
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNaphthalene-1,8-dicarboxylate based zinc coordination polymers: a photophysical study†
Pablo
Guerrero-García
a,
Javier
Cepeda
 b,
Mariano
Ortega-Muñoz
c,
Jose Angel
García
d,
Amparo
Navarro
e,
Duane
Choquesillo-Lazarte
f,
Sara
Rojas
a,
Antonio Rodríguez
Diéguez
a,
María Mar
Quesada-Moreno
*e and
Iñigo J.
Vitórica-Yrezábal
*a
b,
Mariano
Ortega-Muñoz
c,
Jose Angel
García
d,
Amparo
Navarro
e,
Duane
Choquesillo-Lazarte
f,
Sara
Rojas
a,
Antonio Rodríguez
Diéguez
a,
María Mar
Quesada-Moreno
*e and
Iñigo J.
Vitórica-Yrezábal
*a
aDepartamento de Química Inorgánica, Facultad de Ciencias, Universidad de Granada, 18071, Granada, Spain. E-mail: vitorica@ugr.es
bDepartamento de Química Aplicada, Facultad de Química, Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU), 20018, Donostia, Spain
cDepartamento de Química Orgánica, Facultad de Ciencias, Universidad de Granada, 18071, Granada, Spain
dDepartamento de Física, Facultad de Ciencia y Tecnología, Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU), 48940, Leioa, Spain
eDepartamento de Química Física y Analítica, Facultad de Ciencias Experimentales, Universidad de Jaén, Campus Las Lagunillas, 23071 Jaén, Spain. E-mail: mqmoreno@ujaen.es
fLaboratorio de Estudios Cristalograficos, IACT, CSIC-Universidad de Granada, Av. De las palmeras 4, Armilla, Granada E-18100, Spain
First published on 30th January 2024
Abstract
Herein, we report the synthesis and photoluminescence properties of a new 1,3-dioxo-2-(1H-tetrazol-5-yl)-2,3-dihydro-1H-benzo[de]isoquinoline-6,7-dicarboxylic (H3L) ligand and three coordination polymers (CPs). Compound [Zn(ntca)DMF]n·DMF (1) (ntca2− = 1,4,5,8-naphthalenetetracarboxylate 1,8-monoanhydride, DMF = N,N-dimethylformamide) formed 1D coordination polymer chains packed by hydrogen bonding interactions. On its part, compounds namely [(CH3)2NH2](Zn2(μ-OH)(L)(5-NH2-tetrazolate)n·2H2O (2) and [(CH3)2NH2](ZnL)n (3) (where H3L stands for 1,3-dioxo-2-(1H-tetrazol-5-yl)-2,3-dihydro-1H-benzo[de]isoquinoline-6,7-dicarboxylic acid) crystallize as 2D-layered and 3D anionic frameworks containing dimethylammonium cations occupying the voids. Photoluminescence (PL) measurements have been performed in the solid state on all CPs and ligands to characterize their emission properties, including variable-temperature spectra, lifetime and efficiency. Moreover, the photophysical properties have been studied from the theoretical viewpoint by means of time dependent density functional theory (TD-DFT) in order to elucidate the mechanisms and electronic transitions governing the process. Compounds 1 and 3 present an intense blue luminescence which was originated in the electronic transitions of the ntca2− and the L3− ligands. Compound 2 displays a lower quantum yield which could be tentatively attributed to the weak π–π interactions of the aromatic clouds of L3− or the molecular vibrations and/or possible rotational motions of the 5-amino-tetrazolate co-ligand.
Introduction
The development of metal–organic materials based on the combination of inorganic coordination chemistry with organic ligands, resulting in structures with variable dimensionality, has been extensively studied in the past decades.1 The structural and functional properties of metal–organic materials can be rationally designed and modulated by an appropriate selection of metal centres and organic ligands.2 Coordination polymers (CPs) are covalently linked metal–organic centres forming expanded networks in 1, 2 and 3 dimensions, with a great diversity of geometries which arises from not only the coordination sphere of the metal centres and clusters, but also from the spatial arrangement and coordination modes of donor atoms in ligands.3 Due to their characteristic modularity and resulting amenability to design, metal–organic materials4 such CPs,5 offer promise as energy-efficient materials with applications in gas storage and separation,6–8 magnetism,9,10 catalysis,11 spintronics,12 electronics,13 energy storage and conversion,14 agriculture15 and photoluminescence.16,17 Especially, luminescent CPs are subject of an increasing research effort due to their tunable emissions not only derived from CPs components but also from their interplay with the surrounding medium (guest molecules, anlytes, composites, etc.).18,19 As a consequence, luminescent CPs has been shown to have potential applications as chemical sensors to detect and quantify ions, biomolecules, pH, temperature and pressure.20,21Luminescence properties in CPs are usually derived from the metal ions and/or organic ligands or a charge transfer between them. The emission capacity in CPs is often improved by the selection of metal ions with an electronic closed-shell configuration, as these cations (i) avoid the emission quenching derived from d–d transitions, and (ii) diminish the non-radiative vibrational relaxation in the framework.16 Thus, photoluminescence emission can be originated from π-electron-rich aromatic clouds present in organic ligands or due to a charge transfer processes between neighboring ligands. For this reason, in the last years we have worked with perylene derivative ligands to design new materials with interesting properties such as humidity sensors.22 However, crystallisation of these materials is complicated due to the large size of the ligand backbone. For this reason, we have decided to decrease the size of the skeleton by working with naphthalimide and naphthalene derivative ligands. The families of organic ligands containing a 1,4,5,8-naphthalenepolycarboxylic and 1,4,5,8-naphthalenediimides (NDI) core have been extensivetely studied due to their large conjugated plane and their rigid structure, presenting photophysical properties in CPs as photochromisim and photoluminiscence.23–25 The synthesis of a new 1D Zn(II) coordination polymer, namely [Zn(ntca)DMF]n·DMF (ntca = 1,4,5,8-naphthalenetetracarboxylate 1,8-monoanhydride) (1) has been described, where the naphthalene-1,4,5,8-tetracarboxylic acid dianhydride precursor has been partially hydrated in situ in order to form the ntca2− ligand at 368 K degrees of temperature.
In parallel, our group has specialized in the in situ synthesis of new ligands, through a Demko-Sharpeless [2 + 3] cycloaddition reaction of organonitriles with metal azides,26–28 during the formation tetrazolate containing CPs.29,30
In the present work we have also accomplished the synthesis of a novel 1,3-dioxo-2-(1H-tetrazol-5-yl)-2,3-dihydro-1H-benzo[de]isoquinoline-6,7-dicarboxylic acid ligand (H3L) by reacting naphthalene-1,4,5,8-tetracarboxylic acid dianhydride molecule 5-aminotetrazole molecule in acetic acid solution (Scheme 1).
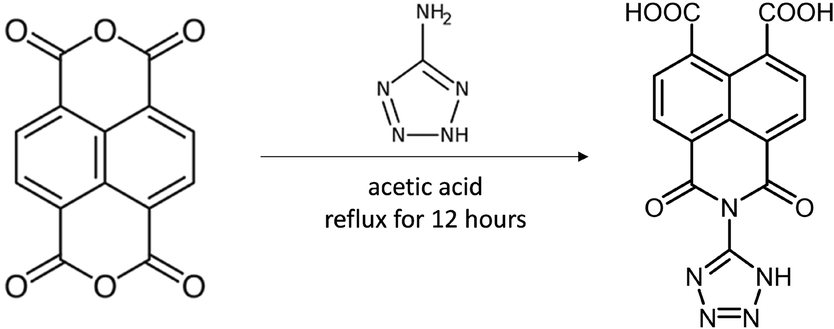 | ||
| Scheme 1 Synthesis of H3L. | ||
The resulting ligand has two carboxylic and one tetrazole groups, allowing the coordination of multiple metal ions in different coordination modes.31 The naphthalene derivate L3− ligand was used to synthetize two new anionic CPs, namely [(CH3)2NH2](Zn2(μ-OH)(L)(5-NH2-tetrazolate)n·2H2O (2) and [(CH3)2NH2](ZnL)n (3). The photophysical properties of the new ligand and CPs 1–3 have been characterized through diffuse reflectance, solid-state photoluminescence and DFT quantum chemical calculations.
Experimental section
Materials: all chemical reagents and solvents were purchased commercially and were used as received without further purification.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The mixture was homogenized and placed in an oven at 368 K. After 24 h of heating, the reaction vessel was slowly cooled down to room temperature during a period of about 3 h. Red crystals of the compound 3 were obtained with 21% (12 mg, 0.026 mmol) yield. Purity was assessed by PXRD (Fig. S20†).
:1). The mixture was homogenized and placed in an oven at 368 K. After 24 h of heating, the reaction vessel was slowly cooled down to room temperature during a period of about 3 h. Red crystals of the compound 3 were obtained with 21% (12 mg, 0.026 mmol) yield. Purity was assessed by PXRD (Fig. S20†).
X-ray diffraction data collection and structure determination
Single crystal X-ray diffraction data were collected at 100 K on a Bruker D8 Venture and Bruker APEX-II diffractometers using Cu Kα (λ = 1.5418 Å) and Mo Kα (λ = 0.71073 Å) equipped with a PHOTON 3 and APEX II detectors, and an Oxford cryosystem. Data were collected and processed using APEX II and APEX III software. Adsorption correction was applied using SADABS software by empirical methods measuring symmetry equivalent reflections at different azimuthal angles. All structures were solved using the SHELXT program and refined using least squares refinement methods on all F2 values as implemented within SHELXL.32,33 Both SHELXT and SHELXL were operated through the Olex2 (v1.5) interface.34The X-ray powder diffraction (XRPD) patterns were acquired on a Philips X'PERT diffractometer using Cu Kα (λ = 1.5418 Å) radiation in the 5 < 2θ < 50° range, with a fixed step size of 0.026° and an acquisition time of 2.5 s per step at 25 °C. Indexation of the diffraction profiles was done with FULLPROF software,35 using the space group and cell parameters obtained from single-crystal X-ray data.
Photoluminescence measurements
Photoluminescence spectra were recorded on an Edinburgh Instruments FLS920 spectrometer equipped with a closed cycle helium cryostat and under high vacuum (of ca. 10−9 mbar) to avoid the presence of oxygen or water in the sample holder. The steady-state emission spectra were acquired with an IK3552R-G HeCd continuous laser (325 nm) in the 350–750 nm range whereas the excitation spectra were collected with a Müller-Elektronik-Optik SVX1450 Xe lamp (in the 200–470 nm range). Luminescence collected in the UV-vis region was analysed with photomultiplier tube (PMT) coupled to the spectrometer. The emission decay curves were measured using a pulsed laser diode LDH-P-C-375 (λ = 375 nm) from PicoQuant as excitation source. The absolute quantum yield (QY %) was measured in solid state at room temperature on a Horiba Quanta-φ integrating sphere coupled to an Oriel Instruments MS257 continuous lamp as excitation source and an iHR550 spectrometer from Horiba to analyse the emission. MicroPL photographs were taken on an Olympus optical microscope equipped with a CCD detector over polycrystalline illuminated with an Hg lamp.Computational details
Density functional theory (DFT) calculations were performed with Gaussian 16 program package (revision A.03)36 using the hybrid exchange-correlation functional CAM-B3LYP.37 6-31G** basis set was used for C, N, O, and H atoms and LANL2DZ for Zn atom.38 The vibrational frequencies were computed in the ground and excited states to check the nature of the minima and the absence of imaginary frequencies. Different finite fragments were built from the X-ray diffraction structure and the molecular geometries were optimized in the ground state and several excited states. To mimic the structure of the MOF, the positions of some atoms were fixed to their corresponding crystallographic coordinates to avoid large deformations from the original structure, and all other atoms of the fragment were allowed to relax during the optimization process. Time dependent DFT (TD-DFT) calculations at the same level of theory were performed from the optimized molecular geometry of the ground state and 100 vertical electronic transitions were computed. The fluorescence emission energy from the optimized excited state was calculated Eem(Sn) = ESn(GSn) − ES0(GSn) at the same level of theory, where ESn(GSn) is the energy of the Sn excited state at its equilibrium geometry (GSn), and ES0(GSn) corresponds to the energy of the S0 ground state at the Sn excited state geometry (GSn). D3 version of Grimme's dispersion correction (GD3zero damping) was included in the calculation.39 Table S1† lists the maximum absorption wavelength and the theoretical vertical electronic transitions. Fig. S21† shows the molecular structure of the optimized fragments highlighting in red and blue the ‘active’ and ‘frozen’ atoms, respectively. Fig. S22 and S23† represents the molecular orbitals involved in the main electronic transitions.Results and discussion
Synthesis and crystal structure of compounds H3L and compounds 1–3
Ligand H3L was originally detected as an impurity in the synthesis of ditetrazole-naphthalenediimide by reacting naphthalene-1,4,5,8-tetracarboxylic acid dianhydride with two equivalents of 5-aminotetrazole monohydrate in DMF at 413 K. The reaction of stoichiometric amounts of naphthalene-1,4,5,8-tetracarboxylic acid dianhydride and 5-aminotetrazole in acidic conditions at 373 K yields a 1,3-dioxo-2-(1H-tetrazol-5-yl)-2,3-dihydro-1H-benzo[de]isoquinoline-6,7-dicarboxylic acid and 1,4,5,8 naphthalenetetracarboxylic acid mixture. Pure H3L ligand was obtained after separation of the reaction mixture by flash column chromatography.Yellow crystals of compound 1 were obtained by slowly decreasing the temperature of a DMF solution after 24 hours at 368 K. Crystal structure of compound 1 comprises Zn2(ntca)4(DMF)2 paddlewheels, with two five coordinate Zn(II) atoms showing a square pyramidal coordination geometry. The Zn(II) centres are bridged by four coordinated carboxylate ligands with Zn–O bonds distances ranging from 2.0446(19) to 2.0607(17) Å in the equatorial plane. The remaining axial positions are occupied by two disordered N,N-dimethylformamide (DMF) solvent molecules with Zn–O distances of 1.983(11) Å. The paddlewheels were interlinked by pairs of ntca2− ligands, forming a 1D-chain coordination polymer which expands in the [010] direction (Fig. 1a). The plane formed by the Zn(II) metal centres of the 1D coordination polymer (Fig. 1a) is rotated 21.94° respect the (100) plane, allowing the overlap of the disordered axially coordinated DMF molecules of the adjacent coordination polymers. The ntca2− ligands interact with the neighbouring ligands forming a C–H⋯O hydrogen bond (2.799 Å) and short contact between one of the carbon atoms of the anhydride group and the adjacent oxygen atom (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C⋯O 2.871(5) Å). This interaction is consequence of the partially positive electrostatic potential distribution around the carbon atom and its attractive interaction with the oxygen lone pair of electrons. The crystal structure contains channels filled with a disordered DMF molecules along the (010) direction, representing the 26.6% of the volume of the crystal (Fig. 1b). The presence of ntca2− ligands confirms the partial hydration of the naphthalene-1,4,5,8-tetracarboxylic acid dianhydride starting material during the in situ formation of compound 1. The mechanism of the partial and full hydration of naphthalene-1,4,5,8-tetracarboxylic acid dianhydride has been previously studied and reported by M. J. Politi and co-workers,40 involving the reaction of each anhydride group with a water molecule. The in situ formation of ntca2− during the synthesis of coordination polymers has been previously reported showing catalytic and photoluminescence properties.41–44 Yellow crystals of compound 2 were obtained by slowly decreasing the temperature of a DMF solution after 24 hours at 368 K. Crystal structure of 2 is formed by pairs of tetrahedrally coordinated Zn(II) atoms bridged by μ2-OH groups (Zn–O distances 1.893(8)–1.931(9) Å) and by L3− ligand through two monodentate carboxylates (Zn–O distances 1.958(8)–1.99(1)). The pairs of Zn(II) atoms are further linked in the (010) direction to other Zn pairs by 5-amino-tetrazolate molecules with Zn–N distances ranging from 1.98(1)–2.02(1) Å. Tetrazolate groups of an adjacent L3− ligand complete the tetrahedral Zn(II) coordination sphere (Zn–N distances 2.02(1)–2.05(1) Å), forming an anionic 2D tape coordination polymer expanded in the (110) plane (Fig. 2).
C⋯O 2.871(5) Å). This interaction is consequence of the partially positive electrostatic potential distribution around the carbon atom and its attractive interaction with the oxygen lone pair of electrons. The crystal structure contains channels filled with a disordered DMF molecules along the (010) direction, representing the 26.6% of the volume of the crystal (Fig. 1b). The presence of ntca2− ligands confirms the partial hydration of the naphthalene-1,4,5,8-tetracarboxylic acid dianhydride starting material during the in situ formation of compound 1. The mechanism of the partial and full hydration of naphthalene-1,4,5,8-tetracarboxylic acid dianhydride has been previously studied and reported by M. J. Politi and co-workers,40 involving the reaction of each anhydride group with a water molecule. The in situ formation of ntca2− during the synthesis of coordination polymers has been previously reported showing catalytic and photoluminescence properties.41–44 Yellow crystals of compound 2 were obtained by slowly decreasing the temperature of a DMF solution after 24 hours at 368 K. Crystal structure of 2 is formed by pairs of tetrahedrally coordinated Zn(II) atoms bridged by μ2-OH groups (Zn–O distances 1.893(8)–1.931(9) Å) and by L3− ligand through two monodentate carboxylates (Zn–O distances 1.958(8)–1.99(1)). The pairs of Zn(II) atoms are further linked in the (010) direction to other Zn pairs by 5-amino-tetrazolate molecules with Zn–N distances ranging from 1.98(1)–2.02(1) Å. Tetrazolate groups of an adjacent L3− ligand complete the tetrahedral Zn(II) coordination sphere (Zn–N distances 2.02(1)–2.05(1) Å), forming an anionic 2D tape coordination polymer expanded in the (110) plane (Fig. 2).
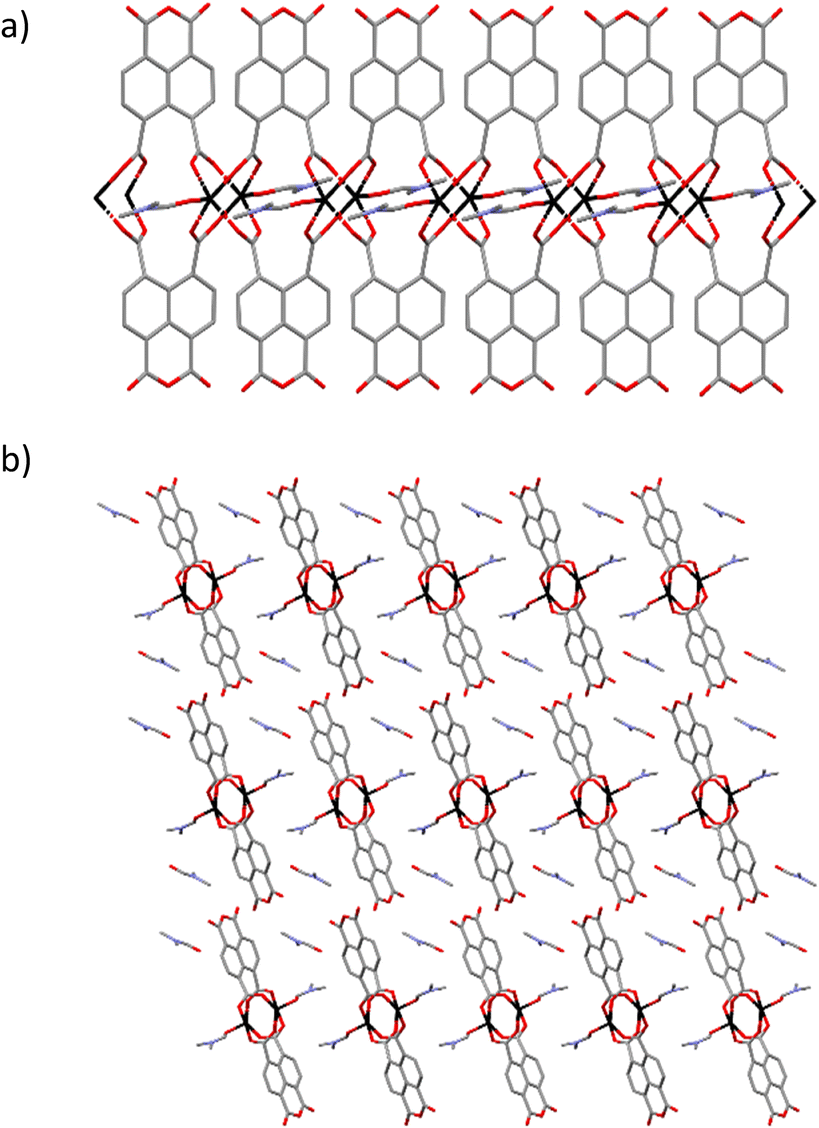 | ||
| Fig. 1 Crystal structure of compound 1 showing a) the 1D tape structure, and b) the packing of the 1D coordination polymers. Colour code: zinc atoms were black, oxygen in red, carbons in grey and nitrogen in blue. Hydrogens were omitted for clarity. | ||
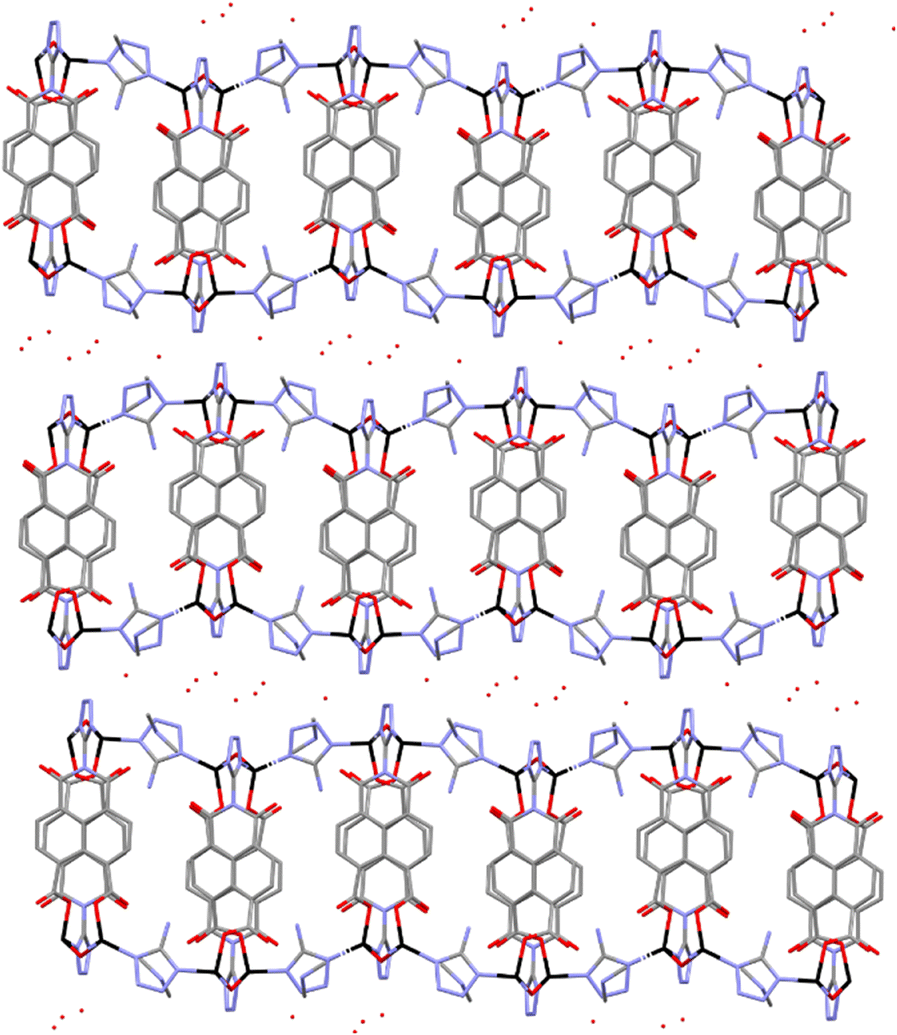 | ||
| Fig. 2 Crystal structure of compound 2 showing the packing of the 2D coordination polymers. Colour code: zinc atoms were black, oxygen in red, carbons in grey and nitrogen in blue. | ||
Neighbouring L3− ligands are forming parallel displaced π⋯π interactions with centroid–centroid distance of 3.940 Å, shift distance of 0.865 Å and at angle between planes of 21.906°. The presence of one dimethylammonium (DMA) cation per formula unit balances the charge of the anionic framework. The DMA molecules are forming (N–H⋯O) hydrogen bonds with the monodentate carboxylate groups and the water solvent molecules. The interlayer space is filled by water molecules forming an intricate hydrogen bond network with the DMA and the tetrazolate groups.
Crystal structure of 3 is formed by pairs of tetrahedral Zn(II) metal centres bridged by pairs of monodentate carboxylates from two L3− ligands (Zn–O distances range from 1.89(1)–2.036(3) Å) (Fig. 3). The pairs of Zn(II) atoms are further connected to other pairs by two tetrazolate groups from another two adjacent L3− ligands (Zn–N distances 2.046(2)–2.058(2) Å). Neighbouring L3− ligands are forming weak parallel displaced π⋯π interactions with centroid–centroid distance of 3.999 Å, but a large shift distance of 1.211 Å and an 0° angle between planes, resulting a weaker interaction than in compound 2. The resulting 3D anionic framework contains pores in the [100] direction filled with DMA cations forming N–H⋯O hydrogen bonds with the oxygens of the carboxylate and ketone groups.
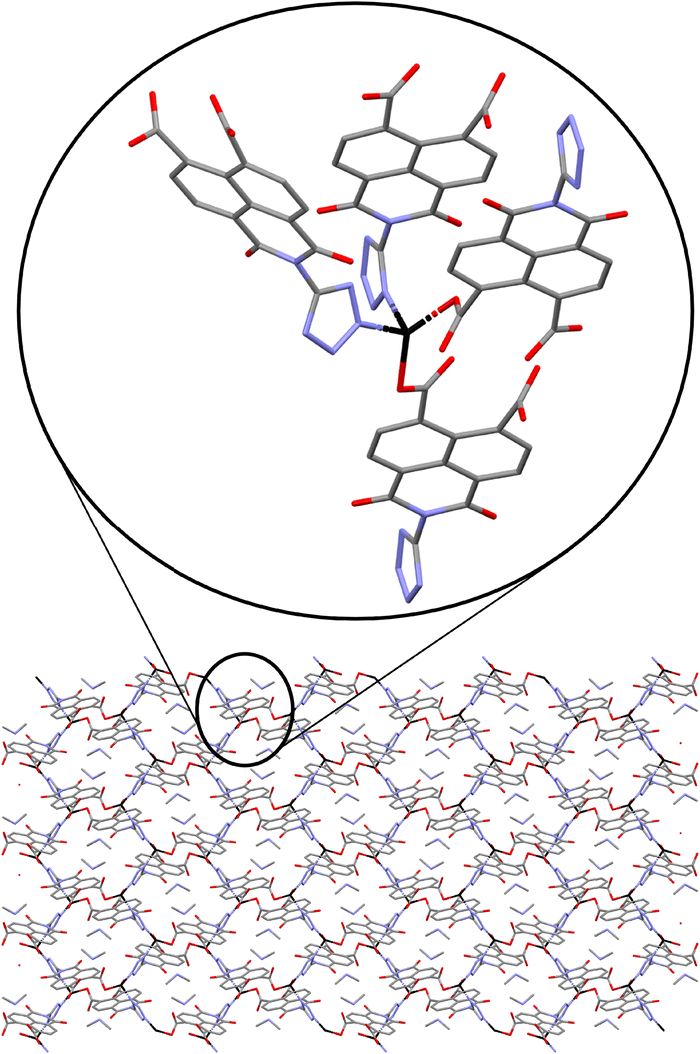 | ||
| Fig. 3 Crystal structure of 3 showing the packing in the (100) view with the enlargement of the coordination geometry around the Zn metal centres. Colour code: zinc atoms were black, oxygen in red, carbons in grey and nitrogen in blue. | ||
Photophysical properties
The diffuse reflectance absorption spectra of compounds 1, 2 and 3 show similar profiles governed by a strong band centered at 333, 336 and 340 nm, respectively (see Fig. S17†). They are very similar to those of the ligands naphthalene-1,4,5,8-tetracarboxylic acid dianhydride and H3L (see Fig. S16†), suggesting the null or small participation of the metallic Zn atom in the absorption process. DFT and TD-DFT calculations were performed to analyze the electronic transitions involved in the absorption spectra. Fig. 4 represents the oscillator strength of the calculated vertical electronic transitions along with the experimental absorption spectra of compounds 1, 2 and 3. Compound 1 presents the main band at 333 nm, and it could be assigned to the electronic transition calculated at 333 nm (f ∼ 0.6) which corresponds to S0 → S39 with contribution of HOMO-12 → LUMO + 1 (10%) and HOMO-14 → LUMO + 1 (10%). The weak band at 606 nm could be assigned to the transition calculated at 621 nm (S0 → S1) (see Fig. 4a). For compound 2, there are three theoretical transitions that could be involved in the resulting spectral profile of the band observed at 336 nm: the electronic transition S0 → S36 calculated at 332 nm (f ∼ 0.1) with contribution of HOMO-14 → LUMO + 1 (18%) and HOMO-13 → LUMO + 1 (58%); the transition S0 → S39 calculated at 326 nm (f ∼ 0.1) with contribution of HOMO-12 → LUMO + 4 (82%) and the stronger transition S0 → S46 calculated at 309 nm (f ∼ 0.2) with contribution of HOMO-14 → LUMO + 2 (26%), HOMO-13 → LUMO + 2 (26%) and HOMO-7 → LUMO + 2 (26%) (see Fig. 4b). In compound 3, the strongest band observed at 340 nm could be assigned to the S0 → S47 transition calculated at 346 nm (f ∼ 0.3) with contributions of HOMO-14 → LUMO + 2 (24%), HOMO-10 → LUMO + 4 (17%), HOMO-9 → LUMO + 4 (18%), along with the transition S0 → S62 calculated at 318 nm (f ∼ 0.2) with contribution of HOMO-13 → LUMO + 1 (43%) (see Fig. 4c). In addition, the weak band observed at 613 nm could be assigned to the electronic transitions calculated at 530 nm (f ∼ 0.2). The molecular orbitals involved in the main electronic transitions have been represented in Fig. 5, and are being localized on the ntca2− and L3− ligands, suggesting that the intraligand and interligand transitions could be present during the photoexcitation process. The absence of the metal influence in the calculated transitions suggests a small or negligible contribution of the ligand to metal (LMCT) or metal to ligand (MLCT) charge transfer transitions.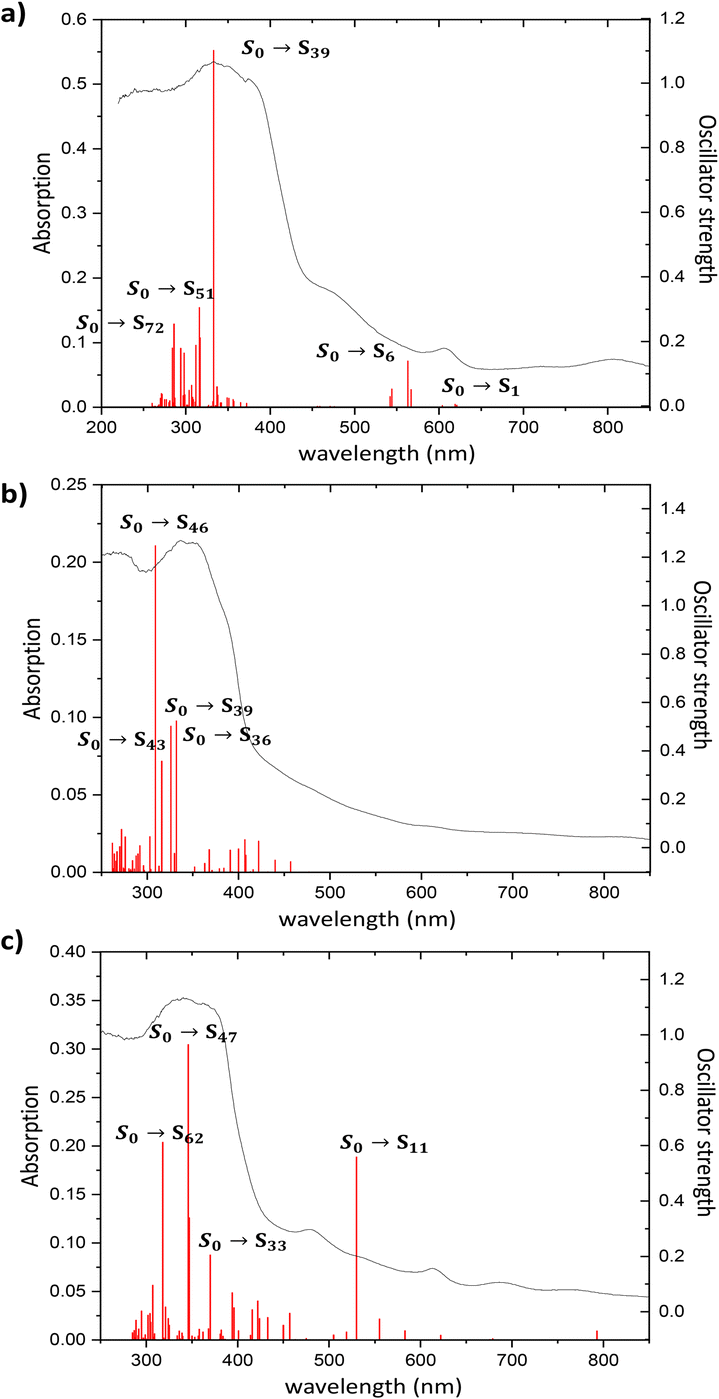 | ||
| Fig. 4 Experimental absorption spectrum of compounds (a) 1, (b) 2 and (c) 3, along with the oscillator strength of the vertical electronic transitions calculated at the TD-CAM-B3LYP-D3/6-31G** + LANL2DZ level of theory in gas phase. | ||
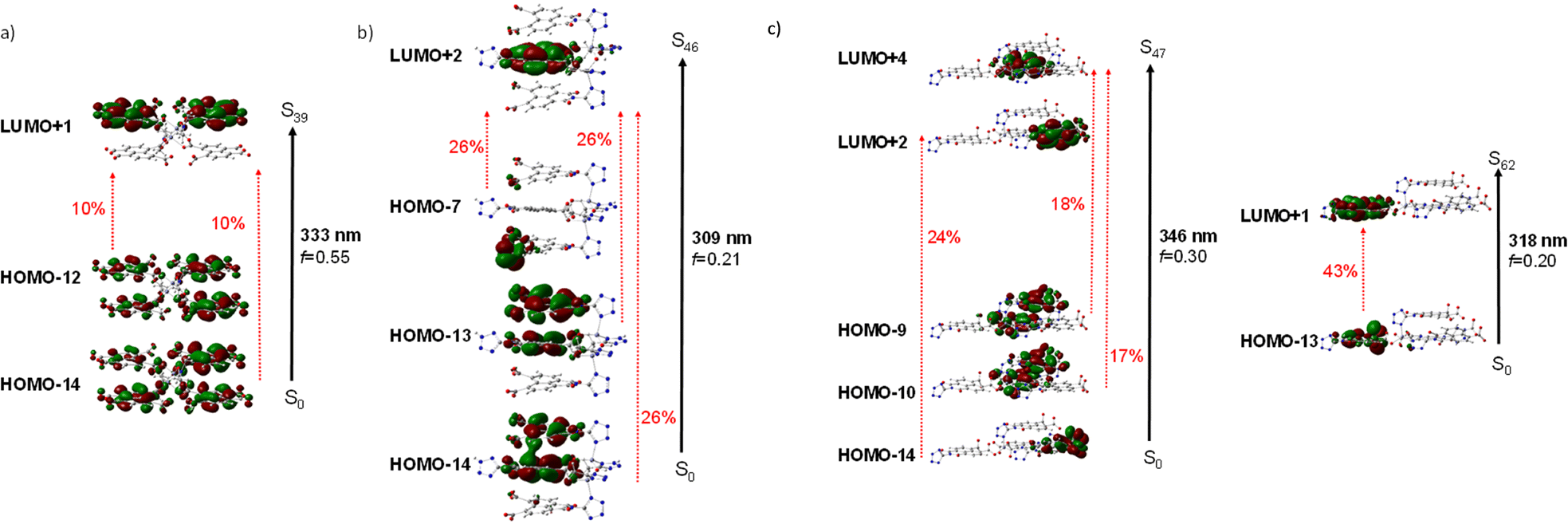 | ||
| Fig. 5 Schematic representation of a) the S0 → S39 electronic transition in compound 1, b) S0 → S46 electronic transition in compound 2 and c) of the S0 → S47 and S0 → S62 electronic transitions in compound 3 (isocontour plots 0.02 a.u.). | ||
Photoluminescence properties were studied for the polycrystalline samples of coordination polymers 1–3 as well as naphthalene-1,4,5,8-tetracarboxylic acid dianhydride and H3L ligand at room and low (20 K) temperature to explore their emission capacity. The formation ntca2− ligand was obtained in situ during the synthesis of compound 1, and could not be isolated from the reaction mixture, so the PL properties of its precursor naphthalene-1,4,5,8-tetracarboxylic acid dianhydride and the reported emission spectra were studied instead.44 As previously mentioned, these coordination polymers are expected to present photoluminescence properties under UV excitation given that their ligands framework contains an extended aromaticity linked to metal ions with closed-shell electronic configuration.24 Under monochromatic UV laser excitation (λex = 325 nm), all compounds present a main band peaking at the 430–465 nm range in addition to a second weak band at ca. 530 nm (Fig. 6), although their emission capacity is quite different (vide infra). In more detail, the emission bands of compound 1 cover the 400–650 nm with the band maxima sited at 465 nm and a wide shoulder peaking at 525 nm. Accordingly, the compound presents a dark blue emission under UV irradiation (λex = 365 nm, see photograph in Fig. 6). The shape of the spectrum is not unexpected as it resembles that of the free naphthalene-1,4,5,8-tetracarboxylic acid dianhydride precursor, in which both a main band and a shoulder at high wavelength are also observed (Fig. S1†), although a red-shift of around 1750 cm−1 may be claimed for 1 respect to the naphthalene-1,4,5,8-tetracarboxylic acid dianhydride. The emission spectra of free H2ntca ligand was previously reported by M. Hong and co-workers showing a band maxima at 487 and a broad peak at 550 nm under 346 nm radiation, in agreement with that shown in the present work.44 Since the emission spectra of compound 1, naphthalene-1,4,5,8-tetracarboxylic acid dianhydride and H2ntca are similar, the emission of 1 can be assigned to the intraligand (π–π*) fluorescence. The excitation spectra recorded for both of the emission maxima (λem = 465 and 525 nm) are similar except for a small excitation band peaking at 420 nm, which is only observed for the spectrum recorded at the highest wavelength λem = 525 nm (Fig. S9†). These excitation spectra are indeed consistent with the diffuse reflectance spectrum of the compound 1 (Fig. S16†), in which the wide absorption band peaks at 335 nm (with a shoulder at 380 nm) and covers the main excitation of 420 nm. According to the TD-DFT calculations (see Table S2†), the band observed at 465 nm in compound 1 could be assigned to the electronic transition S13 → S0 calculated at 437 nm. The second band at 525 nm could be assigned to the S9 → S0 electronic transition which is predicted at 511 nm. Therefore, the fact that both emissions come from different excited states explains well their shapes. In view of these results, it can be estimated that compound 1 presents a Stokes shift of ca. 8300 cm−1. The emission lifetimes were also estimated for this compound by means of the deconvolution of the whole decay curve with respect to the instrument reference file measured at the pulse wavelength. Surprisingly, the compound presents shorter lifetimes than free ligand sample at both emission maxima (1.44(1) and 3.32(1) ns respectively for λem = 460 nm and 525 nm vs. 5.61(1) ns for H3L ligand (see Table 1)). The lifetimes are of the same order in both cases and in the typical range of fluorescence, which discards the occurrence of intersystem crossing in the complexes.45,46 On the other hand, compounds 2 and 3 exhibit a very similar emission pattern, with the maximum of the most intense band sited at 430 nm, which is not surprising given that both compounds contain L3− ligand in their structures. In fact, the free H3L ligand fluoresces with a narrow emission band centred at 460 nm, in such a way that the emission of 2 and 3 is slightly blue-shifted. In any case, the spectrum of 2 presents a significant shoulder at 535 nm whereas the latter band is absent in 3. The existence of a minor emission band at 535 nm in 2 could be incorrectly assigned to the presence of 5-amino-tetrazolate co-ligand, as tetrazole-based compounds usually present an emission band at 325 nm.47 Consequently, the shoulder at 535 nm could correspond to an interligand charge transfer. In concordance to that observed for H3L, compound 2 neither shows a clear excitation band before the emission, meaning that the absorptions observed in the diffuse reflectance spectra lead to weak photoluminescent emissions. According to the absorption band maximum, compound 2 features a Stokes shift of ca. 6600 cm−1. Therefore, the emission capacity of the sample is comparatively lower than 1 as suggested by the photograph taken under UV irradiation (see Fig. 6), which is also supported by its low QY (vide infra). The lower intense emission shown by 2 might be attributed to the presence of weak π–π interactions taking place between the electron deficient aromatic clouds of the inner rings of the naphthalimide ligand in the ground state, known to partially quench the emission, or the possible rotational motions of the 5-amino-tetrazolate co-ligand around the Zn–N bond. To get deeper insight into the lower quantum yield of compound 2, it would be necessary to calculate the electronic couplings resulting from π–π interactions in the ground state, or to investigate the formation of excimers in the excited state which could quench the emission.48,49 However, the observed π–π stacking distances are 3.940 Å, somewhat larger than typical values (3.5 Å) for this type of interactions. This could suggest that other non-radiative relaxation mechanisms could be responsible for the lower emission of compound 2. Considering the molecular structure of each compound, a possible explanation could be that the molecular vibrations, and possible rotational motions of the 5-amino-tetrazolate co-ligand around the Zn–N bond in 2, may provide with an additional non-radiative relaxation pathway, justifying the lower quantum yield value observed in compound 2. In any case, it is worth highlighting that H3L ligand presents a similar absorption profile in view of the diffuse reflectance spectra of compounds 2 and 3 (see Fig. S17†), in which the main band has its maximum at ca. 345 nm and shows almost no more excitation bands above 400 nm. On its part, it must be highlighted that the main band of compound 3 (λem,max = 430 nm) is indeed composed of a shoulder peaking at 450 nm. The excitation spectra recorded over both wavelengths clearly shows the same pattern, characterized for a main excitation band with the maximum at 380 nm (Fig. S14†). Therefore, compound 3 presents the lowest Stokes shift for all herein reported compounds, being of ca. 6100 cm−1.
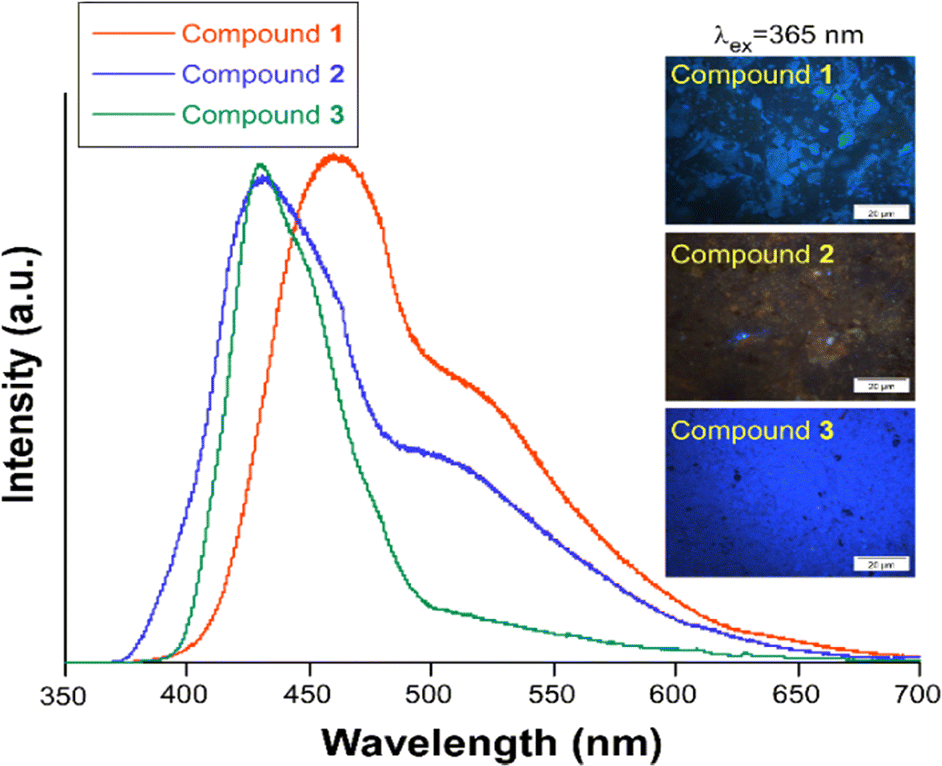 | ||
| Fig. 6 Emission spectra of compounds 1–3 at RT measured with λex = 325 nm. The micro-PL photographs taken over irradiated (λex = 365 nm) polycrystalline samples is also shown. | ||
The band observed at 430 nm could be assigned to the electronic transitions S15 → S0 and S20 → S0, calculated at 450 and 426 nm in compounds 2 and 3, respectively. The second band at 535 nm in compound 2 would correspond to the electronic transition S7 → S0 predicted at 539 nm. Finally, in the case of compound 3, the shoulder observed at 450 nm could be assigned to the transition S18 → S0 calculated at 452 nm. Fig. S23† represents a selection of the molecular orbitals involved in the electronic relaxation. As for the absorption process, these molecular orbitals are localized on the ntca2− and L3− ligands without participation of the metallic Zn(II) atom (Fig. S23†).
The mean emission lifetimes recorded for compounds 2 0.89(3) ns (λem = 430 nm) and 2.79(2) ns (λem = 535 nm) and 3 3.25(1) ns (λem = 430 nm) and 0.47(3) ns (λem = 450 nm) are in the same order than shown by the H3L ligand (τ = 1.00(3) ns, λem = 460 nm), as usually occurs for most of CPs owing to the restricted molecular vibrations that reduce the non-radiative quenching.46 As previously observed for compound 1, the lifetimes are short and in the range of few nanoseconds, which confirms the fluorescent nature of the process. Phosphorescence emission has been previously reported for NDI ligand based Zn(II) coordination polymers,50 where sorption of aromatic guest molecules lead to the formation of complexes with charge transfer nature, producing fluorescence from the exciplex or the stabilisation of the triplet state of the NDI to generate phosphorescence. Thus, the phosphorescence emission in compounds 1–3 could be inhibited by the electron deficient nature of both ncta2− and L3− ligands and absence of electron rich π-donors guest molecules in the framework. Also, no significant intersystem crossing occurs in compounds 1–3 upon ligand coordination to Zn(II), known to afford heavy-atom effect,51 which may be related to the fact that the main excitations and emissions occur between molecular orbitals located on the inner rings of the ligands and thus, quite far from the metal centre. The PL quantum yields measured on solid state for the three compounds with λex = 325 nm monochromatic light are in line with the micro-PL images presented before. The most efficient emission is observed for 1 > 3 > 2 with the values shown in Table 1. In this regard, it is worth mentioning that the low QY of compound 2 is concordant with the lack of excitation bands in the UV region, although its QY is increased up to 1.1(5)% when the sample is excited with λex = 400 nm.
Conclusions
The synthesis of three novel photoluminescent coordination polymers and a new H3L ligand has been described. Novel H3L is formed by benzo[de]isoquinoline core linked to a tetrazolate and two carboxylate groups. Compound 1 comprises Zn2(ntca)4(DMF)2 paddlewheels linked by ntca2− ligands in a 1D coordination polymer. Compound 2 grows up from pairs of tetrahedrally coordinated Zn centres linked by μ2-OH groups, which are further bridged by novel L3− ligands and 5-amino-tetrazolate. L3− ligands coordinate to the Zn(II) metal centres through the tetrazolate and the two monodentate carboxylate groups. The resulting anionic 2D layered coordination polymer is packed by means of hydrogen bonds formed by the carboxylate groups with the DMA cations and crystallization water molecules. Compound 3 is formed by tetrahedrally coordinated Zn atoms linked by bridging tetrazolate and monodentate carboxylate groups, resulting in an anionic 3D coordination polymer, whose pores are filled by DMA cations. The photoluminescence properties were experimentally characterized by measuring the UV excitation and emission spectra, and evaluated by TD-DFT quantum chemical calculations, concluding that the PL emission in compounds 1–3 is originated in the electronic transitions of the ligands with no participation of the Zn(II) atoms. The compounds show quite distinct PL properties, summarized by traceable blue luminescence of compounds 1 and 3 but a poor emission in 2. The lower quantum yield observed in 2 is tentatively attributed to weak π–π interactions of the aromatic clouds of L3− or the molecular vibrations and/or possible rotational motions of the 5-amino-tetrazolate co-ligand around the Zn–N bond. The observed PL emission possesses an intraligand character as confirmed by TD-DFT calculations. Nonetheless, herein described CPs present more long-lived emissions in the main band maxima (λem,max ≈ 430–465 nm) as well as a second weaker emission band, not present in the free ligands and coming from a low-lying excited state, which is sited at higher wavelengths (λem,max ≈ 525–535 nm for 1 and 2, a shoulder with λem,max ≈ 450 nm for 3) and may be related to an interligand charge transfer. Among all CPs, compound 1 is characterized for the highest emission quantum yield.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Gobierno Vasco/Eusko Jaurlaritza (IT1755-22, IT1500-22) and Junta de Andalucía (FQM-394 and P21_00386). The authors thank the technical and human support provided by SGIker of UPV/EHU and European funding (ERDF and ESF)). Junta de Andalucía (PAIDI-FQM-337) is thanked for supporting the research described in this article and the Centro de Servicios de Informática y Redes de Comunicaciones (CSIRC, Universidad de Granada) for providing the computer time that made this work possible. The authors also thank to Universidad de Jaén, FEDER_UJA_2020 (project 2021/00627/001) for supporting the research described in this article. M. M. Q.-M. and S. R. thank Ministerio de Ciencia e Innovación for a Ramón y Cajal contracts (the publication is part of the grants RYC2021-034288-I and RYC2021-032522-I respectively, funded by MCIN/AEI/10.13039/501100011033 and by the European Union “Next Generation-EU/PRTR”).Notes and references
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Introduction to Metal–Organic Frameworks, Chem. Rev., 2012, 112(2), 673–674 CrossRef CAS PubMed.
- J. J. Perry, J. A. Perman and M. J. Zaworotko, Design and synthesis of metal–organic frameworks using metal–organic polyhedra as supermolecular building blocks, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC.
- A. Y. Robin and K. M. Fromm, Coordination polymer networks with O- and N-donors: What they are, why and how they are made, Coord. Chem. Rev., 2006, 250, 2127–2157 CrossRef CAS.
- Z. Chen, M. C. Wasson, R. J. Drout, L. Robison, K. B. Idrees, J. G. Knapp, F. A. Son, X. Zhang, W. Hierse, C. Kühn, S. Marx, B. Hernandez and O. K. Farha, The state of the field: from inception to commercialization of metal–organic frameworks, Faraday Discuss., 2021, 225, 9–69 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The chemistry and applications of metal–organic frameworks, Science, 2013, 341, 6149 CrossRef PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Flexible metal–organic frameworks, Chem. Soc. Rev., 2014, 43, 6062–6069 RSC.
- G. Férey, Hybrid porous solids: past, present, future, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- C. R. Murdock, B. C. Hughes, Z. Lu and D. M. Jenkins, Approaches for synthesizing breathing MOFs by exploiting dimensional rigidity, Coord. Chem. Rev., 2014, 258–259, 119–136 CrossRef CAS.
- G. Mínguez Espallargas and E. Coronado, Magnetic functionalities in MOFs: from the framework to the pore, Chem. Soc. Rev., 2018, 47, 533–557 RSC.
- A. E. Thorarinsdottir and T. D. Harris, Metal–Organic Framework Magnets, Chem. Rev., 2020, 120(16), 8716–8789 CrossRef CAS PubMed.
- D. Yang and B. C. Gates, Catalysis by Metal Organic Frameworks: Perspective and Suggestions for Future Research, ACS Catal., 2019, 9(3), 1779–1798 CrossRef CAS.
- X. Yan, X. Su, J. Chen, C. Jin and L. Chen, Two-Dimensional Metal-Organic Frameworks Towards spintronics, Angew. Chem., 2023, 62, 10 Search PubMed.
- L.-T. Zhang, Y. Zhou and S.-T. Han, The Role of Metal–Organic Frameworks in Electronic Sensors, Angew. Chem., 2021, 60, 15192–15212 CrossRef CAS PubMed.
- T. Qiu, Z. Liang, W. Guo, H. Tabassum, S. Gao and R. Zou, Metal–Organic Framework-Based Materials for Energy Conversion and Storage, ACS Energy Lett., 2020, 5(2), 520–532 CrossRef CAS.
- S. Rojas, A. Rodríguez-Diéguez and P. Horcajada, Metal-Organic Frameworks in Agriculture, ACS Appl. Mater. Interfaces, 2022, 14, 16983–17007 CrossRef CAS PubMed.
- J. Cepeda and A. Rodríguez-Diéguez, Tuning the luminescence performance of metal-organic frameworks based on d10 metal ions: from an inherent versatile behaviour to their response to external stimuli, CrystEngComm, 2016, 18, 8556 RSC.
- Y. Cui, J. Zhang, H. He and G. Qian, Photonic functional metal–organic frameworks, Chem. Soc. Rev., 2018, 47, 5740 RSC.
- M. Gutiérrez, Y. Zhang and J.-C. Tan, Confinement of Luminescent Guests in Metal−Organic Frameworks: Understanding Pathways from Synthesis and Multimodal Characterization to Potential Applications of LG@MOF Systems, Chem. Rev., 2022, 122, 10438–10483 CrossRef PubMed.
- W. Yang, G. Chang, H. Wang, T.-L. Hu, Z. Yao, K. Alfooty, S. Xiang and B. Chen, A Three-Dimensional Tetraphenylethene-Based Metal–Organic Framework for Selective Gas Separation and Luminescence Sensing of Metal Ions, Eur. J. Inorg. Chem., 2016, 27, 4470–4475 CrossRef.
- Y. Zhao, H. Zeng, X.-W. Zhu, W. Lu and D. Li, Metal–organic frameworks as photoluminescent biosensing platforms: mechanisms and applications, Chem. Soc. Rev., 2021, 50, 4484 RSC.
- Z. Li, F. Jiang, M. Yu, S. Li, L. Chen and M. Hong, Achieving gas pressure-dependent luminescence from an AIEgen-based metal-organic framework, Nat. Commun., 2022, 13, 2142 CrossRef CAS PubMed.
- J. M. Seco, E. San Sebastián, J. Cepeda, B. Biel, A. Salinas-Castillo, B. Fernández, D. P. Morales, M. Bobinger, S. Gómez-Ruiz, F. C. Loghin, A. Rivadeneyra and A. Rodríguez-Diéguez, Framework based on Perylene-3,4,9,10-tetracarboxylate as Sensing Layer for Humidity Actuators, Sci. Rep., 2018, 8, 14414 CrossRef PubMed.
- Y.-X. Xie, W.-N. Zhao, G.-C. Li, P.-F. Liu and L. Han, A Naphthalenediimide-Based Metal−Organic Framework and Thin Film Exhibiting Photochromic and Electrochromic Properties, Inorg. Chem., 2016, 55, 549–551 CrossRef CAS PubMed.
- Y. Zhou and L. Han, Recent advances in naphthalenediimide-based metal-organic frameworks: Structures and applications, Coord. Chem. Rev., 2021, 430, 213665 CrossRef CAS.
- Y. Zhou, L. Han and W.-J. Chenc, Inter-ligand charge-transfer interactions in a photochromic and redox active zinc–organic framework, CrystEngComm, 2021, 23, 5982–5988 RSC.
- Z. P. Demko and K. B. Sharpless, Preparation of 5-Substituted 1H-Tetrazoles from Nitriles in Water, J. Org. Chem., 2001, 66, 7945–7950 CrossRef CAS PubMed.
- Z. P. Demko and K. B. Sharpless, An Expedient Route to the Tetrazole Analogues of α-Amino Acids, Org. Lett., 2002, 4, 2525–2527 CrossRef CAS PubMed.
- Z. P. Demko and K. B. Sharpless, An intramolecular [2 + 3] cycloaddition route to fused 5-heterosubstituted tetrazoles, Org. Lett., 2001, 3, 4091–4094 CrossRef CAS PubMed.
- A. J. Calahorro, A. Salinas-Castillo, J. M. Seco, J. Zuñiga, E. Colacio and A. Rodríguez-Diéguez, Luminescence and magnetic properties of three metal–organic frameworks based on the 5-(1H-tetrazol-5-yl)isophthalic acid ligand, CrystEngComm, 2013, 15, 7636–7639 RSC.
- A. Rodríguez-Diéguez, A. J. Mota, J. M. Seco, M. A. Palacios, A. Romerosa and E. Colacio, Influence of metal ions, coligands and reaction conditions on the structural versatility and properties of 5-pyrimidyl-tetrazolate containing complexes, Dalton Trans., 2009, 9578–9586 RSC.
- H. Zhao, Z.-R. Qu, H.-Y. Ye and R.-G. Xiong, In situ hydrothermal synthesis of tetrazole coordination polymers with interesting physical properties, Chem. Soc. Rev., 2008, 37, 84–100 RSC.
- G. M. Sheldrick, SHELXT – Integrated Space-Group and Crystal Structure Determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- J. Rodríguez-Carvajal, FULLPROF 2000,version 2.5d, Lab. Léon Brillouin (CEA-CNRS), Cent. d'Études Saclay, Gif sur Yvette Cedex, Fr., 2000 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- T. H. Dunning Jr. and P. J. Hay, Modern Theoretical Chemistry, Plenum Press, New York, 1977 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- T. C. Barros, I. M. Cuccovia, J. P. S. Farah, J. C. Masini, H. Chaimovichc and M. J. Politi, Mechanism of 1,4,5,8-naphthalene tetracarboxylic acid dianhydride hydrolysis and formation in aqueous solution, Org. Biomol. Chem., 2006, 4, 71–82 RSC.
- G.-Q. Kong and C.-D. Wu, A self-assembled supramolecular solid for catalytic application, Inorg. Chem. Commun., 2009, 12, 731–734 CrossRef CAS.
- M. Usman, G. Haider, S. Mendiratta, T.-T. Luo, Y.-F. Chen and K.-L. Lu, Continuous broadband emission from a metal–organic framework as a human-friendly white light source, J. Mater. Chem. C, 2016, 4, 4728 RSC.
- R. Shankar and A. Dubey, Hydrothermal Approach for Reticular Synthesis of Coordination Assemblies with Dicarboxylatotetramethyldistannoxanes as the Secondary Building Units, Eur. J. Inorg. Chem., 2020, 3877–3883 CrossRef CAS.
- Y. Xu, D. Yuan, B. Wu, F. Jiang, Y. Zhou and M. Hong, Two photoluminescent coordination polymers based on naphthalene-1,4,5,8-tetracarboxylic acid 4,5-anhydride Inorg, Chem. Commun., 2005, 651–655 CAS.
- J. Cepeda, S. Pérez-Yáñez and A. Rodríguez-Diéguez, Adv. Mater. Sci. Res., 2017, 28, 93–150 Search PubMed.
- E. San Sebastian, A. Rodríguez-Diéguez, J. M. Seco and J. Cepeda, Coordination Polymers with Intriguing Photoluminescence Behavior: The Promising Avenue for Greatest Long-Lasting Phosphors, Eur. J. Inorg. Chem., 2018, 2155–2174 CrossRef CAS.
- X. W. Wang, J. Z. Chen and J. H. Liu, Photoluminescent Zn(II) Metal−Organic Frameworks Built from Tetrazole Ligand: 2D Four-Connected Regular Honeycomb (4363)-net, Cryst. Growth Des., 2007, 7, 1227–1229 CrossRef CAS.
- J. M. G. Martinho, A. T. Reis e Sousa, M. E. Oliveira Torres and A. Fedorov, Fluorescence quenching of pyrene monomer and excimer by CH3I, Chem. Phys., 2001, 264, 111–121 CrossRef CAS.
- P. Conlon, C. J. Yang, Y. Wu, Y. Chen, K. Martinez, Y. Kim, N. Stevens, A. A. Marti, S. Jockusch, N. J. Turro and W. Tan, Pyrene Excimer Signaling Molecular Beacons for Probing Nucleic Acids, J. Am. Chem. Soc., 2008, 130, 336–342 CrossRef CAS PubMed.
- Y. Takashima, V. Martínez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Molecular decoding using luminescence from an entangled porous framework, Nat. Commun., 2011, 2, 168 CrossRef PubMed.
- Z. Xiong, W. Gong, P. Xu, M. Jiang, X. Cai, Y. Zhu, X. Ping, H. Feng, H. Ma and Z. Qian, Reexamining the heavy-atom-effect: The universal heavy-atom-induced fluorescence enhancement principle for through-space conjugated AIEgens, Chem. Eng. J., 2023, 451, 139030 CrossRef CAS.
- Due to the weak signal obtained, the wavelength of the peaks maximum could not be estimated.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2312265–2312267. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce01233f |
| This journal is © The Royal Society of Chemistry 2024 |