
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShortening C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) N⋯Br–Csp3 halogen bonds via π-stacking†
N⋯Br–Csp3 halogen bonds via π-stacking†
Kamil
Kupietz
 a,
Rosa M.
Gomila
b,
Thierry
Roisnel
a,
Antonio
Frontera
*b and
Rafael
Gramage-Doria
*a
a,
Rosa M.
Gomila
b,
Thierry
Roisnel
a,
Antonio
Frontera
*b and
Rafael
Gramage-Doria
*a
aUniv. Rennes, CNRS, ISCR – UMR6226, F-35000 Rennes, France. E-mail: rafael.gramage-doria@univ-rennes1.fr
bDepartament de Quimica, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, Spain. E-mail: toni.frontera@uib.es
First published on 1st April 2024
Abstract
The X-ray structure of 2-(dibromomethyl)benzonitrile, featuring unexpectedly short C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N⋯Br halogen bond distances between a nitrile group and a C(sp3)-linked bromine atom, is presented. Despite the weak Lewis base nature of the nitrile N atom and absence of strong electron-withdrawing groups on the Br atom, π-stacking significantly enhances both the electron donor and acceptor capability of the interacting partners. As such, and beyond trivial (hetero)aromatic systems, the rationale of CN⋯Br halogen bonding can also be employed in purely aliphatic systems.
N⋯Br halogen bond distances between a nitrile group and a C(sp3)-linked bromine atom, is presented. Despite the weak Lewis base nature of the nitrile N atom and absence of strong electron-withdrawing groups on the Br atom, π-stacking significantly enhances both the electron donor and acceptor capability of the interacting partners. As such, and beyond trivial (hetero)aromatic systems, the rationale of CN⋯Br halogen bonding can also be employed in purely aliphatic systems.
Halogen bonding is increasingly recognized for its significance in various fields, including materials science,1 catalysis,2,3 and medicinal chemistry.4,5 This interaction typically involves a heavy halogen atom, such as bromine or iodine, acting as an electrophile to form a highly directional noncovalent bond with a nucleophilic counterpart.6 Typically, this nucleophile is an atom like nitrogen, oxygen, or sulfur, possessing a lone pair of electrons.6,7 Halogens are amenable for participating in such interactions due to their unique electronic characteristics, which notably include a region of positive potential, termed the σ-hole, situated along the extension of their covalent bond.8–10 In the realm of supramolecular chemistry and crystal engineering, halogen bonding stands out as a potent tool for designing and synthesizing novel materials.11 Its appeal lies in the predictability, directionality, and tunability of these interactions, facilitating the precise organization of molecular components within crystal lattices. This precision is crucial for generating materials with unique properties and targeted functionalities.12,13 The application of halogen bonding in crystal engineering not only enhances our comprehension of molecular interactions but also paves the way for novel methods in the design and development of advanced materials. In particular, the use of nitrile functional groups is appealing due to their intrinsic unidirectionality.14–18 Herein, we detail the synthesis and X-ray characterization of 2-(dibromomethyl)benzonitrile (1, Fig. 1), which exhibits highly directional and unprecedentedly short C
N⋯Br halogen bonds (HaBs) of 3.11 Å in the solid-state structure (Fig. 1). This is remarkable considering that compound 1 is not ionic and that the bromine atoms do not belong to a (hetero)aromatic system (Csp2-linked) but to an aliphatic one (Csp3-linked).14,15 To elucidate the formation of these HaBs, in-depth density functional theory (DFT) calculations were employed. These calculations reveal that the antiparallel π-stacking arrangement of the aromatic rings amplifies both the electron-donating characteristics of the nitrile groups and the electron-accepting properties of the C–Br groups. Note that extremely short CN⋯Br halogen bonds (<3.2 Å) have been identified for purely (hetero)aromatic or π-conjugated systems (Fig. 1)16 including those presenting metal ions or ionic species that significantly influence the natural electron density of the HaB system.17,18 In the broad context, it is relevant to emphasize that ultrashort N⋯Br distances involving no nitrile groups, but more basic amines and pyridines are known.19
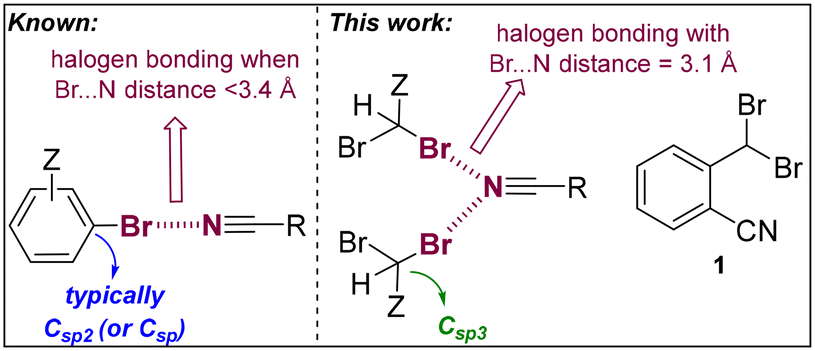 | ||
| Fig. 1 Considerations of N⋯Br halogen bonding in aromatic systems (left) versus the current system (1, right) involving a non-aromatic bromide group. Z = any substitution pattern. | ||
During the course of our studies in the development of supramolecular catalysis,20 we synthesized and obtained single crystals suitable for X-ray diffraction studies of 1 (Fig. 1). Most notably, the experimental CN⋯Br distance of 3.111 Å is significantly shorter than the sum of the van der Waals radii (∑Rvdw = 3.40 Å), and represents an unprecedented finding for a Br–Csp3 σ-hole donor group.14,15 The theoretical study herein, based on DFT calculations, analyzes the short (CN⋯Br–Csp3) halogen bonding interactions observed in the solid state of 1. Initially, the unit cell of compound 1 was optimized using periodic boundary conditions, and the resultant geometry was then compared with the experimentally determined structure. As depicted in Fig. 2, both geometries exhibit nearly identical arrangements. The only difference lies in the position of the hydrogen atoms (the C–H bonds are marginally elongated in the DFT-optimized geometry). Intriguingly, the DFT calculations also reveal a significantly short (3.093 Å) halogen bond. Within the unit cell, beyond the formation of the halogen bond, an antiparallel π-stacking of compound 1 is evident, in good agreement between the experimental and theoretical π⋯π distances. The antiparallel π-stacking of compound 1 in the solid state is obvious from the X-ray molecular packing (Fig. S4 in the ESI†).
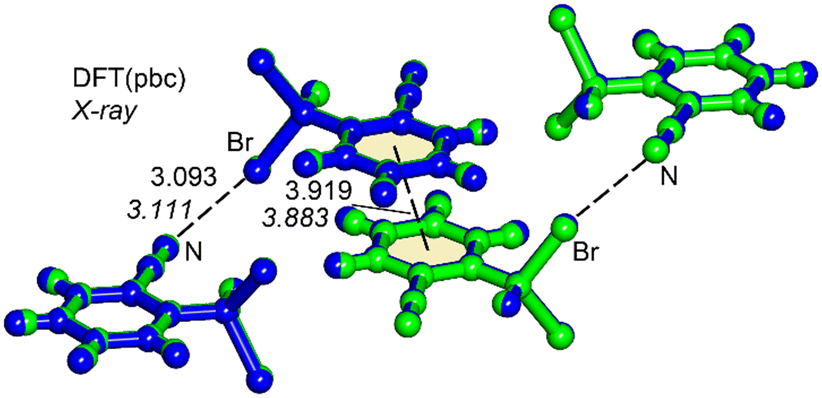 | ||
| Fig. 2 Overlap of the X-ray (in green) and DFT (in blue) fully optimized unit cells of compound 1. Distances in Å. The experimental distances are indicated in italics. | ||
The unique short distance of the CN⋯Br halogen bond in 1 is somewhat surprising with respect to precedents in the literature,14–18 especially considering that the sp-hybridized N atom of the nitrile group is not a strong Lewis base, and the σ-hole donor Br atom is not directly bonded to a potent electron-withdrawing group. We anticipated that the formation of π-stacked dimers might influence both the electron donor and acceptor groups. To investigate this hypothesis, we computed molecular electrostatic potential (MEP) surfaces for both the monomeric and dimeric forms of species 1. As illustrated in Fig. 3, the σ-hole value at the Br atom notably increases by 70% from 14 kcal mol−1 in the monomer to 20.1 kcal mol−1 in the dimer. Additionally, the MEP value at the N atom of the nitrile group shifts from −33.8 kcal mol−1 in the monomer to −35.7 kcal mol−1 in the dimer. These observations clearly suggest a synergistic or cooperative effect between the π-stacking interaction and the halogen bonding. It is also noteworthy that in the monomer, the MEP maximum is located at the aromatic H atom para with respect to the nitrile substituent, marked at 27.6 kcal mol−1, which decreases in the dimer down to 23.8 kcal mol−1. Consequently, the difference between the MEP values at the maximum and the σ-hole is reduced in the dimer, indicating a complex interplay of interactions within the molecular structure.
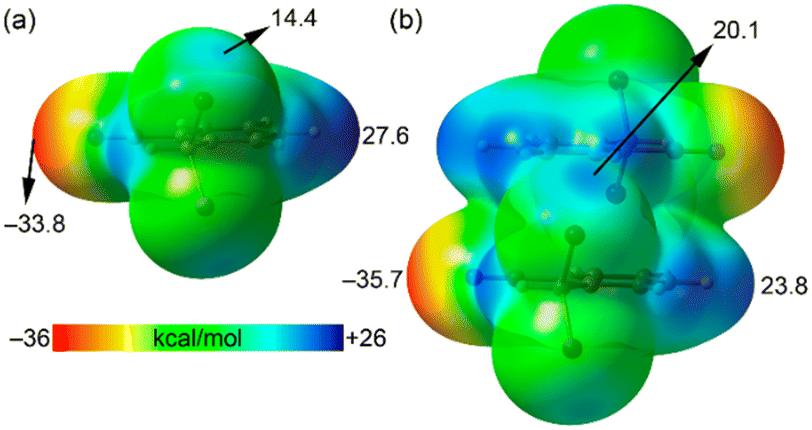 | ||
| Fig. 3 MEP surfaces of 2-(dibromomethyl)benzonitrile (a) and its π-stacked dimer (b). The energies at selected points are given in kcal mol−1. Isovalue 0.001 a.u. | ||
Next, we conducted a thorough analysis of the halogen bonding, hydrogen bonding, and π-stacking interactions, both energetically and using a combination of quantum theory of atoms in molecules (QTAIM) and non-covalent interaction (NCI) plot analyses. The results are presented in Fig. 4(a). Initially, we examined the assembly observed in the unit cell, where the QTAIM analysis reveals a complex array of interactions. The π-stacking interaction is characterized by two bond critical points (BCPs) and bond paths connecting two carbon atoms of the aromatic ring. This interaction is further delineated by a green reduced density gradient (RDG) isosurface enveloping much of the region between the π-systems. Secondly, we identified two symmetrically equivalent halogen bonds, each marked by a BCP and a bond path linking the nitrogen atom to the bromine atom. These halogen bonds are additionally characterized by disk-shaped RDG isosurfaces, coinciding with the BCP locations. Furthermore, several CH⋯Br contacts also play a role in stabilizing the assembly, each indicated by a corresponding BCP, bond path, and green RDG isosurface. The total formation energy of this assembly is calculated to be −19.9 kcal mol−1, with a significant contribution from the π-stacking interaction (−9.6 kcal mol−1, calculated as a π-stacked dimer). The contributions from the halogen bonding and hydrogen bonding interactions were estimated using the QTAIM method, based on the potential energy density values at the BCPs (see theoretical methods in ESI† for details). Interestingly, the contribution of the two halogen bonds (−5.0 kcal mol−1) is slightly more substantial than that of the hydrogen bonds (−4.6 kcal mol−1). The involvement of bromine atoms in hydrogen bonds enhances the positive MEP value at the σ-hole, thereby explaining the more pronounced σ-holes in the dimer compared to the monomer.
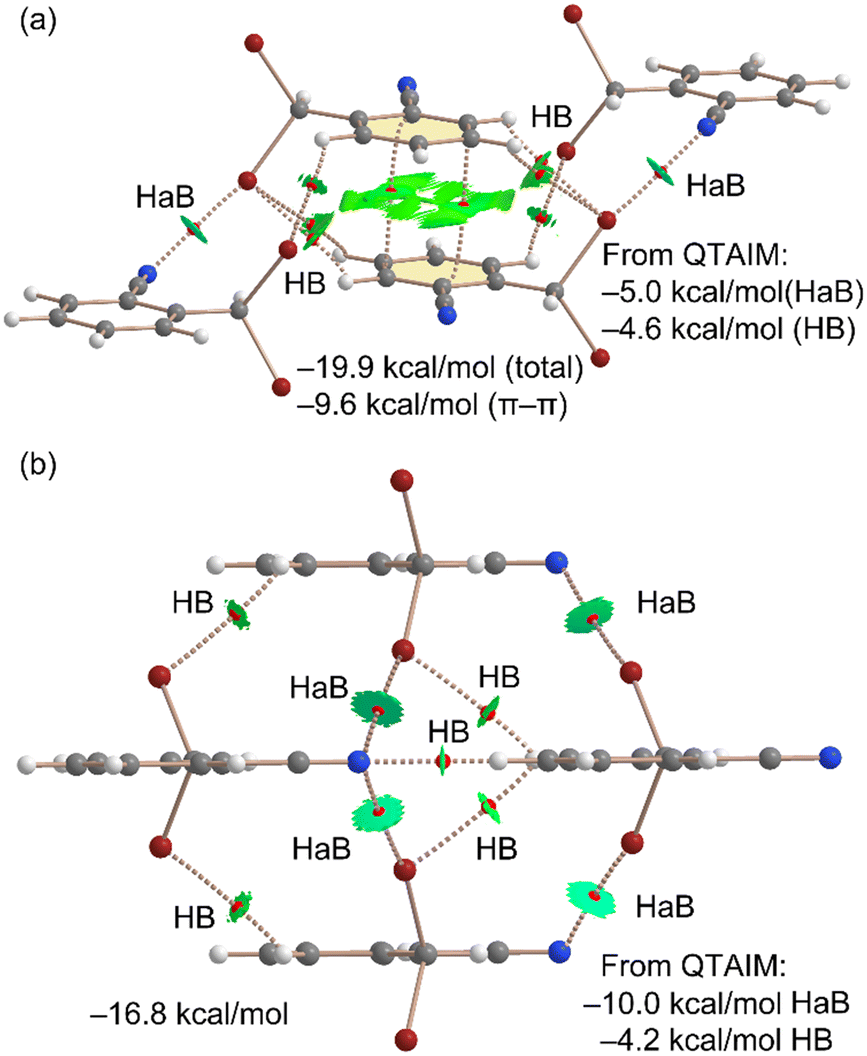 | ||
| Fig. 4 QTAIM/NCIplot analyses of the unit cell (a) and HAB/HB (b) tetrameric assemblies of compound 1. The formation energies and the contribution of the HaB and HBs derived from QTAIM are indicated (see computational methods in ESI†). | ||
Furthermore, a tetrameric assembly from 1 was analyzed using the QTAIM/NCIplot methodology, as shown in Fig. 4(b). In this tetramer, the absence of π-stacking leaves only halogen and hydrogen bonding available for consideration. As a result, a hydrogen bond is formed between the nitrile group and the hydrogen atom para with respect to the nitrile group of an adjacent molecule of 1, aligning well with the MEP analysis, given the maximum and minimum MEP values of the interacting atoms. This nitrogen atom also forms two additional halogen bonds, resulting in a bifurcated halogen bond characterized by two CPs, bond paths, and green RDG isosurfaces. The contribution of the halogen bonds in this structure is −10.0 kcal mol−1 since a total of four HaBs are formed, and that of the hydrogen bonds is −4.6 kcal mol−1, underscoring the significance of halogen bonds in the solid-state structure of compound 1. The formation energy of this tetramer is −16.8 kcal mol−1, slightly more negative than the summed QTAIM contributions of the halogen and hydrogen bonds (−14.2 kcal mol−1). This discrepancy suggests either a minor underestimation by the QTAIM method or the presence of additional long-range van der Waals interactions among the monomers. The estimation of the hydrogen bond (HB) contribution utilizes the formula proposed by Espinosa et al.,21a expressed as E = 0.5 × Vb, while the halogen bond contribution is calculated using the equation recommended by Bartashevich et al. (E = 0.58 × Vb)21b where Vb represents the potential energy density at the bond CP.
To gain a deeper understanding of the interactions at play, an energy decomposition analysis (EDA) was performed on the halogen-bonded dimer depicted in Fig. 5. This analysis breaks down the total energy (Etot) into its constituent parts: electrostatic (Eel), exchange-repulsion (Eex-rep), orbital (Eorb), dispersion (Edisp), and correlation (Ecor). The findings, presented in Fig. 5, show that while all components contribute similarly, the dispersion effect stands out as slightly more significant (−3.05 kcal mol−1, green bar) compared to the others, which range from −2.1 to −2.8 kcal mol−1. Notably, the exchange-repulsion factor (indicated by the red bar) represents the most substantial contribution of 6.50 kcal mol−1, yet it is effectively balanced by the attractive components. This detailed EDA sheds light on the complex and multifaceted nature of halogen bonding interaction in 1.
 | ||
| Fig. 5 EDA analysis of the dimer of 1 at the PBE0-D3/def2-TZVP level. Energies in kcal mol−1. | ||
Finally, we investigated whether orbital effects play a significant role in the bifurcated halogen bonds observed in the solid state of 1. For this purpose, we utilized natural bond orbital (NBO) analysis, which is particularly effective for analyzing charge transfer effects. The donor–acceptor orbitals are depicted in Fig. 6, demonstrating a charge transfer from the lone pair (LP) orbital on the nitrogen atom to the antibonding σ*(Br–C) orbital, a characteristic feature of halogen bonding interactions. However, the stabilization energy associated with the LP(N) → σ*(Br–C) transfer is relatively modest, amounting to only 0.44 kcal mol−1. This finding suggests that orbital effects are not the predominant factor driving the Br⋯N⋯Br interaction in this case.
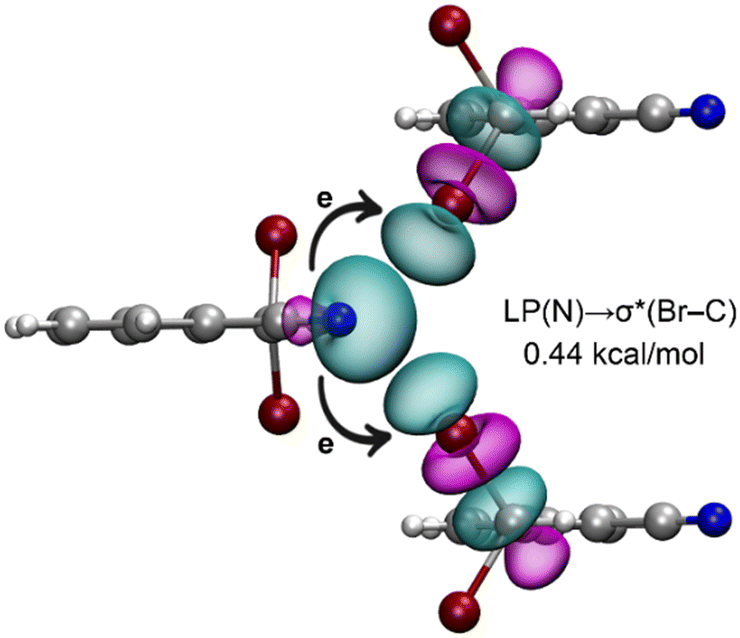 | ||
| Fig. 6 NBOs involved in the bifurcated halogen bonds in a trimeric fragment of compound 1. The E(2) energy is indicated. | ||
In conclusion, the X-ray structure of 2-(dibromomethyl)benzonitrile (1) is reported herein, evidencing an intricate interplay of halogen bonding, hydrogen bonding, and π-stacking interactions in its solid-state structure. Through a combination of QTAIM, NCI plot, EDA, and NBO analyses, we demonstrated the significant role of these noncovalent interactions in stabilizing the molecular assembly. Our findings highlight the unexpectedly strong and short halogen bonds, taking into consideration the relatively weak Lewis base nature of the sp-hybridized N atom and the absence of strong electron-withdrawing groups directly bonded to the σ-hole donor Br atom. The π-stacking interaction has a crucial role in enhancing the strength of the HaBs electrostatically, as demonstrated using MEP surface analysis. This study provides valuable insights into the feasibility of CN⋯Br(Csp3-linked) halogen bonds that may find applications beyond crystal engineering.
Financial support from the CNRS and University of Rennes is acknowledged. A. F. thanks the MICIU/AEI from Spain for financial support (PID2020-115637GB-I00, FEDER funds). This project has received funding from the European Union's Horizon 2020 research program under the Marie Skłodowska-Curie grant agreement no. 899546 (K. K.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, in Halogen Bonding I: Impact on Materials Chemistry and Life Sciences, ed. P. Metrangolo and G. Resnati, Springer International Publishing, Cham, 2015, vol. 1, pp. 1–19 Search PubMed.
- (a) J. Bamberger, F. Ostler and O. G. Mancheño, ChemCatChem, 2019, 11, 5198–5211 CrossRef CAS PubMed; (b) C. Keuper, K. Fengler, F. Ostler, T. Danelzik, D. G. Piekarski and O. García Mancheño, Angew. Chem., Int. Ed., 2023, 62, e202304781 CrossRef PubMed.
- W. Wang, Y. Wang, Y. Zhang, B. Ji, Y. Zhang and R. Wang, Expert Opin. Drug Discovery, 2019, 14, 561–573 CrossRef PubMed.
- (a) P. Metrangolo and G. Resnati, Chem. – Eur. J., 2001, 7, 2511–2519 CrossRef CAS PubMed; (b) M. Malinska, I. Fokt, W. Priebe and K. Wozniak, Cryst. Growth Des., 2015, 15, 2632–2642 CrossRef CAS.
- P. Politzer, P. Lane, M. C. Concha, Y. G. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed.
- J. S. Murray, Z. P. Shields and P. Politzer, Int. J. Quantum Chem., 2010, 110, 2823–2832 CrossRef.
- D. M. P. Mingos, Halogen Bonding, Fundamentals and Applications, Springer-Verlag, Berlin, 2008 Search PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- E. Parisini, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Chem. Soc. Rev., 2011, 40, 2267–2278 RSC.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- (a) P. Metrangolo and G. Resnati, Chem. – Eur. J., 2001, 7, 2511–2519 CrossRef CAS PubMed; (b) D. Yan, A. Delori, G. O. Lloyd, T. Friscic, G. M. Day, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2011, 50, 12483–12486 CrossRef CAS PubMed; (c) J. Wudarczyk, G. Papamokos, V. Margaritis, D. Schollmeyer, F. Hinkel, M. Baumgarten, G. Floudas and K. Mullen, Angew. Chem., Int. Ed., 2016, 55, 3220–3223 CrossRef CAS PubMed; (d) S. Basurto, S. Garcia, A. G. Neo, T. Torroba, C. F. Marcos, D. Miguel, J. Barbera, M. B. Ros and M. R. de la Fuente, Chem. – Eur. J., 2005, 11, 5362–5376 CrossRef CAS PubMed.
- For CN⋯Br distances longer (3.12–3.19 Å) than the current report involving a non-aromatic system, see:
(a) M. Shi and C.-J. Wang, Tetrahedron, 2002, 58, 9063–9074 CrossRef CAS;
(b) A. Mori, H. Kawakami, N. Kato, S.-P. Wu and H. Takeshita, Org. Biomol. Chem., 2003, 1, 1730–1736 RSC;
(c) K. Wright, B. Drouillat, L. Menguy, J. Marrot and F. Couty, Eur. J. Org. Chem., 2017, 7195–7201 CrossRef CAS;
(d) C. J. Salomon, O. A. Mascaretti, C. E. Strouse and G. Punte, Can. J. Chem., 1991, 69, 578–583 CrossRef CAS.
- For a rare example with absence of discussion featuring a CN⋯Br distance of 3.0 Å in which the bromine atom is part of a highly electrophile (i.e. C5Br6), see: S. M. Rupf, P. Prohm and M. Malischewski, Chem. Commun., 2020, 56, 9834–9837 RSC.
- For selected examples of short CN⋯Br distances involving (hetero)aromatic or π systems, see:
(a) S. V. Baykov, S. I. Presnukhina, A. S. Novikov, A. A. Shetnev, V. P. Boyarskiy and V. Y. Kukushkin, Cryst. Growth Des., 2021, 21, 2526–2540 CrossRef CAS;
(b) A. D. Bond, J. Griffiths, J. M. Rawson and J. Hulliger, Chem. Commun., 2001, 2488–2489 RSC;
(c) A. S. Mikherdov, R. A. Popov, A. S. Smirnov, A. A. Eliseeva, A. S. Novikov, V. P. Boyarskiy, R. M. Gomila, A. Frontera, V. Y. Kukushkin and N. A. Bokach, Cryst. Growth Des., 2022, 22, 6079–6087 CrossRef CAS;
(d) L. Lai, B. Fang, M. Fan, W. Cheng and M. Yin, J. Phys. Chem. C, 2021, 125, 16350–16357 CrossRef CAS;
(e) K. Liu, S. Li, L. Fu, Y. Lei, Q. Liao and H. Fu, Nanoscale, 2022, 14, 6305–6311 RSC;
(f) R. Mariaca, N.-R. Behrnd, P. Eggli, H. Stoeckli-Evans and J. Hulliger, CrystEngComm, 2006, 8, 222–232 RSC;
(g) X. Yuan, Y. Zhao, T. Zhan, J. Oh, J. Zhou, J. Li, X. Wang, Z. Wang, S. Pang, P. Cai, C. Yang, Z. He, Z. Xie, C. Duan, F. Huang and Y. Cao, Energy Environ. Sci., 2021, 14, 5530–5540 RSC;
(h) P.-T. T. Pham and M. M. Bader, Cryst. Growth Des., 2014, 14, 916–922 CrossRef CAS;
(i) F. Frausto, Z. C. Smith, T. E. Haas and S. W. Thomas III, Chem. Commun., 2015, 51, 8825–8828 RSC;
(j) Y. Imai, K. Kamon, S. Kido, T. Harada, N. Tajima, T. Sato, R. Kuroda and Y. Matsubara, CrystEngComm, 2009, 11, 620–624 RSC;
(k) A. D. Finke, S. Haberland, W. Bernd Schweizer, P. Chen and F. Diederich, Angew. Chem., Int. Ed., 2013, 52, 9827–9830 CrossRef CAS PubMed;
(l) H. Usta, C. Risko, Z. Wang, H. Huang, M. K. Deliomeroglu, A. Zhukhovitskiy, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 5586–5608 CrossRef CAS PubMed;
(m) R. A. Wiscons, V. Coropceanu and A. J. Matzger, Chem. Mater., 2019, 31, 6598–6604 CrossRef CAS;
(n) T. Bjorvatten, Acta Chem. Scand., 1968, 22, 410–420 CrossRef CAS.
- For selected examples of short CN⋯Br distances involving metal-free ionic species, see:
(a) J. Harloff, K. C. Laatz, S. Lerch, A. Schulz, P. Stoer, T. Strassner and A. Villinger, Eur. J. Inorg. Chem., 2020, 2457–2464 CrossRef CAS;
(b) P. Arsenyan, J. Vasiljeva, S. Belyakov, E. Liepinsh and M. Petrova, Eur. J. Org. Chem., 2015, 5842–5855 CrossRef CAS;
(c) S. V. Rosokha, C. L. Stern, A. Swartz and R. Stewart, Phys. Chem. Chem. Phys., 2014, 16, 12968–12979 RSC;
(d) V. Figala, T. Gessner, R. Gompper, E. Hadicke and S. Lensky, Tetrahedron Lett., 1993, 34, 6375–6378 CrossRef CAS.
- For selected examples of short CN⋯Br distances involving the presence of metal ions, see:
(a) S. Ohkoshi, S. Takano, K. Imoto, M. Yoshikiyo, A. Namai and H. Tokoro, Nat. Photonics, 2014, 8, 65–71 CrossRef CAS;
(b) C. Rumo, A. Stein, J. Klehr, R. Tachibana, A. Prescimone, D. Haussinger and T. R. Ward, J. Am. Chem. Soc., 2022, 144, 11676–11684 CrossRef CAS PubMed;
(c) J. E. Ormond-Prout, P. Smart and L. Brammer, Cryst. Growth Des., 2012, 12, 205–216 CrossRef CAS;
(d) C. Ohde, T. Kusamoto, K. Nakabayashi, S.-I. Ohkoshi and H. Nishihara, Cryst. Growth Des., 2017, 17, 2203–2210 CrossRef CAS;
(e) S. Derossi, L. Brammer, C. A. Hunter and M. D. Ward, Inorg. Chem., 2009, 48, 1666–1677 CrossRef CAS PubMed;
(f) Y. Tsunobuchi, S. Kaneko, K. Nakabayashi and S. Ohkoshi, Cryst. Growth Des., 2011, 11, 5561–5566 CrossRef;
(g) N. Jakupec, L. Fotovic and V. Stilinovic, CrystEngComm, 2020, 22, 8142–8150 RSC;
(h) B. Guo, X. Zhang, J.-H. Yu and J.-Q. Xu, Dalton Trans., 2015, 44, 11470–11481 RSC;
(i) H. Petzold, P. Djomgoue, G. Horner, S. Heider, C. Lochenie, B. Weber, T. Ruffer and D. Schaarschmidt, Dalton Trans., 2017, 46, 6218–6229 RSC.
- For selected examples of ultrashort N⋯Br distances involving no nitrile groups, see: (a) S. C. Blackstock and J. K. Kochi, J. Am. Chem. Soc., 1987, 109, 2484–2496 CrossRef CAS; (b) J. A. Zerkowski, J. C. MacDonald and G. M. Whitesides, Chem. Mater., 1994, 6, 1250–1257 CrossRef CAS; (c) R. Liantonio, P. Metrangolo, T. Pilati, G. Resnati and A. Stevenazzi, Cryst. Growth Des., 2003, 3, 799–803 CrossRef CAS; (d) P.-H. Liu, L. Li, J. A. Webb, Y. Zhang and N. S. Goroff, Org. Lett., 2004, 6, 2081–2083 CrossRef CAS PubMed; (e) L. Catalano, S. Perez-Estrada, H.-H. Wang, A. J.-L. Ayitou, S. I. Khan, G. Terraneo, P. Metrangolo, S. Brown and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2017, 139, 843–848 CrossRef CAS PubMed; (f) C. Weinberger, R. Hines, M. Zeller and S. V. Rosokha, Chem. Commun., 2018, 54, 8060–8063 RSC; (g) A. M. Abeysekera, B. B. Averkiev, P. Le Magueres and C. B. Aakeröy, Org. Biomol. Chem., 2021, 19, 6671–6681 RSC.
- (a) P. Zardi, T. Roisnel and R. Gramage-Doria, Chem. – Eur. J., 2019, 25, 627–634 CrossRef CAS PubMed; (b) N. Abuhafez and R. Gramage-Doria, Faraday Discuss., 2023, 244, 186–198 RSC.
- (a) E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS; (b) E. V. Bartashevich and V. G. Tsirelson, Russ. Chem. Rev., 2014, 83, 1181–1203 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, X-ray data and cartesian coordinates. CCDC 2330354. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00307a |
| This journal is © The Royal Society of Chemistry 2024 |