
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Uranyl ion coordination polymers with the dibenzobarrelene-based rac- and (R,R)-trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylate ligands†
Young Hoon
Lee
 a,
Sotaro
Kusumoto
b,
Youssef
Atoini
c,
Yoshihiro
Koide
b,
Shinya
Hayami
*d,
Yang
Kim
*d,
Jack
Harrowfield
*e and
Pierre
Thuéry
*f
a,
Sotaro
Kusumoto
b,
Youssef
Atoini
c,
Yoshihiro
Koide
b,
Shinya
Hayami
*d,
Yang
Kim
*d,
Jack
Harrowfield
*e and
Pierre
Thuéry
*f
aDepartment of Chemistry, University of Ulsan, Tekeunosaneop-ro 55beon-gil, Nam-gu, Ulsan 44610, Republic of Korea
bDepartment of Material & Life Chemistry, Kanagawa University, 3-27-1 Rokkakubashi, Kanagawa-ku, Yokohama 221-8686, Japan
cTechnical University of Munich, Campus Straubing, Schulgasse 22, 94315 Straubing, Germany
dDepartment of Chemistry, Graduate School of Science and Technology, Institute of Industrial Nanomaterials (IINa), Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto 860-8555, Japan. E-mail: hayami@kumamoto-u.ac.jp; ykim@kumamoto-u.ac.jp
eUniversité de Strasbourg, ISIS, 8 allée Gaspard Monge, 67083 Strasbourg, France. E-mail: harrowfield@unistra.fr
fUniversité Paris-Saclay, CEA, CNRS, NIMBE, 91191 Gif-sur-Yvette, France. E-mail: pierre.thuery@cea.fr
First published on 1st February 2024
Abstract
trans-9,10-Dihydro-9,10-ethanoanthracene-11,12-dicarboxylic acid (deadcH2), in its racemic or R,R enantiomeric forms, has been used to synthesize eight uranyl ion complexes under solvo-hydrothermal conditions. [UO2(deadc)]·1.5CH3CN (1) and [H2NMe2]2[(UO2)2(deadc)3]·2H2O (2) crystallize as monoperiodic coordination polymers in which deadc2− forms both 4- and 7-membered chelate rings. Although synthesized in the same conditions as 2, the enantiomerically pure complex [H2NMe2]4[(UO2)2(O)(R,R-deadc)2]2 (3) is a discrete tetranuclear complex containing two μ3-oxo anions. Association with the zwitterion Ni(tpyc)2, where tpyc− is 2,2′:6′,2′′-terpyridine-4′-carboxylate, gives [(UO2)2(deadc)(deadcH)(NO3)Ni(tpyc)2]·CH3CN·2H2O (4), a rake-shaped monoperiodic assembly. [UO2(deadc)(DMA)] (5), [UO2(deadc)] (6) and [PPh4]2[(UO2)2(R,R-deadc)3] (7) crystallize as diperiodic networks with the fes, sql and hcb topologies, respectively, the thick layers formed being coated on both sides by protruding, hydrophobic dibenzobarrelene groups. Finally, [(UO2)2Ag2(deadc)3(CH3CN)2]·0.5H2O (8) contains monoperiodic uranyl–deadc2− subunits which are assembled into a triperiodic framework by bridging silver(I) cations, the latter interacting with both carboxylate groups and aromatic rings. Except for 6, all these complexes are emissive with photoluminescence quantum yields of 2–26%, and most spectra display the usual vibronic fine structure of uranyl emission.
Introduction
trans-9,10-Dihydro-9,10-ethanoanthracene-11,12-dicarboxylic acid (deadcH2, Scheme 1) has been the subject of various studies of its capacity to form, sometimes selectively, crystalline clathrates with small organic molecules.1–4 Less well established are the roles of its conjugate anions in coordination chemistry, though both deadcH− and deadc2− have been shown to be useful counteranions for crystallization of organometallic cations5 and structural studies of complexes formed with CoII and CdII have shown deadc2− to be suited to the formation of coordination polymers.6 The acid deadcH2 is chiral and while several methods are known for its resolution into enantiomers,7 only in one instance has resolved deadc2− been used as a ligand in an osmium complex, not structurally characterized, employed as a catalyst for asymmetric synthesis.8 | ||
| Scheme 1 The diacid deadcH2. | ||
The ligand deadc2− has carboxylate donor groups in a similar spatial disposition to those of trans-1,2-cyclohexanedicarboxylate (t-1,2-chdc2−). The latter ligand has been shown, both as its racemate and pure enantiomers, to give uranyl ion complexes of several forms including closed tetranuclear species as well as mixed-ligand complexes,9 with one of them involving both anionic (R,R-t-1,2-chdc2−) and zwitterionic carboxylate donors.9g Modelling of the isolated diacid deadcH2 (at MM2 level using Chem3D10) provides a disposition of the carboxyl groups (C⋯C distance, 3.52 Å) intermediate between those found experimentally, for example, in crystal structures of the racemic acid (3.62 Å)1a and its S,S isomer (3.40 Å)1b (as its hexane adduct), indicating, along with moderate torsion angle differences, that there is rather limited flexibility of the molecule. The same distance and torsion angles found in organometallic complexes of deadcH− and deadc2− anions not directly coordinated to a metal ion5 show that deprotonation has little obvious effect on this degree of flexibility. The carboxylate C⋯C separation for deadc2− here is intermediate between those of the diaxial isomer of t-1,2-chdc2− (3.86–3.88 Å)9c and its more commonly observed diequatorial form (2.96–3.12 Å)9a,b but closer to the former, indicating that it should favour polymer formation over that of closed oligomers. In the known transition metal ion complexes of deadc2−, which are in fact both polymers,6 the values for this separation (3.35–3.54 Å) fall within the range of the values given above. Another obvious difference between deadc2− and t-1,2-chdc2− is the much greater bulkiness of the dibenzobarrelene compared to the cyclohexane platform, and the presence of aromatic rings with the variety of interactions that they entail.
Although an impressive variety of polycarboxylates have been used for the synthesis of coordination polymers and frameworks based on the uranyl ion,11 there has been no report of a complex involving deadc2−. Since the peculiarities of this ligand, i.e. both the closeness of its coordination sites to those of 1,2-chdc2−, and the hydrophobicity of the dibenzobarrelene platform, led us to anticipate novel types of arrangements, we synthesized eight uranyl ion complexes involving either rac- (racemic) or R,R-deadc2− under solvo-hydrothermal conditions. As in previous work, we have used various additional species, either neutral in the form of coordinating cosolvents, or cationic as structure-directing counterions. Additional metal cations were also used in two cases, silver(I) for its ability to interact with aromatic rings, and nickel(II) in association with 2,2′:6′,2′′-terpyridine-4′-carboxylic acid (tpycH), a combination producing the zwitterionic, “expanded” ligand Ni(tpyc)2. All these complexes have been characterized by their crystal structure and emission properties in the solid state.
Experimental
Synthesis
11R*,12R*-9,10-Dihydro-9,10-ethanoanthracene-11,12-dicarboxylic acid (rac-deadcH2). This racemate was prepared by a literature method12 (see ESI† for details). 1H NMR (DMSO-d6): δ 7.42–7.40 (m, 2H), 7.28–7.26 (m, 2H), 7.14–7.07 (m 4H), 4.73 (s, 2H), 3.13 (s, 2H) (Fig. S1†).
(11R,12R)-(+)-9,10-Dihydro-9,10-ethanoanthracene-11,12-dicarboxylic acid (R,R-deadcH2). The racemate was resolved into each enantiomer as in the literature by formation of its diastereomeric inclusion complexes with S-proline7c (see ESI† for details).
[UO2(NO3)2(H2O)2]·4H2O (RP Normapur, 99%), Ni(NO3)2·6H2O, and AgNO3 were purchased from Prolabo; 2,2′:6′,2′′-terpyridine-4′-carboxylic acid (tpycH) and guanidinium nitrate were from Alfa-Aesar. Elemental analyses were performed by MEDAC Ltd. For all syntheses, the solutions were placed in 10 mL tightly closed glass vessels (Pyrex culture tubes with SVL15 stoppers and Teflon-coated seals, provided by VWR) and heated at 140 °C in a sand bath (Harry Gestigkeit ST72). The crystals were grown in the hot, pressurized solutions and not as a result of a final return to ambient conditions.
[UO2(deadc)]·1.5CH3CN (1). deadcH2 (15 mg, 0.05 mmol), [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol), and guanidinium nitrate (12 mg, 0.10 mmol) were dissolved in a mixture of water (0.4 mL) and acetonitrile (0.2 mL). Yellow crystals of complex 1 were obtained within one week (11 mg, 36%). Chemical analysis indicates the loss of 0.5 acetonitrile molecule with respect to the formula derived from crystal structure determination. Anal. calcd for C20H15NO6U: C, 39.81; H, 2.51; N, 2.32. Found: C, 39.50; H, 2.50; N, 2.26%.
[H2NMe2]2[(UO2)2(deadc)3]·2H2O (2). deadcH2 (15 mg, 0.05 mmol) and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.5 mL) and N,N-dimethylformamide (0.2 mL). Yellow crystals of complex 2 were obtained overnight (19 mg, 49%). Anal. calcd for C58H56N2O18U2: C, 45.09; H, 3.65; N, 1.81. Found: C, 44.76; H, 3.67; N, 1.82%.
[H2NMe2]4[(UO2)2(O)(R,R-deadc)2]2 (3). R,R-deadcH2 (15 mg, 0.05 mmol) and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.5 mL) and N,N-dimethylformamide (0.2 mL). Yellow crystals of complex 3 were obtained within four days (7 mg, 23%). Chemical analysis indicates the presence of about three water molecules in excess of the formula derived from crystal structure determination. Anal. calcd for C80H80N4O26U4 + 3H2O: C, 38.14; H, 3.44; N, 2.22. Found: C, 38.06; H, 3.22; N, 2.15%.
[(UO2)2(deadc)(deadcH)(NO3)Ni(tpyc)2]·CH3CN·2H2O (4). deadcH2 (15 mg, 0.05 mmol), [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol), Ni(NO3)2·6H2O (10 mg, 0.03 mmol), and tpycH (14 mg, 0.05 mmol), were dissolved in a mixture of water (0.6 mL) and acetonitrile (0.2 mL). Orange crystals of complex 4 were obtained within four days (40 mg, 85%). Anal. calcd for C70H52N8NiO21U2: C, 44.82; H, 2.79; N, 5.97. Found: C, 44.20; H, 2.84; N, 5.86%.
[UO2(deadc)(DMA)] (5). deadcH2 (15 mg, 0.05 mmol) and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.5 mL) and N,N-dimethylacetamide (0.2 mL). Yellow crystals of complex 5 were obtained overnight (22 mg, 68%). Anal. calcd for C22H21NO7U: C, 40.69; H, 3.26; N, 2.16. Found: C, 40.71; H, 3.27; N, 2.10%.
[UO2(deadc)] (6). deadcH2 (15 mg, 0.05 mmol), [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol), and CsNO3 (20 mg, 0.10 mmol) were dissolved in a mixture of water (0.6 mL) and acetonitrile (0.2 mL). Yellow crystals of complex 6 were obtained within three days (20 mg, 71%). Chemical analysis indicates the presence of 0.5 acetonitrile in excess of the formula derived from crystal structure determination. Anal. calcd for C18H12O6U + 0.5CH3CN: C, 39.16; H, 2.33; N, 1.20. Found: C, 39.16; H, 2.36; N, 1.12%.
[PPh4]2[(UO2)2(R,R-deadc)3] (7). R,R-deadcH2 (15 mg, 0.05 mmol), [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol), and PPh4Br (21 mg, 0.05 mmol) were dissolved in a mixture of water (0.5 mL) and N,N-dimethylacetamide (0.2 mL). Yellow crystals of complex 7 were obtained overnight (15 mg, 43%). Anal. calcd for C102H76O16P2U2: C, 58.46; H, 3.66. Found: C, 58.11; H, 3.71%.
[(UO2)2Ag2(deadc)3(CH3CN)2]·0.5H2O (8). deadcH2 (15 mg, 0.05 mmol), [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol), and AgNO3 (17 mg, 0.10 mmol) were dissolved in a mixture of water (0.6 mL) and acetonitrile (0.2 mL). Yellow crystals of complex 8 were obtained within four days (20 mg, 46%). Chemical analysis indicates the possible presence of one water molecule in excess of the formula derived from crystal structure determination. Anal. calcd for C58H43Ag2N2O16.5U2 + H2O: C, 40.00; H, 2.60; N, 1.61. Found: C, 39.78; H, 2.48; N, 1.38%.
Crystallography
Data collections were performed at 100(2) K on a Bruker D8 Quest diffractometer using an Incoatec Microfocus Source (IμS 3.0 Mo) and a PHOTON III area detector, and operated with APEX3.13 The data were processed with SAINT,14 and empirical absorption corrections were made with SADABS.15 The structures were solved by intrinsic phasing with SHELXT,16 and refined by full-matrix least-squares on F2 with SHELXL,17 using the ShelXle interface.18 The hydrogen atoms bound to oxygen and nitrogen atoms in 2 and 4 were retrieved from residual electron density maps and they were refined with geometric restraints. All other hydrogen atoms in all compounds were introduced at calculated positions and treated as riding atoms with an isotropic displacement parameter equal to 1.2 times that of the parent atom (1.5 for CH3). In 1, one of the solvent acetonitrile molecules has been given an occupancy factor of 0.5 to account for its closeness to its image by symmetry. In 3, one dimethylammonium cation is disordered over two positions which have been refined with occupancy parameters constrained to sum to unity and with restraints on bond lengths and displacement parameters. In 8, the solvent water molecule has been given partial occupancy so as to retain an acceptable displacement parameter. For compounds 3 and 4, the SQUEEZE19 software was used to subtract the contribution of disordered solvent molecules to the structure factors; in compound 3, about 15 electrons per formula unit were thus added in the solvent-accessible volume, which is less than the number corresponding to the three water molecules found from elemental analysis (see above), the volume being however sufficiently large to accommodate them (121 Å3 per formula unit); in compound 4, about 25 electrons per formula unit were added in a volume of 118 Å3, possibly corresponding to about 2.5 water molecules, these being apparently lost during drying of the crystals before elemental analysis. Crystal data and structure refinement parameters are given in Table 1. Drawings were made with ORTEP-3,20 and VESTA,21 and topological analyses were performed with ToposPro.22| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|---|
| Chemical formula | C21H16.5N1.5O6U | C58H56N2O18U2 | C80H80N4O26U4 | C70H52N8NiO21U2 | C22H21NO7U | C18H12O6U | C102H76O16P2U2 | C58H43Ag2N2O16.5U2 |
| M/g mol−1 | 623.89 | 1545.10 | 2465.60 | 1875.96 | 649.43 | 562.31 | 2095.62 | 1723.74 |
| Crystal system | Triclinic | Monoclinic | Orthorhombic | Triclinic | Monoclinic | Monoclinic | Orthorhombic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c | P21212 |
P |
C2/c | P21/c | P21212 | C2/c |
| a/Å | 8.7652(4) | 15.7139(5) | 17.5195(6) | 8.6348(3) | 28.9120(13) | 8.4166(3) | 23.3097(6) | 15.4596(4) |
| b/Å | 10.7608(5) | 15.0080(5) | 26.5815(7) | 14.3691(5) | 11.9986(5) | 21.8953(7) | 13.3655(3) | 15.1930(4) |
| c/Å | 11.2060(5) | 22.3323(6) | 8.8808(3) | 27.8539(11) | 13.6561(5) | 8.4203(3) | 13.5379(3) | 21.8735(5) |
| α/° | 103.3336(18) | 90 | 90 | 93.9567(17) | 90 | 90 | 90 | 90 |
| β/° | 92.5999(18) | 96.4976(11) | 90 | 98.1776(17) | 95.2025(17) | 93.6517(12) | 90 | 95.5571(9) |
| γ/° | 107.6526(17) | 90 | 90 | 94.8470(16) | 90 | 90 | 90 | 90 |
| V/Å3 | 972.43(8) | 5232.9(3) | 4135.7(2) | 3397.1(2) | 4717.8(3) | 1548.58(9) | 4217.68(17) | 5113.5(2) |
| Z | 2 | 4 | 2 | 2 | 8 | 4 | 2 | 4 |
| Reflections collected | 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 533 533 |
85794 |
53405 |
137913 |
63094 |
19826 |
96094 |
90293 |
| Independent reflections | 3696 | 6774 | 7839 | 12911 |
4484 | 3990 | 10917 |
6585 |
| Observed reflections [I > 2σ(I)] | 3431 | 6126 | 6652 | 12122 |
4047 | 3814 | 10575 |
6542 |
| R int | 0.061 | 0.057 | 0.118 | 0.061 | 0.063 | 0.039 | 0.051 | 0.043 |
| Parameters refined | 282 | 375 | 533 | 935 | 283 | 226 | 551 | 371 |
| R 1 | 0.021 | 0.019 | 0.047 | 0.036 | 0.034 | 0.016 | 0.019 | 0.018 |
| wR2 | 0.046 | 0.043 | 0.106 | 0.080 | 0.089 | 0.039 | 0.041 | 0.047 |
| S | 1.151 | 1.059 | 1.035 | 1.208 | 1.174 | 1.119 | 1.038 | 1.073 |
| Δρmin/e Å−3 | −1.73 | −0.84 | −1.07 | −2.32 | −1.31 | −1.07 | −0.47 | −0.88 |
| Δρmax/e Å−3 | 0.95 | 0.69 | 1.43 | 2.07 | 2.16 | 1.28 | 0.44 | 1.56 |
| Flack parameter | 0.016(17) | −0.012(4) |
Luminescence measurements
Emission spectra were recorded on solid samples using an Edinburgh Instruments FS5 spectrofluorimeter equipped with a 150 W CW ozone-free xenon arc lamp, dual-grating excitation and emission monochromators (2.1 nm mm−1 dispersion; 1200 grooves per mm) and an R928P photomultiplier detector. The powdered compounds were pressed to the wall of a quartz tube, and the measurements were performed using the right-angle mode in the SC-05 cassette. An excitation wavelength of 420 nm was used in all cases and the emission was monitored between 450 and 600 nm. Gaussian deconvolution of the spectrum of 4 was made with the Origin software. The quantum yield measurements were performed by using a Hamamatsu Quantaurus C11347 absolute photoluminescence quantum yield spectrometer and exciting the samples between 300 and 400 nm.Results and discussion
Synthesis
All the complexes presently described were obtained from reaction mixtures in which the UVI/deadcH2 ratio was 1:1 and in 5 of the 8 crystalline solids obtained, this stoichiometry was retained, though in a considerable variety of structures. If it is assumed that crystal deposition is not kinetically controlled, the crystals deposited have to be simply those of the least soluble component of the reaction mixture. This would explain why, when additional non-coordinating cations (H2NMe2+, PPh4+) are present in the mixture, it is possible to obtain crystals incorporating these cations in which UVI is present in an anionic component where the ratio U/deadc2− is 1:1.5 (complexes 2 and 7). Where an additional cation is present, such as C(NH2)3+ in the synthesis of complex 1 and Cs+ in that of complex 6, but is not incorporated in the precipitated crystals, any influence it may have on the solution equilibria is uncertain but it does not prevent the formation of crystals of a neutral 1:1 complex.
In general, it is unsurprising to find that different solvates may have different structures and this is seen when complex 5 is compared to 1 and 6 (see structural discussion ahead), even though all have a U/deadc2− ratio of 1:1. Where AgI is the additional cation and interacts, along with UVI, with the deadc2− ligand, its incorporation in the isolated crystals (complex 8) leads to a U/deadc2− ratio of 1:1.5, formally because its insolubility is favoured by cation⋯anion attraction. In contrast, where NiII is the additional cation but is cloaked within an additional ligand (tpyc−) so as to be a neutral (zwitterionic) component not in direct interaction with deadc2−, the 1:1 ratio is regained (complex 4). In the case of complexes 2 and 3, the cation is generated through hydrolysis of the cosolvent N,N-dimethylformamide (DMF), a reaction which must result in buffering of the solution pH and thus modification of the solution equilibria. These two complexes display a marked difference in composition, although they were prepared under identical conditions except that for the former the deadcH2 reactant was the racemate and for the latter the R,R enantiomer. The structure of 2 (see ahead) shows both enantiomers to be present, i.e. that the complex is racemic, so clearly the same structure could not be obtained with a single enantiomer. It appears that a tetranuclear species present as a result of the partial hydrolysis of uranyl ion under the reaction conditions forms the least soluble species involving coordination to R,R-deadc2−.
Crystal structures
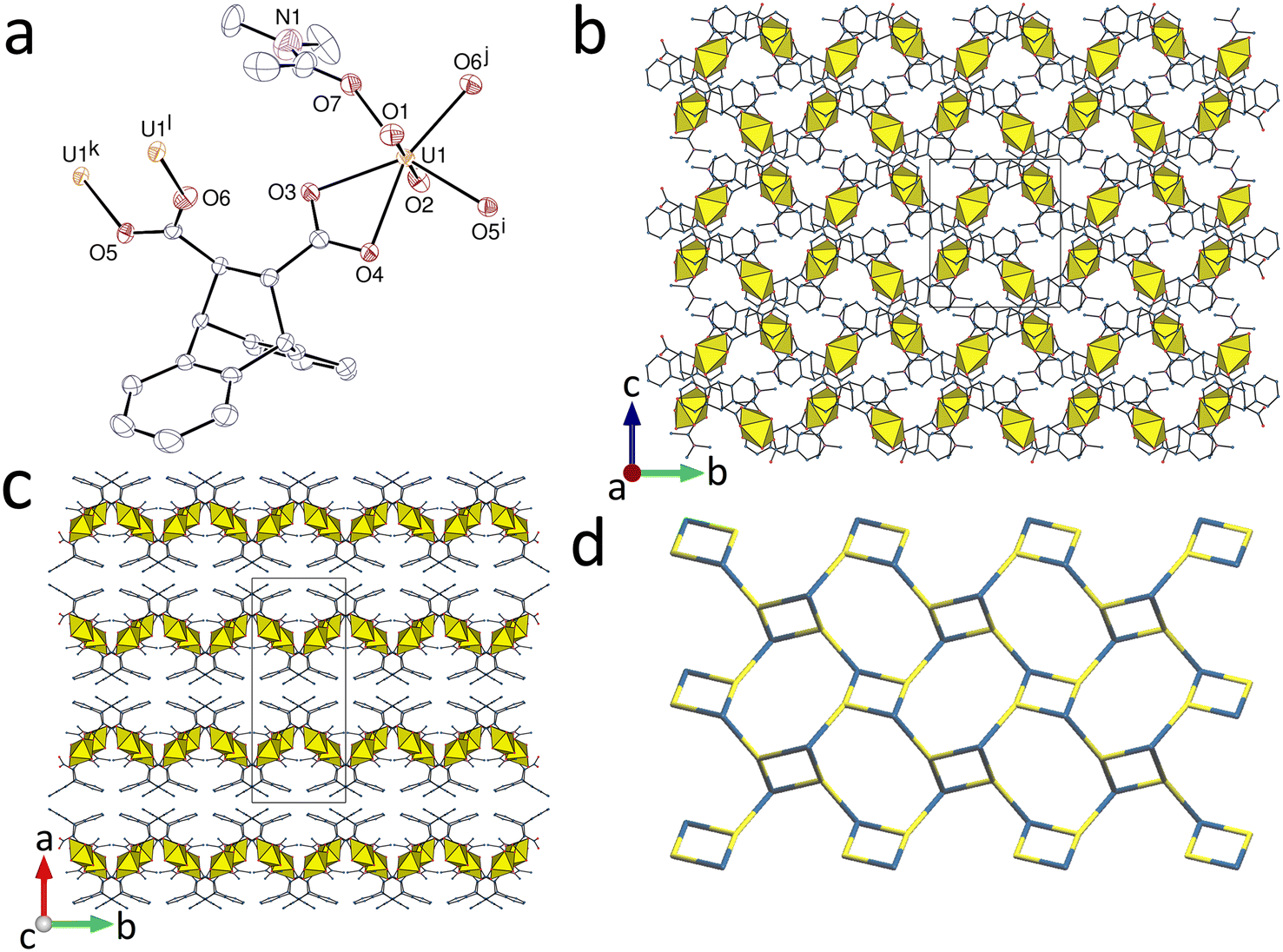
The complex [UO2(deadc)]·1.5CH3CN (1), obtained with acetonitrile as organic cosolvent, is the simplest case in this series. The unique uranyl cation is κ2O,O′-chelated by one carboxylate group from one ligand, chelated between the two carboxylate groups of a second ligand (seven-membered chelate ring) and bound to one more carboxylate oxygen atom from a third ligand (Fig. 1). The uranium environment is thus pentagonal-bipyramidal [U–O(oxo), 1.757(3) and 1.762(3) Å; U–O(carboxylate), 2.474(3) and 2.506(3) Å for the chelating group, 2.304(3)–2.404(3) Å for the others]. The deadc2− ligand has thus one carboxylate group chelating and bridging in the μ2-κ2O,O′:κ1O mode, and the other syn/anti μ2-κ1O:κ1O′-bridging. Both metal and ligand are thus three-coordinated (3-c) nodes, and the coordination polymer formed is monoperiodic and directed along [100]. The uranium centers are assembled into centrosymmetric, edge-sharing dimers arranged into ribbons with flanking dihydroanthracenyl groups. | ||
| Fig. 1 (a) View of complex 1 with displacement ellipsoids shown at the 50% probability level. Solvent molecules and hydrogen atoms are omitted. Symmetry codes: i = 1 − x, 1 − y, 1 − z; j = 2 − x, 1 − y, 1 − z. (b) and (c) Two views of the monoperiodic assembly showing uranium coordination polyhedra. (d) Packing with chains viewed edge-on. | ||
The carboxylate C⋯C separation is 3.445(6) Å, showing that the separation in phthalate (close to 3.1 Å),23 for which such chelate rings are well known,23,24 does not represent a limit for 7-membered chelate ring formation on uranyl ion. The small variations of the torsional parameters in the aliphatic part of the ligand are indicative of the rigidity of deadc2− (Table S1, ESI†). The chains lie side-by-side in sheets parallel to (011) in such a way that enantiomeric dihydroanthracenyl entities confront one another, thus creating cavities. This feature of the structure has a clear similarity to those seen in inclusion complexes of deadcH2 alone.1–4 The acetonitrile molecules are involved in multiple weak interactions. One parallel-displaced π-stacking interaction between two aromatic rings pertaining to adjacent chains may be present [centroid⋯centroid distance, 3.870(3) Å; dihedral angle, 0°] and the Kitaigorodsky packing index (KPI, evaluated with PLATON25) is 0.66 (with disordered acetonitrile molecule excluded).
A dicarboxylate/U ratio of 3:2 commonly results in the formation of [tris(κ2O,O′-carboxylate)UO2]− centres of hexagonal-bipyramidal geometry11 but yet another exception to this occurs in the complex [H2NMe2]2[(UO2)2(deadc)3]·2H2O (2). Here, the monoperiodic polymer present has unique UVI centres with a pentagonal-bipyramidal coordination geometry and an environment similar to that in 1 [U–O(oxo), 1.7740(17) and 1.7811(17) Å; U–O(carboxylate), 2.4368(17) and 2.5298(16) Å for the chelating group, 2.3354(17)–2.3656(17) Å for the others] (Fig. 2). The deadc2− ligands adopt two different coordination modes, both of them bridging. The first ligand is involved in 7-membered chelate ring formation and further bridging (μ2-κ1O:κ1O′;κ1O′′ mode), and the second, which has twofold rotation symmetry, is bis(κ2O,O′-chelating). Formation of a 7-membered chelate ring shows again deadc2− to have some resemblance to t-1,2-chdc2−.9
 | ||
| Fig. 2 (a) View of complex 2 with displacement ellipsoids shown at the 50% probability level. Carbon-bound hydrogen atoms are omitted, and hydrogen bonds are shown as dashed lines. Symmetry codes: i = 1/2 − x, 1/2 − y, 1 − z; j = 1 − x, y, 1/2 − z. (b) and (c) Two views of the monoperiodic assembly showing uranium coordination polyhedra. (d) Packing with chains viewed edge-on. | ||
The carboxylate C⋯C distances are 3.468(3) Å in the ligand forming the 7-membered ring and 3.713(4) in the other, showing again that deadc2− does have some flexibility, if considerably less than that of t-1,2-chdc2−. The dimethylammonium counterion, produced in situ from hydrolysis of DMF, is hydrogen bonded to the uncoordinated atom O4 and to the water molecule. The metal cation is here also a 3-c node while the ligands are simple edges, and the coordination polymer formed is monoperiodic and directed along [10]. Packing of the zigzag polymer strands involves some degree of interdigitation within the layers parallel to (010). One parallel-displaced π-stacking interaction between two aromatic rings pertaining to adjacent chains may be present here also [centroid⋯centroid distance, 4.0874(15) Å; dihedral angle, 6.26(12)°], as well as two CH⋯π edge-to-face interactions involving the same two ligands and resulting in their dimerization. This interpenetration produces a very compact packing with no evidence of significant porosity (KPI, 0.72).
The complex [H2NMe2]4[(UO2)2(O)(R,R-deadc)2]2 (3), produced under conditions identical to those giving 2 from the racemic diacid, has a structure which shows that chirality here is in fact an important influence. Hydrolysis of DMF and the concomitant buffering of the reaction mixture may explain the partial hydrolysis of uranyl ion to give a slightly twisted tetranuclear uranate cluster. The latter has twofold rotation symmetry and it is built around two μ3-oxo anions and bounded by an up-down alternating garland of four R,R-deadc2− ligands (Fig. 3). The cluster, a coordination oligomer only, is an example of a U4O2 motif with additional peripheral ligands which has previously been observed in several cases,23,24i,26 with in particular one instance involving t-1,2-chdc2− in the diequatorial conformation.9e U1 is part of two 7-membered chelate rings and bound to one oxo anion (pentagonal-bipyramidal environment), and U2 is bis(κ2O,O′-chelated) and bound to two oxo anions (hexagonal-bipyramidal environment) [U–O(oxo), 1.748(12)–1.783(13) Å; U–O(carboxylate), 2.526(10)–2.646(11) Å for the chelating groups, 2.315(10)–2.439(11) Å for the others; U–O(μ3-oxo), 2.243(10)–2.278(11) Å]. The sum of the three U–O–U angles around the oxo atom O13 is 353.4°, indicating that the three U–O bonds around this atom are nearly coplanar. The sum of bond valence parameters calculated with PLATON25 for atom O13 is 1.99, thus confirming that it is an oxo and not a hydroxo anion. The uranium coordination polyhedra share either two or three edges with their neighbours, giving one of the more compact arrangements among those found in uranyl tetranuclear clusters.11c
 | ||
| Fig. 3 (a) View of compound 3 with displacement ellipsoids shown at the 30% probability level. The disordered counterion and carbon-bound hydrogen atoms are omitted, and hydrogen bonds are shown as dashed lines. Symmetry code: i = 1 − x, 1 − y, z. (b) Side view of the tetranuclear complex. (c) and (d) Two views of the packing with counterions omitted. | ||
While there are two inequivalent ligands in each cluster, the differences between them are very minor, the carboxylate C⋯C separation being 3.46(2) Å for both, showing again that the conformation of deadc2− is quite insensitive to its environment. The tetranuclear anions lie in stacks aligned parallel to [001], with two disordered H2NMe2+ cations sandwiched between every two clusters. The ordered H2NMe2+ cation forms two bifurcated hydrogen bonds with two carboxylate groups bound to the same uranyl group. While no significant π-stacking interaction is present, each of the two methyl groups of the ordered counterion forms two CH⋯π interactions involving the two rings of one ligand. These counterions are thus included in the cavity formed by the concave parts of two dihydroanthracenyl units from adjacent cluster columns (KPI, 0.65).
To explore the consequences of the combination of deadc2− with carboxylate zwitterions, [Ni(tpyc)2], in which tpyc− is 2,2′:6′,2′′-terpyridine-4′-carboxylate, was used to synthesize [(UO2)2(deadc)(deadcH)(NO3)Ni(tpyc)2]·CH3CN·2H2O (4), providing the first instance in the present study of a complex involving both deadc2− and deadcH−. The two independent uranium atoms are both tris(κ2O,O′-chelated) and in hexagonal-bipyramidal environments. While U1 is bound to two carboxylate groups from two deadc2− ligands and one from Ni(tpyc)2, U2 is bound to one carboxylate from deadcH−, one from Ni(tpyc)2, and one nitrate anion [U–O(oxo), 1.765(4)–1.779(4) Å; U–O(carboxylate), 2.410(3)–2.523(4) Å; U–O(nitrate), 2.500(4) Å for both] (Fig. 4). Complex 4 is a monoperiodic polymer in which {UO2(deadcH)(NO3)Ni(tpyc)2} units can be considered as pendent substituents on a {UO2(deadc)} main chain directed along [100]. However, if hydrogen bonding between the carboxylic group of deadcH− and the uranyl oxo group O4 is recognized, the polymer has a ladder-like, double stranded form. The essential structural unit is a U4Ni2 metallacycle closely similar to that seen in several other mixed-ligand complexes involving the [Ni(tpyc)2] zwitterion.9g There is also a parallel with the complex [(UO2)2(1,2-pda)(1,2-pdaH)Ni(tpyc)2(NO3)]·CH3CN (1,2-pda2− = 1,2-phenylenediacetate),9g considered as a monoperiodic meander polymer where hexagonal-bipyramidal UVI centres bound to the two non-zwitterion species 1,2-pda2− and 1,2-pdaH− alternate along the chain rather than forming separate chains as here. Unlike 1,2-pda2− and 1,2-pdaH−, which can adopt chiral but labile conformations,9g,27 deadc2− and deadcH− exist as kinetically stable enantiomers (as shown indirectly by the isolation of the cis and trans isomers,5 as well as through chiral resolution7). The carboxylate C⋯C separations are 3.673(7) Å in deadc2− and 3.468(7) Å in deadcH−, much longer than those (∼2.97 Å) found for the inequivalent, non-disordered t-R,R-chdc2− units in the complex [(UO2)4(t-R,R-chdc)4Ni2(tpyc)4]9g where the conformation is diequatorial, indicating that deadc2− and deadcH− could be considered as rigidified analogues of diaxial t-1,2-chdc2−. The quite different compositions and structures of the complexes of the anthracene and cyclohexane derivatives must be at least in part a reflection of this conformational factor. Although there are examples for t-1,2-chdc2− where chirality does not seem to be an important influence,9a complexes 2 and 3 show that this is not necessarily the case with deadc2−. Neighbouring Ni(tpyc)2 units in 4 are possibly associated by one intrachain parallel-displaced π-stacking interaction [centroid⋯centroid distance, 3.607(3) Å; dihedral angle, 9.4(2)°], while two weaker ones involving deadcH− anions and tpyc− may be found between adjacent chains. Both intra- and interchain CH⋯π interactions are present as well, and the packing has a KPI of 0.67.
 | ||
| Fig. 4 (a) View of compound 4 with displacement ellipsoids shown at the 50% probability level. Carbon-bound hydrogen atoms are omitted and hydrogen bonds are shown as dashed lines. Symmetry codes: i = x + 1, y, z; j = x − 1, y, z. (b) The heterometallic monoperiodic assembly with uranium coordination polyhedra yellow and those of nickel blue, and hydrogen bonds shown as dotted blue lines. (c) Packing with ribbon-like chains viewed end-on. | ||
Direct coordination of a cosolvent molecule, here N,N-dimethylacetamide (DMA), to UVI produces a significant structural change in [UO2(deadc)(DMA)] (5). The unique uranium centre is κ2O,O′-chelated by one carboxylate group and bound to two more carboxylate donors from two different ligands, and to the DMA molecule (Fig. 5). The uranium environment is thus pentagonal-bipyramidal [U–O(oxo), 1.769(5) and 1.773(5) Å; U–O(carboxylate), 2.421(5) and 2.462(5) Å for the chelating group, 2.316(5) and 2.320(5) Å for the others; U–O(DMA), 2.374(5) Å]. The deadc2− ligand is chelating and syn/anti bridging (μ3-κ2O,O′;κ1O′′:κ1O′′′), and the carboxylate C⋯C separation [3.456(9) Å] is essentially unchanged, as are the torsional parameters. Both metal and ligand are thus 3-c nodes, and the coordination polymer formed, which is diperiodic and parallel to (100), has the {4·82} point symbol and the common fes topological type. When viewed down [001], the thick layers display a central, zigzag layer of uranyl cations surrounded on both sides by dihydroanthracenyl units pointing outward, each surface comprising a racemic mixture of ligands. Enantiomeric ligands in adjacent sheets are united pairwise through one parallel-displaced π-stacking interaction [centroid⋯centroid distance, 3.808(4) Å; dihedral angle, 0°] and two edge-to-face CH⋯π interactions. The KPI of 0.62 shows that disordered and unresolved solvent molecules are possibly present.
 | ||
| Fig. 5 (a) View of compound 5 with displacement ellipsoids shown at the 50% probability level and hydrogen atoms omitted. Symmetry codes: i = x, 1 − y, z + 1/2; j = 3/2 − x, y − 1/2, 3/2 − z; k = x, 1 − y, z − 1/2; l = 3/2 − x, y + 1/2, 3/2 − z. (b) View of the fes diperiodic assembly. (c) Packing with layers viewed edge-on. (d) Nodal representation of the network (yellow, uranium nodes; blue, deadc2− nodes; view down [100] with [001] horizontal). | ||
Although its composition differs from that of 1 only by the latter incorporating acetonitrile solvent molecules, the complex [UO2(deadc)] (6) has a different structure, possibly as a result of subtle effects of the additional reagents, guanidinium nitrate for 1 and cesium nitrate here, although these species are not present in the final compound (the difference in concentration of the reactants may also play a part). The single, hexagonal-bipyramidal uranium atom is κ2O,O′-chelated by two carboxylate groups (one of them very unsymmetrical) and bound to two more carboxylate donors in trans positions [U–O(oxo), 1.7586(17) and 1.7652(17) Å; U–O(carboxylate), 2.4440(19)–2.5874(17) Å for the chelating groups, 2.4582(17) and 2.4727(17) Å for the others] (Fig. 6). The ligand is bis(chelating/bridging) [bis(μ2-κ2O,O′:κ1O)] and is thus a 4-c node, as the metal cation. The diperiodic, uninodal coordination polymer formed is parallel to (010) and it has the {44·62} point symbol and the simple sql topological type. The layers contain strands of edge-sharing UO6 groups running parallel to [001] which are crosslinked by the ligands. Here also, the dihydroanthracenyl units form upper and lower faces to each sheet. The packing displays dimerization of enantiomeric ligands pertaining to adjacent sheets similar to that found in 5, with both one parallel-displaced π-stacking interaction [centroid⋯centroid distance, 3.7429(15) Å; dihedral angle, 0°] and two edge-to-face CH⋯π interactions. The packing is however more compact here and no solvent-accessible space is present (KPI, 0.71).
 | ||
| Fig. 6 (a) View of compound 6 with displacement ellipsoids shown at the 50% probability level and with hydrogen atoms omitted. Symmetry codes: i = x, 3/2 − y, z − 1/2; j = x − 1, y, z; k = x − 1, 3/2 − y, z − 1/2; l = x, 3/2 − y, z + 1/2; m = x + 1, y, z; n = x + 1, 3/2 − y, z + 1/2. (b) View of the sql diperiodic assembly. (c) Packing with layers viewed edge-on. | ||
In the enantiomerically pure complex [PPh4]2[(UO2)2(R,R-deadc)3] (7), the uranium atom is tris(κ2O,O′-chelated) by three carboxylate groups [U–O(oxo), 1.776(3) and 1.781(3) Å; U–O(carboxylate), 2.458(2)–2.484(2) Å] (Fig. 7). As often found in complexes in which the uranyl cation is a 3-c node and the ligand a simple edge, the diperiodic network formed, parallel to (001) has the {63} point symbol and the hcb topological type. The sheets are strongly puckered, with PPh4+ cations embedded within due to multiple CH⋯O interactions, and there is no significant free space (KPI, 0.67). In the case of complexes of the enantiomers of t-1,2-chdc2− prepared under conditions of 2:3 U/dicarboxylate stoichiometry,9c these materials have a 1:1 stoichiometry, though the ratio U/carboxylate of 1:3 is achieved due to the coordination of formate deriving from DMF hydrolysis; they are, nonetheless, monoperiodic polymers in which the t-1,2-chdc2− ligands have diaxial conformations with inter-carboxylate C⋯C separations of 3.86–3.88 Å, significantly longer than those in 7, 3.672(5) and 3.575(7) Å. The rigidity of deadc2− thus appears to severely limit the extent to which it may be considered an analogue of diaxial t-1,2-chdc2−.
 | ||
| Fig. 7 (a) View of compound 7 with displacement ellipsoids shown at the 50% probability level and with hydrogen atoms omitted. Symmetry codes: i = 3/2 − x, y − 1/2, 2 − z; j = 1 − x, 1 − y, z; k = 3/2 − x, y + 1/2, 2 − z. (b) View of the hcb diperiodic assembly. (c) Packing with layers viewed edge-on and counterions omitted for clarity. (d) Nodal representation of the network (yellow, uranium nodes; blue, deadc2− edges; view down [001] with [010] horizontal). | ||
The second heterometallic complex in this series, [(UO2)2Ag2(deadc)3(CH3CN)2]·0.5H2O (8), crystallizes in the same space group as complex 2 and with very close unit cell parameters, so that these two species can be considered to be isomorphous, although not isostructural. The pentagonal-bipyramidal uranium atom is κ2O,O′-chelated by one carboxylate group, chelated by two groups of another ligand (7-membered ring), and bound to one more carboxylate donor from a third ligand [U–O(oxo), 1.7732(17) and 1.7778(17) Å; U–O(carboxylate), 2.4214(17) and 2.5071(17) Å for the chelating group, 2.3107(17)–2.3680(17) Å for the others] (Fig. 8). The metal centre is thus a 3-c node, while both ligands are only bound to two uranium atoms, the unsymmetrical one in a bridging mode and that with twofold rotation symmetry in the bis(chelating) mode. Ignoring interactions with AgI, the monoperiodic polymer formed is essentially identical with that present in 2. As might be expected of AgI,28 its coordination sphere is irregular and heteroleptic, interactions here involving two adjacent aromatic carbon atoms, the acetonitrile nitrogen donor, one carboxylate donor and possibly one water molecule (with partial occupancy) [Ag–C, 2.367(2) and 2.464(2) Å; Ag–N, 2.167(3) Å; Ag–O(carboxylate), 2.3389(17) Å; Ag–O(water), 2.493(9) Å]. While it may appear that AgI and H2NMe2+ are two very different cations, the latter is involved in weak interactions in addition to the obvious NH⋯O bond formation and these include CH⋯π interactions, so that in sum the interactions of both are quite similar and this may explain the retention of the same form of the associated polymer anion. However, a difference between the weak interactions involving H2NMe2+ and the coordination bonds formed by silver(I) is that the latter connect the monoperiodic, uranyl-only coordination polymers into a compact triperiodic framework (KPI, 0.73). Dimerization of deadc2− ligands involving one parallel-displaced π-stacking interaction [centroid⋯centroid distance, 3.6826(13) Å; dihedral angle, 11.60(11)°], and two CH⋯π edge-to-face interactions is found here also.
 | ||
| Fig. 8 (a) View of compound 8 with displacement ellipsoids shown at the 50% probability level. The water molecule and the hydrogen atoms are omitted. Symmetry codes: i = 1/2 − x, 1/2 − y, 1 − z; j = 1 − x, 1 − y, 1 − z; k = −x, y, 3/2 − z. (b) and (c) Two views of the uranyl-based monoperiodic subunit. (d) View of the triperiodic framework with uranium coordination polyhedra yellow and silver atoms shown as blue spheres. | ||
Notwithstanding its closeness to t-1,2-chdc2− regarding the relative position of the two donor groups, deadc2− appears in the present series as a ligand giving only mono- and diperiodic uranyl ion complexes, except for the tetranuclear species 3 formed with R,R-deadc2− and the triperiodic assembly resulting from silver-bridging of chains in 8. In particular, no cage-like complex has been obtained with deadc2−, while such species are quite common in the case of 1,2-chdc2−, the bulkiness of the dibenzobarrelene group possibly having an effect here. The weak interactions involving the dibenzobarrelene platform are also distinctive and introduce an element of novelty. The carboxylate groups being located in the half of the molecule opposite to the aromatic-lined concave side, the latter is thus naturally pointing outward from the main body of the coordination polymer, resulting in a hydrophobic coating of the chains or networks reminiscent of that found with phosphonates29 or Kemp's tricarboxylate,30 for example. Such an arrangement favors interactions with neighbouring polymeric units or with solvent molecules or counterions, and it may thus have a structure-directing effect. Three of the association modes found in the present complexes are shown in Fig. 9. Interactions with the acetonitrile molecules in 1 seem to be loose at best: the methyl group of the well-ordered acetonitrile molecule is located on the border of the cavity defined by the two facing deadc2− ligands, and the Hirshfeld surface31 does not reveal any significant interaction (the nitrogen atom, in contrast, is involved in two CH⋯N hydrogen bonds with other anions). Interactions with the H2NMe2+ counterion in 3 are much more extensive and they clearly illustrate the hydrophilic/hydrophobic nature of the ligand: the ammonium protons form two bifurcated hydrogen bonds with the carboxylate groups of two ligands, while the methyl groups are involved in four CH⋯π interactions with the aromatic rings of two more ligands facing one another and thus defining a hydrophobic cavity. The arrangement most often found however, in complexes 2, 5, 6 and 8, is that bringing two deadc2− anions in close interaction to form a dimer held by three aromatic⋯aromatic interactions, one parallel and the other two edge-to-face. It is notable that the formation of the only triperiodic framework among these complexes is due to bridging by silver(I), a cation able to interact with both the hydrophilic and hydrophobic parts of the ligand.
 | ||
| Fig. 9 Intermolecular associations of the deadc2− ligand. (a) Loose association with acetonitrile in 1 (only one position of the disordered solvent is shown). (b) NH⋯O (dashed lines) and CH⋯π (dotted lines) interactions with H2NMe2+ in 3. (c) Dimerization through one parallel-displaced π-stacking interaction (dashed line) and two edge-to-face interactions (dotted lines) in 2, 5, 6 and 8. | ||
Luminescence properties
Except for 6, all the present complexes are emissive, with however strong disparities in photoluminescence quantum yields (PLQYs), these being 2% for 1 and 4, 6% for 8, 8% for 7, 12% for 3, 23% for 5 and 26% for 2. These variations might be related to differences in aromatic interactions and intermetallic distances, the presence of solvent molecules also possibly playing a part. The emission spectra measured in the solid state under excitation at 420 nm for complexes 1, 2, 4, 5, 7 and 8 show the typical vibronic progression due to the S11 → S00 and S10 → S0ν (ν = 0–4) transitions of the uranyl ion32 (Fig. 10). Complexes 1, 2, 5 and 8, all with single uranyl sites and pentagonal-bipyramidal uranium environments, have quasi-identical positions for the main four emission maxima, at 491–492, 511–513, 533–536 and 559–561 nm. These values are typical of uranyl carboxylate complexes with O5 equatorial environments.33 Complex 7, with a unique uranyl cation in a tris-chelated, hexagonal-bipyramidal environment, has maxima at 480, 499, 521 and 544 nm, as expected for such an O6 environment.33 Complex 4 contains two inequivalent uranium centres, both in hexagonal-bipyramidal environments, but one of them chelated by three carboxylate groups and the other by two carboxylate and one nitrate groups, and its emission spectrum obviously results from the superposition of two emission series which can be separated by deconvolution (blue and orange lines in Fig. 11), except for the last, very weak and unresolved maximum corresponding to a wavelength near 573 nm. The wavelengths of the four main maxima for the two series are at 480, 499, 523 and 543 nm for the most blue-shifted one, and 489, 511, 532 and 556 nm for the other, major one. The first series matches exactly that for complex 7 and thus may reasonably be attributed to the tris(carboxylate)-coordinated uranyl centre (U1). The other component, which is red-shifted and with maxima positions in the highest range measured for complexes with O6 equatorial environments,33 should thus be attributed to the uranyl cation chelated by two carboxylate and one nitrate groups (U2). It may be noted that the maxima in the spectrum of uranyl nitrate hexahydrate measured under the same conditions are at 486, 508, 532 and 557 nm,34i.e. very close to those of the second component here. Calculation of bond valence parameters35 with PLATON25 gives axial/equatorial values of 3.47/2.589 for U1 and 3.446/2.683 for U2. If these values are taken as an indication of bond strength, the donor strength in the equatorial plane is slightly larger for U2, which induces a decrease of the oxo bond order to uranyl,36 as reflected in the maxima positions. Finally, the spectrum of complex 3, which is reproducible when measured on different samples, displays only a very broad emission peak centered at about 535 nm (Fig. S2, ESI†). Although unusual and of uncertain origin, similar spectra have previously been observed in other uranyl ion complexes with carboxylates.37 | ||
| Fig. 10 Emission spectra of complexes 1, 2, 4, 5, 7 and 8 in the crystalline state upon excitation at 420 nm. | ||
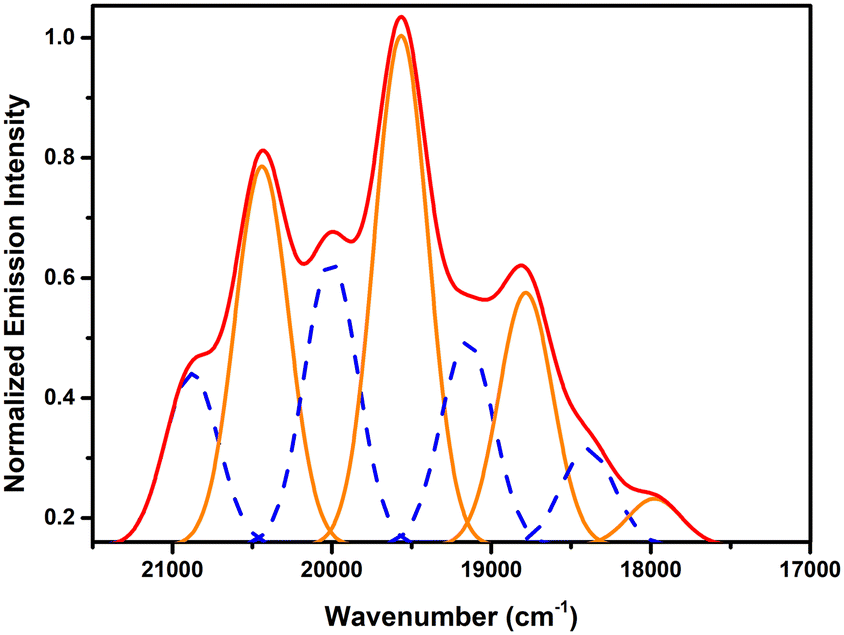 | ||
| Fig. 11 Deconvolution of the emission spectrum of 4 (red) showing the two series of peaks (dashed blue and orange). | ||
Conclusions
We have reported the synthesis, crystal structure and emission spectrum of eight uranyl ion complexes with deadc2−, a bulky aromatic dicarboxylate ligand. Depending on the organic cosolvent and the additional species present, the periodicity of the complexes varies from 0 to 3, the extreme values being obtained for a μ3-oxo-bridged tetranuclear complex and a framework in which additional bridging is provided by silver(I) cations, respectively, while the diperiodic networks present the fes, sql or hcb topologies. All but two complexes have emission spectra in the solid state with maxima positions matching those usual for O5 or O6 equatorial coordination numbers, with a mixture of two distinct components in one case.The deadc2− ligand has the attractive feature of adopting a fairly predictable conformation, though the capacity of the carboxylate groups to adopt various coordination modes still adds variety to the structure of their complexes. The capacity of the acid itself to form inclusion complexes through aggregation defining cavities, seems to be partly retained in the anion complexes but the formation of cage-like oligomeric uranyl ion complexes, anticipated on the basis of similarity of the anions to those derived from trans-1,2-cyclohexanedicarboxylic acid, has not yet been observed. The chirality of the anions appears to be an important influence upon the structures of their complexes, the fact that in all cases so far known, use of the racemic acid always results in structures which are also racemic meaning that the use of isolated enantiomers necessarily results in quite different species.
Segregation of the hydrophobic dibenzobarrelene units on the outside of the coordination polymers formed, together with the existence of weak π-stacking or CH⋯π interactions involving these units, either with one another or with counterions, are common features of these complexes and they play a prominent role in defining the packing of the polymeric moieties. Due to their ability to be bonded to both carboxylates and aromatic rings, silver(I) cations are able to bridge the hydrophilic and hydrophobic parts of different polymeric units and thus increase the overall periodicity, yet another example of the versatility of this cation and its interest in the synthesis of uranyl-based coordination polymers.38
In conclusion, these results show that, although deadc2− is fairly rigid and has a relative positioning of the coordination sites closely similar to that in trans-1,2-cyclohexanedicarboxylate, it has failed up to now to produce cage-like complexes with uranyl ion, a disappointing fact because this was one of the anticipated properties of these ligands. Its amphiphilic nature can lead to enhanced periodicity through the addition of particular metal ions, but here its reduced denticity when compared with the amphiphilic Kemp's tricarboxylate30 prevents formation of closed species such as nanotubes or cages.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2022R1I1A1A01064010) (Y. H. L.), and by Iketani Science and Technology Foundation and KAKENHI Grant-in-Aid for Early-Career Scientists JP22K14698 (S. K.).References
- (a) E. Weber, I. Csöregh, J. Ahrendt, S. Finge and M. Czugler, J. Org. Chem., 1988, 53, 5831 CrossRef CAS; (b) O. Helmle, I. Csöregh, E. Weber and T. Hens, J. Inclusion Phenom. Mol. Recognit. Chem., 1998, 30, 45 CrossRef CAS.
- (a) L. Y. Izotova, D. M. Ashurov, B. T. Ibragimov, E. Weber, M. Perrin and S. A. Talipov, J. Struct. Chem., 2005, 46, S103 CrossRef CAS; (b) B. T. Ibragimov, L. Y. Izotova, D. M. Ashurov, E. Weber and S. A. Talipov, J. Struct. Chem., 2005, 46, S109 CrossRef CAS; (c) B. T. Ibragimov, L. Y. Izotova, D. M. Ashurov, S. A. Talipov, E. Weber, M. Perrin and A. J. Blake, Mol. Cryst. Liq. Cryst., 2005, 440, 107 CrossRef CAS.
- Y. Yang, J. N. Zhang, S. R. Luo, X. H. Song and Q. Li, Acta Chim. Sin., 2006, 64, 1904 CAS.
- (a) B. Barton, U. Senekal and E. C. Hosten, CrystEngComm, 2021, 23, 4560 RSC; (b) B. Barton, M. R. Caira, U. Senekal and E. C. Hosten, Cryst. Growth Des., 2022, 22, 3385 CrossRef CAS; (c) B. Barton, M. R. Caira, U. Senekal and E. C. Hosten, CrystEngComm, 2022, 24, 4573 RSC.
- D. Braga, S. d'Agostino and F. Grepioni, Organometallics, 2012, 31, 1688 CrossRef CAS.
- (a) D. Han, K. Huang, X. Li, M. Peng, L. Jing, B. Yu, Z. Chen and D. Qin, RSC Adv., 2019, 9, 33890 RSC; (b) X. Li, K. Huang, M. Peng, D. Han, Q. Qiu, L. Jing and D. Qin, Polyhedron, 2020, 178, 114349 CrossRef CAS.
- (a) S. Hagishita and K. Kuriyama, Tetrahedron, 1972, 28, 1435 CrossRef CAS; (b) Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1993, vol. 6, p. 331 Search PubMed; (c) C. R. Ramanathan and M. Periasamy, Tetrahedron: Asymmetry, 1998, 9, 2651 CrossRef CAS.
- M. Periasamy, C. R. Ramanathan, N. S. Kumar and M. Thirumalaikumar, J. Chem. Res., 2001, 512 CrossRef CAS.
- (a) P. Thuéry and J. Harrowfield, Inorg. Chem., 2017, 56, 1455 CrossRef PubMed; (b) P. Thuéry and J. Harrowfield, Cryst. Growth Des., 2017, 17, 2881 CrossRef; (c) P. Thuéry, Y. Atoini and J. Harrowfield, Cryst. Growth Des., 2018, 18, 2609 CrossRef; (d) P. Thuéry, Y. Atoini and J. Harrowfield, Cryst. Growth Des., 2018, 18, 3167 CrossRef; (e) P. Thuéry and J. Harrowfield, Cryst. Growth Des., 2018, 18, 5512 CrossRef; (f) P. Thuéry and J. Harrowfield, Cryst. Growth Des., 2020, 20, 262 CrossRef; (g) P. Thuéry and J. Harrowfield, Inorg. Chem., 2022, 61, 9725 CrossRef PubMed.
- Chem3D 15.0, Perkin-Elmer Informatics, 2015 Search PubMed.
- (a) K. X. Wang and J. S. Chen, Acc. Chem. Res., 2011, 44, 531 CrossRef CAS PubMed; (b) M. B. Andrews and C. L. Cahill, Chem. Rev., 2013, 113, 1121 CrossRef CAS PubMed; (c) T. Loiseau, I. Mihalcea, N. Henry and C. Volkringer, Coord. Chem. Rev., 2014, 266–267, 69 CrossRef CAS; (d) J. Su and J. S. Chen, Struct. Bonding, 2015, 163, 265 CrossRef CAS; (e) P. Thuéry and J. Harrowfield, Dalton Trans., 2017, 46, 13660 RSC; (f) K. Lv, S. Fichter, M. Gu, J. März and M. Schmidt, Coord. Chem. Rev., 2021, 446, 214011 CrossRef CAS.
- A. B. Naidu, E. A. Jaseer and G. Sekar, J. Org. Chem., 2009, 74, 3675 CrossRef CAS PubMed.
- APEX3, ver. 2019.1-0, Bruker AXS, Madison, WI, 2019 Search PubMed.
- SAINT, ver. 8.40A, Bruker Nano, Madison, WI, 2019 Search PubMed.
- (a) SADABS, ver. 2016/2, Bruker AXS, Madison, WI, 2016 Search PubMed; (b) L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- (a) M. N. Burnett and C. K. Johnson, ORTEPIII, Report ORNL6895, Oak Ridge National Laboratory, TN, 1996 Search PubMed; (b) L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576 CrossRef CAS.
- I. Mihalcea, N. Henry and T. Loiseau, Cryst. Growth Des., 2011, 11, 1940 CrossRef CAS.
- (a) I. Mihalcea, C. Volkringer, N. Henry and T. Loiseau, Inorg. Chem., 2012, 51, 9610 CrossRef CAS PubMed; (b) J. Olchowka, C. Falaise, C. Volkringer, N. Henry and T. Loiseau, Chem. – Eur. J., 2013, 19, 2012 CrossRef CAS PubMed; (c) A. T. Kerr, S. A. Kumalah, K. T. Holman, R. J. Butcher and C. L. Cahill, J. Inorg. Organomet. Polym. Mater., 2014, 24, 128 CrossRef CAS; (d) X. Gao, C. Wang, Z. F. Shi, J. Song, F. Y. Bai, J. X. Wang and Y. H. Xing, Dalton Trans., 2015, 44, 11562 RSC; (e) X. Gao, J. Song, L. X. Sun, Y. H. Xing, F. Y. Bai and Z. Shi, New J. Chem., 2016, 40, 6077 RSC; (f) W. Xu, Z. X. Si, M. Xie, L. X. Zhou and Y. Q. Zheng, Cryst. Growth Des., 2017, 17, 2147 CrossRef CAS; (g) L. W. Zeng, K. Q. Hu, L. Mei, F. Z. Li, Z. W. Huang, S. W. An, Z. F. Chai and W. Q. Shi, Inorg. Chem., 2019, 58, 14075 CrossRef CAS PubMed; (h) G. Andreev, N. Budantseva, A. Levtsova, M. Sokolova and A. Fedoseev, CrystEngComm, 2020, 22, 8394 RSC; (i) P. Thuéry and J. Harrowfield, Cryst. Growth Des., 2021, 21, 3000 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- (a) I. A. Charushnikova, N. N. Krot, I. N. Polyakova and V. I. Makarenkov, Radiochemistry, 2005, 47, 241 CrossRef CAS; (b) P. Thuéry, Eur. J. Inorg. Chem., 2014, 58 CrossRef; (c) D. V. Kravchuk, A. Blanes Diaz, M. E. Carolan, E. A. Mpundu, D. M. Cwiertny and T. Z. Forbes, Inorg. Chem., 2020, 59, 8134 CrossRef CAS PubMed.
- (a) P. Thuéry, Y. Atoini and J. Harrowfield, Cryst. Growth Des., 2019, 19, 6611 CrossRef; (b) P. Thuéry, Y. Atoini and J. Harrowfield, Inorg. Chem., 2019, 58, 6550 CrossRef PubMed; (c) P. Thuéry, Y. Atoini and J. Harrowfield, Inorg. Chem., 2020, 59, 2503 CrossRef PubMed; (d) P. Thuéry and J. Harrowfield, Eur. J. Inorg. Chem., 2021, 2182 CrossRef.
- B. S. Fox, M. K. Beyer and V. E. Bondybey, J. Am. Chem. Soc., 2002, 124, 13613 CrossRef CAS PubMed.
- (a) D. M. Poojary, D. Grohol and A. Clearfield, Angew. Chem., Int. Ed. Engl., 1995, 34, 1508 CrossRef CAS; (b) M. A. G. Aranda, A. Cabeza, S. Bruque, D. M. Poojary and A. Clearfield, Inorg. Chem., 1998, 37, 1827 CrossRef CAS; (c) M. B. Doran, A. J. Norquist and D. O'Hare, Chem. Mater., 2003, 15, 1449 CrossRef CAS.
- (a) P. Thuéry, Cryst. Growth Des., 2014, 14, 901 CrossRef; (b) P. Thuéry, Cryst. Growth Des., 2014, 14, 2665 CrossRef; (c) J. Harrowfield and P. Thuéry, Eur. J. Inorg. Chem., 2020, 749 CrossRef CAS; (d) P. Thuéry and J. Harrowfield, Inorg. Chem., 2021, 60, 1683 CrossRef PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006 CrossRef CAS PubMed.
- (a) A. Brachmann, G. Geipel, G. Bernhard and H. Nitsche, Radiochim. Acta, 2002, 90, 147 CrossRef CAS; (b) M. Demnitz, S. Hilpmann, H. Lösch, F. Bok, R. Steudtner, M. Patzschke, T. Stumpf and N. Huittinen, Dalton Trans., 2020, 49, 7109 RSC.
- P. Thuéry and J. Harrowfield, Inorg. Chem., 2017, 56, 13464 CrossRef PubMed.
- P. Thuéry, Y. Atoini and J. Harrowfield, Inorg. Chem., 2020, 59, 2923 CrossRef PubMed.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192 CrossRef.
- (a) M. P. Redmond, S. M. Cornet, S. D. Woodall, D. Whittaker, D. Collison, M. Helliwell and L. S. Natrajan, Dalton Trans., 2011, 40, 3914 RSC; (b) L. S. Natrajan, Coord. Chem. Rev., 2012, 256, 1583 CrossRef CAS.
- (a) P. Thuéry, E. Rivière and J. Harrowfield, Inorg. Chem., 2015, 54, 2838 CrossRef PubMed; (b) J. Harrowfield, Y. Atoini and P. Thuéry, CrystEngComm, 2022, 24, 1475 RSC; (c) S. Kusumoto, Y. Atoini, S. Masuda, Y. Koide, J. Y. Kim, S. Hayami, Y. Kim, J. Harrowfield and P. Thuéry, CrystEngComm, 2022, 24, 7833 RSC.
- (a) Z. T. Yu, Z. L. Liao, Y. S. Jiang, G. H. Li and J. S. Chen, Chem. – Eur. J., 2005, 11, 2642 CrossRef CAS PubMed; (b) M. Frisch and C. L. Cahill, Dalton Trans., 2006, 4679 RSC; (c) Z. L. Liao, G. D. Li, M. H. Bi and J. S. Chen, Inorg. Chem., 2008, 47, 4844 CrossRef CAS PubMed; (d) A. T. Kerr and C. L. Cahill, Cryst. Growth Des., 2014, 14, 1914 CrossRef CAS; (e) P. Thuéry and J. Harrowfield, CrystEngComm, 2016, 18, 1550 RSC; (f) P. Thuéry and J. Harrowfield, Cryst. Growth Des., 2017, 17, 2116 CrossRef; (g) G. A. Senchyk, A. B. Lysenko, H. Krautscheid and K. V. Domasevitch, Inorg. Chem. Commun., 2020, 113, 107813 CrossRef CAS; (h) K. P. Carter, M. Kalaj, S. McNeil, A. Kerridge, M. H. Schofield, J. A. Ridenour and C. L. Cahill, Inorg. Chem. Front., 2021, 8, 1128 RSC; (i) D. M. Brager, A. C. Marwitz and C. L. Cahill, Dalton Trans., 2022, 51, 10095 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Fig. S1 and S2, Table S1. CCDC 2290585–2290592. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce01176c |
| This journal is © The Royal Society of Chemistry 2024 |