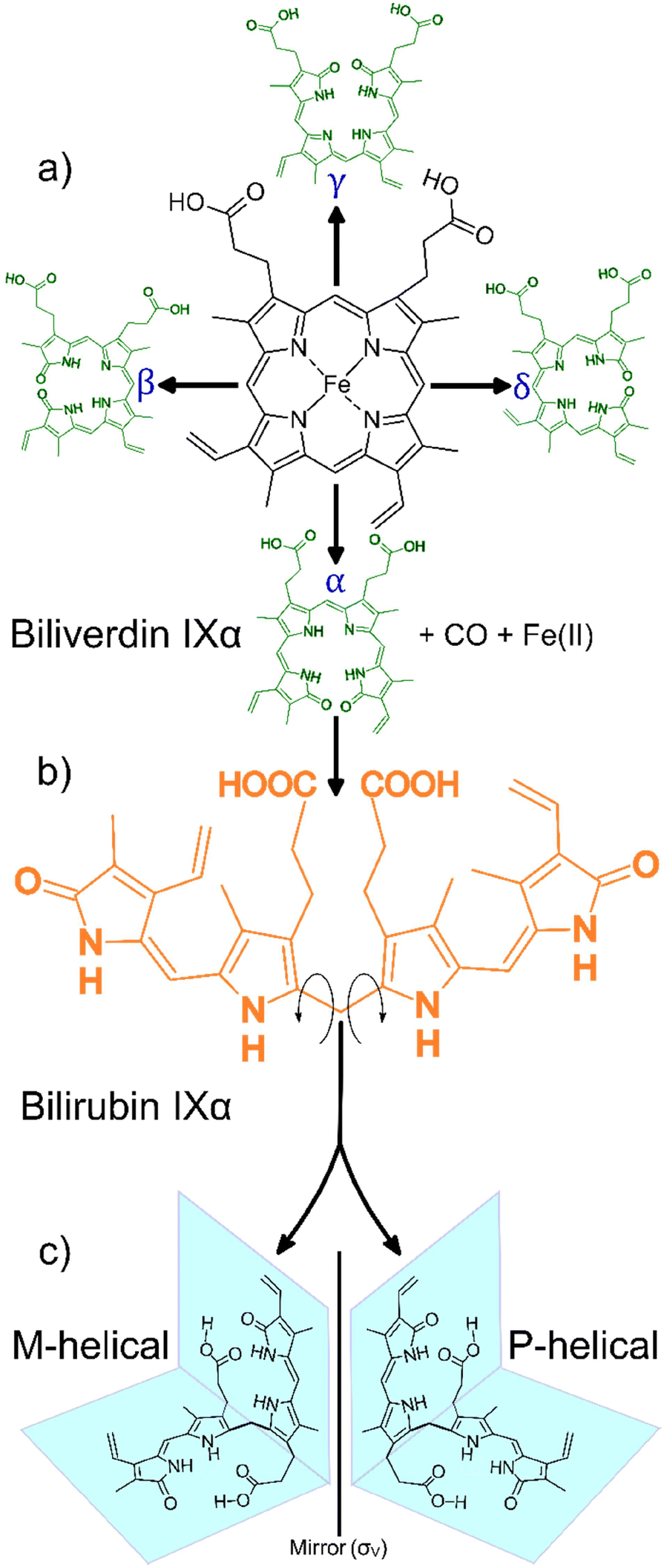 | ||
| Scheme 1 General heme catabolism: (a) heme may be cleaved at one of the four positions on the porphyrin (α, β, γ, δ) to generate one of the four biliverdin isomers (adapted from Takemoto et al., 2019);7 (b) each biliverdin isomer can then generate the corresponding bilirubin isomer (only the bilirubin IXα isomer is shown); (c) bilirubin IXα exhibits two forms, M-helical and P-helical, which are mirror versions of each other, depending on the direction the molecule folds after heme cleavage (adapted from Zunszain et al., 2008).6 | ||
Heme metabolites biliverdin and bilirubin are structurally complex molecules. In principle, one of the four biliverdin isomers may be produced depending on the heme cleavage site (α, β, γ or δ), leading to the corresponding bilirubin isomers.5,8 Interestingly, in mammals there appears to be a strong regioselective drive towards cleavage at the α-position (catalysed by the canonical heme oxygenase-1 isoform). This cleavage yields the most abundant biliverdin isomer in mammals, biliverdin IXα, which in turn produces the only lipophilic bilirubin isomer, bilirubin IXα.7
The lipophilicity of bilirubin IXα is another intriguing feature. Containing several polar moieties, bilirubin IXα is expected to exhibit considerable hydrophilicity at physiological pH. However, solid state data reveal that the molecule assumes a compact “ridge-tile” conformation where the dipyrrinones are in proximity to the opposite propionic groups. This proximity allows for the formation of a robust intramolecular hydrogen bonding network thereby diminishing bilirubin's interaction with water. Further, this rather contorted ensemble exhibits two enantiomeric forms (P or M helicity, Scheme 1c) and the hydrophobicity is still conserved in both forms.6 The aforementioned structural complexities (i.e. the apparent isomeric regioselectivity and drive towards lipophilicity) probe the functional significance of bilirubin, garnering renewed interest in its structure.9,10 Bilirubin is insoluble in aqueous media and is precipitated from the biliary tract as pigmented gall stones. Crystals also form in the urine indicating poor hepatic function, as bilirubin is usually processed by the liver before being excreted. The immature livers of neonatal children may result in hyperbilirubinemia, a condition where crystalline bilirubin accumulates in vivo and can result in neurotoxicity and death.11 Therefore, the crystallization and crystal forms of bilirubin are of biological and medical relevance.
2. Experimental
2.1 Crystallization
Bilirubin was purchased from Sigma Aldrich and used without further purification. Crystals were obtained by vapour diffusion of diethyl ether, hexane, and ethanol into chloroform. Crystals were also obtained by vapour diffusion of water or ethanol into dimethylsulfoxide, as well as diffusion of diethyl ether into pyridine. All crystallizations were kept in the dark, but the crystals obtained this way were small, flawed, and poorly diffracting. Bonnett's original method12 for crystallizing bilirubin was thus adapted as follows: a turbid solution of bilirubin in pyridine was created, and then one or two drops of 25% aqueous ammonia were added to afford a clear solution. Vapour diffusion of diethyl ether into this solution was allowed to occur in the dark and under ambient conditions. Concentrated solutions of bilirubin afforded X-ray quality crystals in 1–2 days (Form II) whilst dilute solutions afforded quality crystals in 2–4 days (Form I). The crystals are shown in Fig. 1. | ||
| Fig. 1 Crystals of Form I and Form II of bilirubin. | ||
2.2 X-ray data collection and refinement
Intensity data for compounds of Forms I and II were collected on a Bruker D8 Venture Bio PHOTON III 28 pixel array area detector (208 ×128 mm2) diffractometer with a Mo Kα IμS DIAMOND source (50 kV, 1.4 mA) at 173 K. The collection method involved ω- and φ-scans, and 1536 × 1024 bit data frames. The unit cell and full data set were collected using APEX4.13 SAINT was used to integrate the data, and SADABS was used to make empirical absorption corrections and scale the data. Space group assignments were made using XPREP. Using OLEX2,14 the crystal structures were solved with the ShelXT15 structure solution program using intrinsic phasing and refined with the ShelXL16 refinement program using least squares minimization. Non-hydrogen atoms were first refined isotropically followed by anisotropic refinement by full matrix least-squares calculations based on F2. All H atoms on bilirubin were positioned geometrically and allowed to ride on their parent atoms, with atom—H bond lengths of 0.95 Å (CH), 0.95 Å (CH2), and 0.88 Å (NH) and isotropic displacement parameters set to 1.2 times Ueq of the parent atom, or 0.98 Å (CH3) and 0.84 Å (OH) and isotropic displacement parameters set to 1.5 times Ueq of the parent atom. Molecules in both Forms I and II are disordered. To solve the disorder, SADI, DFIX, RIGU, and SIMU restraints were used for Form I, where molecular sites were a mixture of M- and P-helical conformations, and SAME, DFIX, RIGU, and SIMU restraints were used for Form II, where molecular sites were a 180° rotational disordered mixture of only one conformer. The disorder of both forms is discussed below. Crystal data and structure refinement details are summarized in Table 1.| Crystals | Form I | Form II | Form III |
|---|---|---|---|
| Moiety formula | C33H36N4O6 | C33H36N4O6 + solvent | C33H36N4O6·CHCl3·CH3OH |
| Temperature/K | 173 | 173 | 223 |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group |
P |
P |
P |
| a/Å | 11.6630(7) | 11.8077(10) | 9.58(4) |
| b/Å | 15.3457(8) | 15.0220(12) | 11.96(4) |
| c/Å | 17.2204(10) | 19.3471(17) | 15.60(5) |
| α/° | 73.515(2) | 98.973(3) | 93.3(1) |
| β/° | 81.274(3) | 104.421(3) | 99.9(1) |
| γ/° | 82.823(3) | 111.029(3) | 84.8(1) |
| Volume/Å3 | 2910.2(3) | 2985.8(4) | 1750 |
| Z | 4 | 4 | 2 |
| ρ calc g cm−3 | 1.334 | 1.301 | — |
| μ mm−1 | 0.093 | 0.090 | — |
| F(000) | 1240.0 | 1240.0 | — |
| Crystal size/mm3 | 0.225 × 0.156 × 0.154 | 0.151 × 0.144 × 0.054 | — |
| Radiation | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | CuKα (λ = 1.54056) |
| 2θ range for data collection/° | 3.188 to 49.998 | 3.242 to 49.996 | — |
| Index ranges | −13 ≤ h ≤ 13, −18 ≤ k ≤ 18, −20 ≤ l ≤ 20 | −14 ≤ h ≤ 14, −17 ≤ k ≤ 17, −22 ≤ l ≤ 22 | — |
| Reflections collected | 127![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 211 211 |
161376 |
2140 |
| Independent reflections | 10254 [Rint = 0.0904, Rsigma = 0.0594] |
10506 [Rint = 0.2197, Rsigma = 0.0903] |
2140 |
| Data/restraints/parameters | 10254/1355/1519 |
10506/180/808 |
— |
| Goodness-of-fit on F2 | 1.086 | 1.029 | — |
| Final R indices [I ≥ 2σ(I)] | R 1 = 0.0851, wR2 = 0.2279 | R 1 = 0.0948, wR2 = 0.2240 | R = 0.11 [I ≥ 4σ(I)] |
| Final R indices [all data] | R 1 = 0.1243, wR2 = 0.2679 | R 1 = 0.1867, wR2 = 0.2974 | — |
| Largest diff. peak/hole/e Å−3 | 0.47/−0.27 | 0.60/−0.58 | — |
| CCDC number | 2331350 | 2331351 | — |
2.3 Calculations
Optimization calculations were performed using Gaussian-16 Rev. C.01 (ref. 18) at the B3LYP/6-31G* level of theory and utilizing the GD3BJ19 empirical dispersion correction. All optimizations were carried out in vacuo and GaussView 6.0.16 (ref. 20) was used to prepare input files and visualize output data. All jobs finished with normal termination.Molecule⋯molecule interaction energies were calculated using CLP-PIXEL21,22 as incorporated in the April 2014 version of the CLP23 package. The calculations were carried out as prescribed within the CLP manual. The calculation was initialized by geometrically normalizing bonds involving H to neutron distances within CLP, followed by the calculation of an ab initio MP2/6-31G** molecular electron density using Gaussian-09 Rev. E.01. This molecular electron density description was then used as an input with CLP-PIXEL to carry out the calculations. Only the molecule⋯molecule interactions between dominant conformations (occupancy more than 80%) were calculated for Form I. However, for Form II, where there is around 50% disorder for the second molecule, calculations on both conformers were carried out. The results of these calculations are included in the ESI.†
Simulated powder patterns and structural voids were calculated using Mercury 2023.3.0.24 The parameters used for the simulated powder patterns were: λ = 1.54056 Å (CuKα), 2 theta range = 5–50° with a 0.02° step size, and FWHM (2 theta) = 0.1°. Voids were calculated using the contact surface with the following parameters: spherical probe = 1.2 Å and approximate grid spacing = 0.3 Å.
3. Results and discussion
The crystal structure of Form I of bilirubin was published in 1978 by Bonnet, Davies, Hursthouse, and Sheldrick.12 In that study, the structure was solved by direct methods and refined using data sets that were collected on a diffractometer in combination with data that were collected using a Weisenberg camera. Bonnet et al. commented about the disorder in the crystal solution, but at that time, the severe disorder was difficult to resolve. Up until today, this remained the only structure of bilirubin to be deposited into the CSD (BILRUB10). Thus, we grew crystals to resolve the disorder and address the lack of high-quality structural data for this medically relevant molecule. The reported crystallographic data for Form I are as follows: space group P, a = 19.439 Å, b = 11.707 Å, c = 15.5 Å, α = 97.19°, β = 100.22°, γ = 118.2°, V = 2969.21 Å3, and Z = 4. Initially, crystals were grown from chloroform and dimethyl sulfoxide solutions, as well as from pyridine in the absence of ammonia. These methods resulted in crystals that had diffraction patterns containing both sharp and diffuse reflections. The sharp reflections were associated with triclinic cell dimensions, where a = 9.720 Å – half the final unit cell – a finding that was also discussed in the paper of Bonnet et al. These crystals were so poorly diffracting that the a-axis was always found to be half of the true unit cell. The paucity and poor intensity of these data sets did not warrant further structure refinement. Using Bonnett's crystal growing conditions (vapour diffusion of diethyl ether into an ammoniacal pyridine solution), we successfully crystallized the original Form I from dilute bilirubin solutions. Additionally, Form II was crystallized from concentrated solutions of bilirubin and found to be a solvate.
On a modern diffractometer, the unit cell parameters for Form I are different, a = 11.6630(7) Å, b = 15.3457(8) Å, c = 17.2204(10) Å, α = 73.515(2)°, β = 81.274(3)°, γ = 82.823(3)°, and V = 2910.2(3) Å3. However, Bonnett's original cell can be transformed into the present unit cell using the following matrix: 0 −1 0/0 0 1/−1 −1 0. The calculated powder patterns using crystallographic coordinates are shown in Fig. 2 and indicate that the crystal structures are identical.
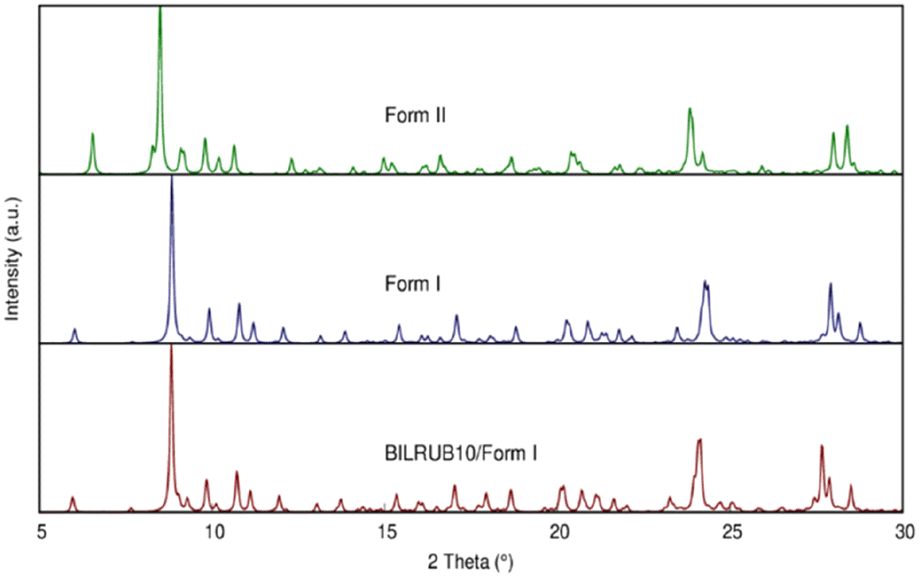 | ||
| Fig. 2 Simulated Cu X-ray powder patterns using the crystal coordinates for Bonnett et al. (1978)12 original data (BILRUB10), Form I, and Form II. | ||
The asymmetric unit for Form I has two disordered molecular sites, each occupied by both the M- and P-helical conformational enantiomers. In the asymmetric unit, the P-helical enantiomer occupies both molecular sites A and B, as shown in Fig. 3, with partial occupancies of 0.828(3) and 0.805(3), respectively, whilst the M-helical enantiomer has partial occupancies of 0.172(3) and 0.195(3). All hydrogen bonding is intramolecular (Table 2), allowing either enantiomer to occupy both sites without causing close contacts that disrupt the crystal packing arrangement. Due to this disordered packing, bilirubin crystals inherently exhibit weak reflections and do not scatter to high resolution. In Bonnett's study, the best data sets were obtained from a crystal that was annealed over several weeks. Attempts were made to improve our current data sets by annealing the crystals at 85 °C for two days and ambiently over two weeks, followed by repeating the data collection. However, heating the samples had no effect on the diffraction quality and repeated exposure to low temperature permanently damaged the crystals.
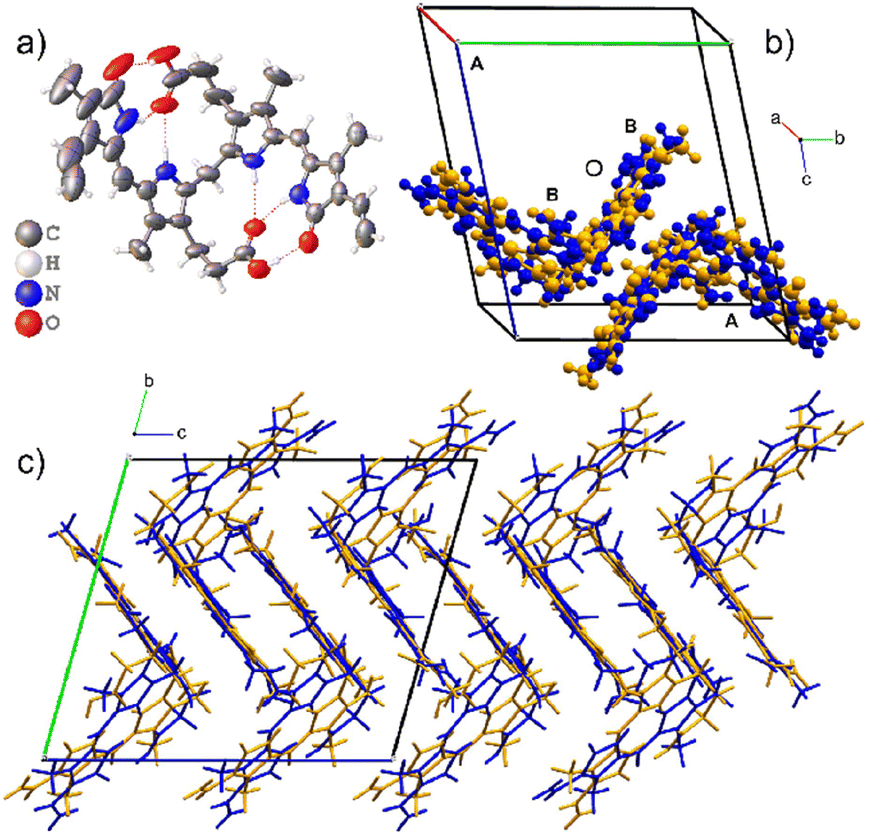 | ||
| Fig. 3 The crystal structure of Form I. The asymmetric unit is composed of two disordered molecular sites. a) ORTEP diagram of the major component of one molecular site and the intramolecular hydrogen bonding. b) The unit cell with the disordered asymmetric unit with P-enantiomers coloured blue and M-enantiomers as orange. c) The zig-zag packing of Form I. | ||
| Crystal | O–H⋯O/N–H⋯O | D–H | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|---|---|
| Form I | O3–H3⋯O1 | 0.84 | 1.80 | 2.620(6) | 166.0 |
| O6–H6⋯O4 | 0.84 | 1.78 | 2.604(6) | 167.5 | |
| N1–H1⋯O2 | 0.88 | 1.95 | 2.800(6) | 162.1 | |
| N2–H2⋯O2 | 0.88 | 2.00 | 2.851(5) | 161.4 | |
| N3–H3A⋯O5 | 0.88 | 2.00 | 2.852(5) | 161.9 | |
| N4–H4⋯O5 | 0.88 | 1.93 | 2.775(6) | 160.0 | |
| O9–H9⋯O7 | 0.84 | 1.86 | 2.641(6) | 154.9 | |
| O12–H12⋯O10 | 0.84 | 1.78 | 2.582(13) | 158.7 | |
| N5–H5⋯O8 | 0.88 | 1.95 | 2.800(7) | 162.8 | |
| N6–H6B⋯O8 | 0.88 | 1.99 | 2.839(6) | 161.2 | |
| N7–H7⋯O11 | 0.88 | 2.03 | 2.877(7) | 160.6 | |
| N8–H8B⋯O11 | 0.88 | 2.00 | 2.823(12) | 155.0 | |
| Form II | O3–H3⋯O1 | 0.84 | 1.79 | 2.614(6) | 168.4 |
| O6–H6A⋯O2 | 0.84 | 1.80 | 2.629(7) | 166.7 | |
| N1–H1⋯O2 | 0.88 | 1.96 | 2.809(7) | 162.9 | |
| N2–H2⋯O2 | 0.88 | 1.99 | 2.842(6) | 161.4 | |
| N3–H3A⋯O5 | 0.88 | 2.00 | 2.857(6) | 162.6 | |
| N4–H4⋯O5 | 0.88 | 1.96 | 2.797(6) | 158.6 | |
| O9–H9⋯O7 | 0.84 | 1.79 | 2.601(6) | 161.5 | |
| O12–H12⋯O10 | 0.84 | 1.80 | 2.623(6) | 166.9 | |
| N5–H5⋯O8 | 0.88 | 1.97 | 2.827(6) | 162.7 | |
| N6–H6⋯O8 | 0.88 | 2.06 | 2.888(6) | 155.5 | |
| N7–H7⋯O11 | 0.88 | 1.98 | 2.819(6) | 159.8 | |
| N8–H8⋯O11 | 0.88 | 1.93 | 2.784(6) | 161.7 | |
Form II also crystallizes in the space group P with two molecules in the asymmetric unit as shown in Fig. 4. Both molecules are M-helical enantiomers where one molecular site is ordered, and the second molecular site is occupied by the M- helical enantiomer in two orientations related by 180° rotation with partial occupancies of 0.543(9) and 0.457(9). The hydrogen bonding is intramolecular (Table 2), allowing facile disorder as dispersion forces are left to facilitate packing.
 | ||
| Fig. 4 The crystal structure of Form II which is a solvate. The asymmetric unit is composed of two molecular sites, one ordered and one disordered. a) ORTEP diagram of the ordered molecule. b) The unit cell of Form II with the ordered molecular site coloured by element. The disordered molecular site is occupied by a disordered M-helical enantiomer of bilirubin which is shown in orange. The two-component disorder is related by 180° rotation. Both sites are occupied by the same helical enantiomer. c) The crystal packing of the solvate contains voids that exist on the corners of the unit cell. | ||
Form II is a solvate with voids occupying a volume of 147 Å3 at the corners of the unit cell, which is 4.9% of the cell volume. Le Bas et al.17 published another form of bilirubin, Form III, which is presented in Fig. 5. It also crystallizes in the space group P (Table 1); however, its data were never deposited into the Cambridge Structural Database25 but we used the non-hydrogen atom coordinates from the original paper to plot the diagrams. The bridging methylene carbon in the original paper is missing, and we needed to add it to the original coordinates by using the crystal structures for Forms I and II as a reference, then added hydrogens geometrically as is usually done with X-ray crystal structures. Form III is a solvate, and in addition to the bilirubin molecule, there are disordered methanol and chloroform molecules crystallizing in channels running along the c-axis in the structure. By deleting the solvent atoms, the void space accounts for 428 Å3 or 24.5% of the unit cell volume, and this is likely the most soluble form of the bile pigment. The bilirubin in this structure is ordered and has the same conformation and intramolecular hydrogen bonding as the other forms.
 | ||
| Fig. 5 Form III is a solvated structure with one ordered bilirubin molecule and disordered chloroform and methanol in solvent channels that run along the c-axis. These voids continue as infinite accessible channels through the crystal, and it is likely that Form III is the most water soluble. | ||
The conformation of bilirubin has been described by Bonnett et al. as a “ridge tile” with intramolecular hydrogen bonding creating a rigid molecule with very little conformational flexibility. In Form II, the fold angle between the two halves of the ordered molecule is 86.1°, and the fold angles of the other molecules in both forms are similar. The conformation of the “ridge tile” was calculated at the B3LYP/GD3BJ/6-31G* level of theory and overlaid by least squares using all the non-hydrogen atoms on the ordered molecule in Form II and one of the molecules in Form I, resulting in a good fit with a correlation of 0.2 Å as shown in Fig. 6.
 | ||
| Fig. 6 DFT calculated structure overlaid with Form I and Form II. The bilirubin conformation was optimized at the B3LYP/GD3BJ/6-31G* level of theory and shows RMSD correlations of 0.30 and 0.26 Å when overlaid with Forms I and II, respectively. The molecular overlay of Forms I and II produces a correlation of 0.20 Å. | ||
Bilirubin is a rigid molecule due to strong intramolecular hydrogen bonding. However, there are no strong hydrogen bonds between molecules. Instead, the molecules aggregate through weak interactions, particularly dispersion forces. Despite this, the crystal packing in all three forms resembles each other. The results of CLP-PIXEL calculation for the three forms (with solvents removed) are depicted on the distance between centres of mass of the interacting molecules vs. energy plot, also known as an R/E plot,26 in Fig. 7. This plot displays the stabilization energy between molecules as a function of distance in the crystal structure. The closer the distance between molecules, the more stable they are, and vice versa. With a greater distance between the molecules, their interaction tends towards 0 kJ mol−1 at 15 Å and beyond.
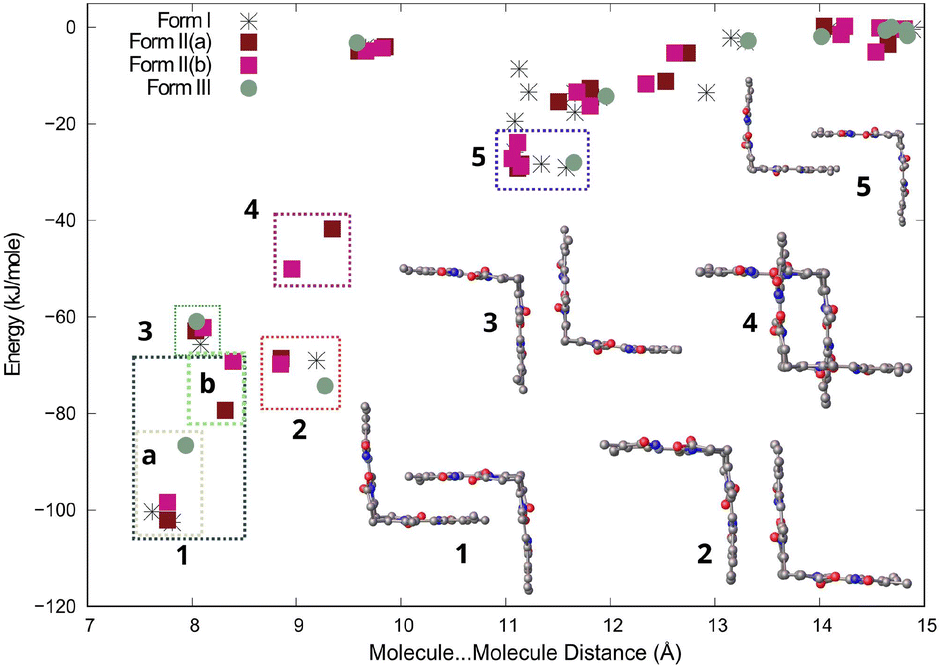 | ||
| Fig. 7 Distance vs. energy plot for molecule–molecule interactions in all three forms of bilirubin. Interactions are labelled from 1 to 5 in decreasing stability, with the interaction type indicated on the right. Rectangles are used to group interactions of the same type. The second molecule in the crystal structure of Form II is disordered around 50%. The plotted energies for Form II(a) and Form II(b) display different energies for both. | ||
The most stable molecule⋯molecule arrangements in all three forms are grouped, ranked in decreasing stability, and displayed in Fig. 7. The most stable molecule⋯molecule arrangement (region 1) can be found in all three forms. The energy and distance of the arrangement vary slightly depending on the form, ranging from −69 to −102 kJ mol−1, with the distance varying between 7.5 and 8.5 Å, indicating its flexibility. The arrangement resembles two clasped hands, but there are actually two molecular orientations that are very similar, which are shown in Fig. S1a in the ESI.† These two orientations are really two molecule⋯molecule interactions (arrangements), but since they look similar, we grouped them together in region 1 in Fig. 7. The most stable orientation, present in Forms I, II, and III, involves the vinyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) groups of both molecules overlapping with the carboxylic group of the neighbouring molecule. The interaction energy for this orientation ranges from −86 to −102 kJ mol−1. These interaction energies are shown in region 1a in Fig. 7. The other molecular orientation is unique to Form II and has the methyl overlapping the carboxylic group of the neighbouring molecule (Fig. S1b†). The interaction energy ranges from −69 to −79 kJ mol−1 and is shown in region 1b in Fig. 7. Both orientations are centrosymmetric (Fig. S1 and Table S1†). The molecule⋯molecule arrangements in regions 2, 3, and 5 are found in all three forms. For these molecule⋯molecule arrangements, energies and distances are similar in all three forms, suggesting that these are more rigid. The molecule⋯molecule arrangement in region 4 is unique to Form II.
C) groups of both molecules overlapping with the carboxylic group of the neighbouring molecule. The interaction energy for this orientation ranges from −86 to −102 kJ mol−1. These interaction energies are shown in region 1a in Fig. 7. The other molecular orientation is unique to Form II and has the methyl overlapping the carboxylic group of the neighbouring molecule (Fig. S1b†). The interaction energy ranges from −69 to −79 kJ mol−1 and is shown in region 1b in Fig. 7. Both orientations are centrosymmetric (Fig. S1 and Table S1†). The molecule⋯molecule arrangements in regions 2, 3, and 5 are found in all three forms. For these molecule⋯molecule arrangements, energies and distances are similar in all three forms, suggesting that these are more rigid. The molecule⋯molecule arrangement in region 4 is unique to Form II.
4. Conclusions
The crystal structures and DFT calculations show that the bilirubin molecule is rigid due to strong intramolecular hydrogen bonding, always resulting in the same solid-state conformation. All three forms crystallize in the triclinic space group P. While Form I is a pure material, Forms II and III are solvates with the solvent occupying 5% and 25% of the unit cell volume, respectively. Despite these supramolecular differences, molecule⋯molecule energy calculations show that the three strongest interactions are geometrically consistent.
Author contributions
M. L. B. was the principal experimentalist and investigator. M. A. F. was the principal crystallographer and investigator. S. M. was the supervisor who conceptualized and funded the project. All contributed to writing the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NRF and the University of the Witwatersrand for generous funding to enable the purchase of a dual wavelength hybrid diamond anode X-ray diffractometer (Bruker D8 Discovery Bio) under NEP Grant No 129920. We are also grateful for funding received under the NRF Grants MATH021 and CPRR 111705. We also thank the Centre for High Performance Computing (Project CHEM1633, CHPC, Cape Town) for the access to the supercomputing infrastructure.Notes and references
- M. Seya, T. Aokage, T. Nojima, A. Nakao and H. Naito, Eur. J. Med. Res., 2022, 27, 224 CrossRef PubMed.
- M. Ilbert and V. Bonnefoy, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 161–175 CrossRef CAS PubMed.
- J. H. Dawson, Science, 1988, 240, 433–439 CrossRef CAS PubMed.
- S. Kumar and U. Bandyopadhyay, Toxicol. Lett., 2005, 157, 175–188 CrossRef CAS PubMed.
- A. F. McDonagh, Nat. Struct. Biol., 2001, 8, 198–200 CrossRef CAS PubMed.
- P. A. Zunszain, J. Ghuman, A. F. McDonagh and S. Curry, J. Mol. Biol., 2008, 381, 394–406 CrossRef CAS PubMed.
- J. Y. Takemoto, C. T. Chang, D. Chen and G. Hinton, Isr. J. Chem., 2019, 59, 378–386 CrossRef CAS.
- N. Blanckaert, K. P. M. Heirwegh and F. Compernolle, Biochem. J., 1976, 155, 405–417 CrossRef CAS PubMed.
- S. Gazzin, L. Vitek, J. Watchko, S. M. Shapiro and C. Tiribelli, Trends Mol. Med., 2016, 22, 758–768 CrossRef CAS PubMed.
- G. Longhi, S. Ghidinelli, S. Abbate, G. Mazzeo, M. Fusè, S. E. Boiadjiev and D. A. Lightner, Molecules, 2023, 28, 2564 CrossRef CAS PubMed.
- J. Fevery, Liver Int., 2008, 28, 592–605 CrossRef CAS PubMed.
- R. Bonnett, J. E. Davies, M. B. Hursthouse and G. M. Sheldrick, Proc. R. Soc. B, 1978, 202, 249–268 CAS.
- APEX4version 2021.4-1 Data Collection Software (includes SAINT Version 8.40B, SADABS-2016/2, and XPREP Version 2014/2), Bruker AXS Inc., Madison, Wisconsin, USA, 2021 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. Le Bas, A. Allegret, Y. Mauguen, C. De Rango and M. Bailly, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 3007–3011 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01. 2016 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView Version 6, 2019 Search PubMed.
- A. Gavezzotti, J. Phys. Chem. B, 2003, 107, 2344–2353 CrossRef CAS.
- A. Gavezzotti, Z. Kristallogr., 2005, 220, 499–510 CAS.
- A. Gavezzotti, New J. Chem., 2011, 35, 1360–1368 RSC.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- J. Bernstein, J. D. Dunitz and A. Gavezzotti, Cryst. Growth Des., 2008, 8, 2011–2018 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal coordinates as CIF files for all the structures reported in Table 1 have been deposited at the CCDC (2331350 and 2331351). The CIF files and the Le Bas et al. (1980) structure as a compressed file are available. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00123k |
| This journal is © The Royal Society of Chemistry 2024 |