
Mimic uracil–uracil base pairing: self-assembly and single crystal structure†
Menglei
Zhang
a,
Zhongkui
Li
a,
Yanhong
Zhu
ab,
Li
Yan
ac,
Xue
Zhong
a,
Yijie
Zhang
a,
Ziyao
Li
a,
Yixiang
Bai
a and
Hui
Li
 *a
*a
aKey Laboratory of Cluster Science of Ministry of Education, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing, 102488, P. R. China. E-mail: lihui@bit.edu.cn
bNorth China University of Science and Technology, School of Pharmacy, Tangshan, Hebei 063210, P. R. China
cAnalysis & Testing Center of Beijing Institute of Technology, Liangxiang Campus, Liangxiang East Road, Beijing 102488, P. R. China
First published on 4th December 2023
Abstract
The controllable synthesis of non-classical nucleobase pairs that can mimic partial biological behavior is very important for the in-depth study of DNA or RNA. Here, three different interaction modes of uracil–uracil base pairs (U-motif) have been explored in the coordination complexes of 2′-deoxyuracil 5′-monophosphate (dUMP) and uracil 5′-monophosphate (UMP) with transition metal ions by single crystal structural analysis and DFT calculations for the first time. The results indicate that the structural diversity of non-classical nucleobase pairs may be a bank of supramolecular interactions, which will contribute to the study of supramolecular chemistry and biochemistry.
Introduction
Nucleobase recognition is a crucial biological phenomenon, which underpins many intricate processes within living systems, including gene transcription and translation. It is the cornerstone of life.1–4 One fundamental question is the recognition mode between base pairs, specifically the type of hydrogen bond between bases that dominates the structure.5–7 In the classical DNA double helix structure, nucleobase recognition is limited to a few choices, namely the complementary adenine:thymine base pair (A·T) and cytosine:guanine base pair (C·G).8,9 Numerous non-classical base pairs, involving at least two hydrogen bonds between any two bases except for the Watson–Crick base pairs, have been discovered based on diverse characterizations, which are of great significance in revealing genetic diseases and DNA rearrangements.10–13 However, among these characterizations, only single crystal X-ray diffraction (SC-XRD) can offer well-defined structural information, enabling the precise determination of the protonation position of the nucleobase, the hydrogen bond geometry, and the interaction mode between bases in the solid state. Unfortunately, the lack of a well-defined structure for non-classical base pairs hinders investigations into this issue.14,15With the growing field of self-assembly chemistry, non-covalent interactions have been employed to construct complicated supramolecular systems.16–18 The precise design and synthesis of self-assembly systems that can partially mimic biological behaviours is an effective way to address the above issue. For decades, scientists have been dedicated to exploring novel base pairing patterns through hydrogen bond directed assembly of nucleotides. The nucleobase edges have been classified into three categories: Watson–Crick, Hoogsteen, and Sugar edges, according to their different roles in hydrogen bond formation (Fig. S1†). And this has greatly increased the possible orientation and the number of interacting bases, leading to the discovery of innovative base-pairing.19
Wobble base pairs in RNA sequences, such as G·U, I·A, I·U and I·C, play a pivotal role in biochemistry.20,21 Particularly, research focused on G·U wobble base pairs has demonstrated the potential to regulate biological processes.22–24 Compared to the widely studied guanine base pairs and cytosine base pairs,25–27 the uracil–uracil base pair (U-motif) has received less attention. Although seven kinds of U-motifs have been reported, most of these are based on planar nucleobase derivatives (Fig. S2†). Additionally, previous research28–30 has identified general U-motifs in RNA tertiary structures. Compared to DNA or RNA macromolecules, the crystal structures of smaller native nucleotide complexes have higher resolution.31 However, there are few experimental and structural data on native nucleotide molecules.10 Here, by using aromatic ligands and cobalt or cadmium transition metals, a hydrophobic space is constructed during the coordination process, which induces the formation of U-motifs.32–34 For the convenience of our study, a structural overview of the five U-motif types is presented in Fig. 1.
 | ||
| Fig. 1 The structural scheme of five types of U-motifs. R represents the sugar ring and monophosphate group of UMP. | ||
Experimental section
Materials and methods
All reagents and organic solvents were obtained from commercial sources and used without further purification. 1H NMR spectra were recorded on a 400 MHz Bruker FT-NMR spectrometer using DMSO-d6. FT-IR spectra (KBr pellet) were performed on a Thermo IS5 FT-IR spectrometer in the range of 400–4000 cm−1. Powder X-ray diffraction (PXRD) patterns were carried out on a Bruker D8 Advanced X-ray diffractometer with a scanning rate of 10° min−1 (2θ angle measurement range: 5–50°). Elemental analysis (C, H, and N) was obtained by using an EA3000 elemental analyser. UV-vis absorption spectra were measured on a Persee TU-1950 spectrophotometer. Circular dichroism spectra in solution were measured in a concentration of 0.025 mmol L−1. The solid circular dichroism (CD) spectra were measured with a KBr pellet (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200) at room temperature using a JASCO J-1500 spectropolarimeter under a stable nitrogen stream purge. Thermogravimetric analysis (TGA) was carried out using a DTG-60H thermal analyzer under nitrogen atmosphere from 50 °C to 600 °C with a heating rate of 10 °C min−1.
:200) at room temperature using a JASCO J-1500 spectropolarimeter under a stable nitrogen stream purge. Thermogravimetric analysis (TGA) was carried out using a DTG-60H thermal analyzer under nitrogen atmosphere from 50 °C to 600 °C with a heating rate of 10 °C min−1.
Synthesis and physical properties
Single-crystal X-ray diffraction
Single crystals of 1–5 with suitable dimensions were selected for single-crystal X-ray diffraction analysis, respectively. Data collection was conducted on a Bruker DUO APEX II-CCD area diffractometer using graphite monochromatic Mo Kα (λ = 0.71073 Å) radiation at room temperature (296.15 K). The XSCAN program is used to find different peaks and determine the unit cell under the condition of 45 KV and 30 mA. Diffraction data collection adopts the ω–2θ scanning mode and all scanned data are subjected to empirical absorption correction. Using Olex2,35 the crystal structure was solved with the SHELXT36 structure solution program using the intrinsic phasing method, and refined with the SHELXL37 refinement program using least squares minimisation. All non-hydrogen atomic positions were obtained from different Fourier maps and refined anisotropically. All hydrogen atoms were located at their geometrically generated positions by different Fourier maps combined with geometric analysis and refined isotropically. In the five heavy-atom structure, (as it was not possible to see clear election-density peaks in different maps, which would correspond with an acceptable location for part of the water oxygen sites), the refinement was completed with no allowance for these H atoms in the model. Crystallographic data are given in Tables S1 and S2.†Computational details
Gaussview 6 is used to establish the calculation model, and Gaussian 16W is used for DFT calculation. The calculation parameters of structural optimization38–40 are as follows: #p opt freq b3lyp/6-311g(d,p) em = gd3bj. The parameters of energy calculation are: #b3lyp/6-311g(d,p) em = gd3bj. The parameters of BSSE (basis set superposition error) corrected energy calculation are: #b3lyp/6-311g(d,p) em = gd3bj counterpoise = 2. The surface electrostatic potential was plotted using Multiwfn41 and VMD programs.42Results and discussion
Crystal structures
In this work, five types of coordination complexes of 2′-deoxyuracil 5′-monophosphate (dUMP) and uracil 5′-monophosphate (UMP) with transition metal ions were designed and synthesized successfully (Scheme 1 and Fig. S3†), and the crystallographic data are shown in Tables S1 and S2.† Three different types of U-motifs, U-motif I, U-motif III, and U-motif IV, with well-defined structures have been reported for the first time based on SC-XRD. Notably, there are two different U-motifs in 5 (U-motif I and U-motif IV) and they form a chain like structure. It is the first example that realizes the fully compensated base-pairing with all hydrogen donors and acceptors of the uracil base being fully utilized.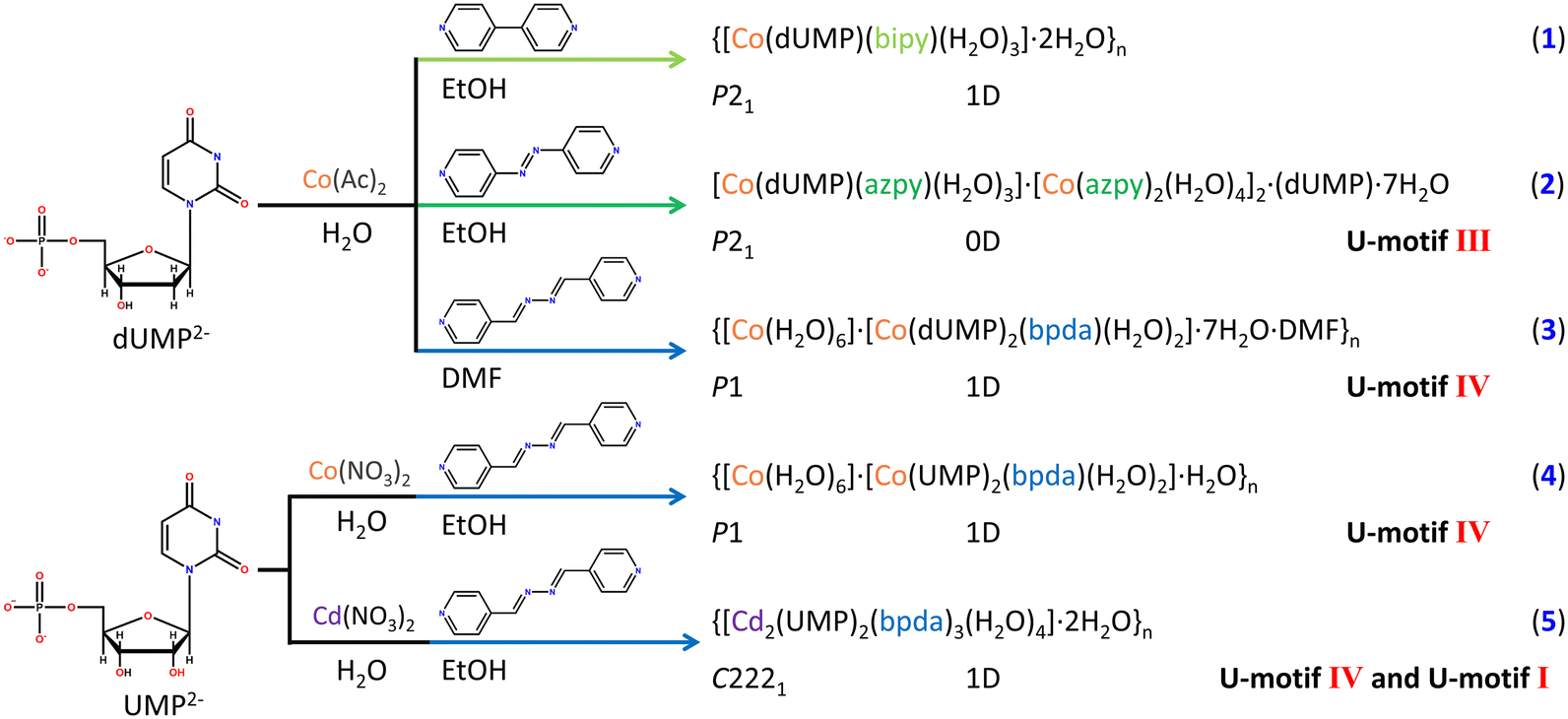 | ||
| Scheme 1 Synthetic procedures of 1, 2, 3, 4, and 5. | ||
1 is a 1D coordination polymer bridged by 4,4′-bipy (Fig. S8 and S10†). The orientation of the uracil base was initially fixed by an intrachain π–π stacking interaction of 4.014(2) Å from the pyridine ring of the auxiliary ligand. And an interchain π–π stacking interaction of 3.837(2) Å was found between adjacent chains, which further stabilized the orientation of the nucleobase and extended the chains into a two-dimensional structure (Fig. S11†). In-depth structural analysis shows that there is no U-motif structure in 1, which may be attributed to the short length of 4,4′-bipy that limited the formation of a U-motif within and between the chains.
Clearly, the length of auxiliary ligand may be an important factor to the construction of U-motifs. Longer auxiliary ligands, azpy and bpda, were used to replace 4,4′-bipy for experiments (Fig. S3†). As expected, U-motifs were successfully constructed in complexes 2–5, and the single crystal structures of the four complexes have been obtained fortunately. 2 is a supramolecular complex containing azpy, and there are both coordinated nucleotides and guest nucleotides in the asymmetry units (Fig. S8b and S12a†). For the auxiliary ligand azpy, only one end participates in the coordination with metal ions, and it doesn't act as a bridging ligand. There are face to face π–π stacking interactions between azpy ligands, which provided the hydrophobic cavity and promoted the formation of the U-motif III (Fig. 2a and d). The U-motif III has been found in adjacent bases from coordinated dUMP and guest dUMP (Fig. S12b and c†). The orientations of uracil bases were stabilized by edge to face π–π stacking interactions The details of the H-bonding in the U-motif III are: N3–H3⋯O4: 1.92 Å, 2.778(8) Å, 173.7° and N3–H3⋯O4: 2.01 Å, 2.868(8) Å, 175.1° (atom names are converted to standard nucleotide atom names), and the dihedral angle involved in the U-motif is 2.2(3)°, which suggest that it is a perfect U-motif structure with high coplanarity.
 | ||
| Fig. 2 The hydrophobic chemical environment formed by the auxiliary ligand in (a) 2, (b) 3, and (c) 5, in which nucleobases were fixed by π–π stacking interaction. The formed U-motifs III and IV in (d) 2 and (e) 3 respectively. The U-motifs I (pink dashed line) and IV (dark blue dashed line) formed in (f) 5. | ||
The length of the auxiliary ligand bpda is longer than azpy and is applied in complexes 3–5 (Fig. S3†). 3 and 4 are 1D coordination polymers bridged by bpda. In 3, strong face to face π–π stacking interactions fixed the orientation of nucleobases within the chain (Fig. 2b). The U-motif IV has been found between adjacent chains (Fig. 2e and S14†). The structural details of the U-motif IV are: N3–H3⋯O2: 1.98 Å, 2.829(10) Å, 170.8° and N3–H3⋯O2: 1.97 Å, 2.825(9) Å, 172.5° (atom names are converted to standard nucleotide atom names), and the dihedral angle involved in the U-motif IV is 4.3(4)°, which is an approximately coplanar structure. This indicates that bpda is suitable for the construction of the U-motif. To further verify the role of bpda in inducing the formation of the U-motif, dUMP was replaced by UMP in 4 (Fig. S15 and S16†). The crystal structural analysis has shown that there are similar U-motif IV existing in 4. The structural details of the U-motif IV in 4 are similar with those in 3 (Fig. S17†). Clearly, bpda is suitable for the construction of the U-motif regardless of dUMP and UMP.
In 1–4, the central metal ions are all Co(II) ions in order to verify the influence of the length of auxiliary ligands. The above studies reveal that the length of auxiliary ligands can significantly influence the formation of the U-motif, which can provide a hydrophobic microenvironment for nucleobases, fix the orientation of nucleobases and induce the formation of non-classical base pairs. So, it is a feasible strategy to regulate the U-motif by changing the length of auxiliary ligands.
To further explore the interaction mode between uracil bases, our investigation focused on the central metal ions. The Cd2+ ion which have stronger bonging ability with nucleotide was used in construction of 5.43 Different from 1–4, the asymmetry unit of 5 was a binuclear Cd(II) species bridged by the phosphate group (μ2-O2) of UMP and extended into a 1D chain by the link of the bpda ligand (Fig. S9b and S18†). Notably, there is an edge-to-face π–π stacking between the nucleobases and the auxiliary ligands, which initially fixes the orientation of the nucleotide bases and induces the formation of the U-motif I within the 1D chain, with the details of C5–H5⋯O4: 2.55 Å, 3.405(6) Å, and 161.2° (Fig. 2c (pink dashed line), f (pink dashed line), and S18†). The U-motif IV was found between adjacent chains (N3–H3⋯O2: 1.94 Å, 2.785(5) Å, 168.1°) (Fig. 2c (blue dashed line), f (dark blue dashed line), and S19†). It is worth noting that two different U-motifs, U-motif I and U-motif IV, appear alternately to form a 1D chain like structure (Fig. S20†), and the dihedral angle between the adjacent uracil bases is 32.8(2)°, whose coplanarity is violently distorted. The edge to face π–π stacking interaction mentioned above stabilizes this structure. It is the first reported nucleobase pairing pattern based on uracil bases, which makes full use of the intrinsic hydrogen bond donors and acceptors of the uracil nucleobase, and this unique structure was names as the full uracil–uracil base pair structure. It provides a basis for discussing the deep interaction between nucleotide bases.
DFT calculations
The electrostatic surface potential (ESP) of the three auxiliary ligands is shown in Fig. S21.†44 The minimum surface electrostatic potentials of 4,4′-bipy, azpy and bpda are concentrated on pyridine nitrogen atoms, and the energy of these are −35.23 kcal mol−1, −33.94 kcal mol−1 and −34.90 kcal mol−1, respectively. So, azpy has a weaker binding ability with metal ions than 4,4′-bipy and bpda. The binding ability of the terminal pyridine nitrogen atom to the metal ion and the size of the auxiliary ligand can regulate the structure of the complexes, which is verified by the coordination mode of azpy in 2. Additionally, the introduction of nitrogen atoms in the middle of azpy and bpda leads to the appearance of new ESP extreme values, which are related to the formation of edge to face π–π stacking interaction between the auxiliary ligand and uracil bases in 2 and 5 and promoted the formation of U-motifs.DFT calculation based on 1-methyluracil reveals the values of interaction energy of the five different U-motifs and shows their thermodynamic stability without considering the effect of sugar ring substitution (Fig. 3). Combined with structure analyses, we found that the U-motif containing the non-classical hydrogen bond (C–H⋯O) have higher interaction energy, while the U-motif containing the classical hydrogen bond (N–H⋯O) have lower one. Clearly, it suggests that classic hydrogen bond is more helpful in stabilizing the formation of base pairs.
 | ||
| Fig. 3 Interaction energy of optimized U-motif structures based on BSSE correction: U-motif I: −21.19565 kJ mol−1; U-motif II: −30.24775 kJ mol−1; U-motif III: −43.61591 kJ mol−1; U-motif IV: −39.81219 kJ mol−1; U-motif V: −41.18331 kJ mol−1 (red square points represent a symmetric structure; blue triangle points represent an asymmetric structure). | ||
Additionally, H-bonding parameters of base pairs obtained by theoretical calculation were very similar to those in crystal structures (Fig. S22 and Table S18†), which indicates that the crystal lattice is suitable for the stable binding between uracil bases. To further visualize the interaction between the uracil bases, DFT calculation was done based on the single crystal structure in the solid state (Fig. S23 and S24†). Strong electrostatic surface potential penetration was observed between the positive electrostatic potential (red colour) and the negative electrostatic potential (green colour) in the two uracil bases of the U-motif, which proves the existence of non-covalent base interaction.
CD spectroscopy
Complexes 1–5 crystallized in the chiral space group, and flack factor indicates that they are all homochiral single crystals (Tables S1 and S2†). The molecular and supramolecular chirality of 1–5 were explored via circular dichroism (CD) spectra in solution and the crystallized solid-state. Two characteristic peaks of the Na2dUMP/Na2UMP ligand were observed in solution: (1) two negative shoulder peaks, 221 nm(−) and 238 nm(−), centered at 229 nm(−), which correspond to the n–π* in nucleotide; (2) positive peaks at 269 nm(+) (Fig. S25†), which correspond to the n–π* and π–π* transition in nucleotide (Fig. S26a†).45 The solution CD spectra for 1–5 revealed that the molecular chirality of 1–5 depended on the intrinsic chirality of nucleotide molecules.In the crystallized solid-state CD spectra, two characteristic peaks of Na2dUMP/Na2UMP were observed: (1) negative shoulder peaks centered at 241/245 nm(−), which correspond to the n–π* and π–π* in nucleotide molecule; (2) positive peaks at 286/289 nm(+) (Fig. 4), which may be caused by intermolecular electron transitions induced by base accumulation (Fig. S26b†).46 The peaks in the range of 200–300 nm of complexes 1–5 are attributed to σ–π*, n–π*, and π–π* transitions in the nucleotide ligands, respectively. The signal flip at 289 nm(−) in 1 is caused by the changes in symmetry of electronic transitions in nucleotide. Two CD peaks at 243 nm(−) and 290 nm(+) in 3 were a characteristic chiral signal caused by exciton coupling.47 The peaks in the range of 300–400 nm appeared in complexes 2–4 are typical EAC (extended axial chirality) induced by the coordination of the auxiliary ligand and nucleotide with metal ions.48 Meanwhile in 5, the peaks at 308 nm(+) and 345 nm(−) suggests that a new chiral source has been generated by the induced coordination of metal ions. However, due to the disorder of the auxiliary ligand, there is no strong chiral signal in the range of 300–400 nm. The coordination processes with metal ions along with the participation of auxiliary ligands can facilitate the extension and transfer of chiral sources, while the interactions between bases in chiral nucleotide molecules can effectively affect the chiral signal of nucleotide complexes.
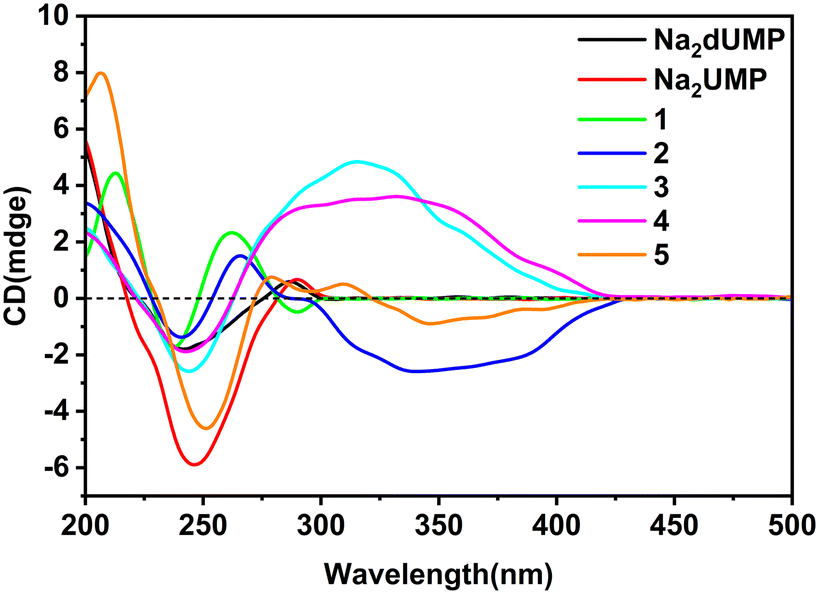 | ||
| Fig. 4 The CD spectra of Na2dUMP, Na2UMP, and 1–5 in solid state (KCl:[sample] = 200:1). | ||
Conclusions
In summary, the construction of U-motifs and the chirality of uracil nucleotide complexes are systematically investigated. On the basis of the high-quality single crystal structure, three types of U-motifs have been successfully constructed by adjusting the auxiliary ligands and metal ions, which provides a feasible route for the construction of base pairs. In particular, an unusual full uracil–uracil base pairing consisted of U-motifs I and IV has been observed. The interaction mode of nucleobases has been confirmed by implementing DFT calculations. Finally, the chirality of these complexes during self-assembly has been examined using solid-state CD spectroscopy in combination with crystal structure analysis.Author contributions
All authors are contributed in work and writing of this manuscript. Prof. Hui Li is the project leader and a corresponding author of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21471017).References
- J. Oh, M. Kimoto, H. Xu, J. Chong, I. Hirao and D. Wang, Nat. Commun., 2023, 14, 195 CrossRef PubMed.
- J. A. Watts, C. Grunseich, Y. Rodriguez, Y. Liu, D. Li, J. T. Burdick, A. Bruzel, R. J. Crouch, R. W. Mahley, S. H. Wilson and V. G. Cheung, Nucleic Acids Res., 2022, 50, 12497–12514 CrossRef PubMed.
- W. Hu, H. Zeng, Y. Shi, C. Zhou, J. Huang, L. Jia, S. Xu, X. Feng, Y. Zeng, T. Xiong, W. Huang, P. Sun, Y. Chang, T. Li, C. Fang, K. Wu, L. Cai, W. Ni, Y. Li, Z. Yang, Q. C. Zhang, R. Chian, Z. Chen, X. Liang and K. Kee, Nat. Commun., 2022, 13, 5114 CrossRef PubMed.
- H. Lu, M. Yang and Q. Zhou, Trends Cell Biol., 2023, 33, 682–694 CrossRef PubMed.
- H. A. Heus and C. W. Hilbers, Nucleosides, Nucleotides Nucleic Acids, 2003, 22, 559–571 CrossRef PubMed.
- R. L. Kumawat and C. D. Sherrill, J. Chem. Inf. Model., 2023, 63, 3150–3157 CrossRef CAS PubMed.
- J. H. Hur, C. Y. Kang, S. Lee, N. Parveen, J. Yu, A. Shamim, W. Yoo, A. Ghosh, S. Bae, C. J. Park and K. K. Kim, Nucleic Acids Res., 2021, 49, 10150–10165 CrossRef CAS.
- J. D. Watson and F. H. Crick, Nature, 1953, 171, 737–738 CrossRef PubMed.
- S. Wain-Hobson, Nature, 2006, 439, 539 CrossRef PubMed.
- M. K. Cabaj and P. M. Dominiak, Nucleic Acids Res., 2020, 48, 8302–8319 CrossRef PubMed.
- J. B. Zhou, E. D. Wang and X. L. Zhou, Cell. Mol. Life Sci., 2021, 78, 7087–7105 CrossRef PubMed.
- S. Mukherjee, L. Blaszczyk, W. Rypniewski, C. Falschlunger, R. Micura, A. Murata, C. Dohno, K. Nakatani and A. Kiliszek, Nucleic Acids Res., 2019, 47, 10906–10913 CrossRef.
- A. K. Chokkalla, S. L. Mehta and R. Vemuganti, Transl. Stroke Res., 2022, 13, 1–11 CrossRef PubMed.
- T. Sawada, M. Yoshizawa, S. Sato and M. Fujita, Nat. Chem., 2009, 1, 53–56 CrossRef PubMed.
- T. Sawada and M. Fujita, J. Am. Chem. Soc., 2010, 132, 7194–7201 CrossRef.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef PubMed.
- G. L. Li, Z. Zhuo, B. Wang, X. L. Cao, H. F. Su, W. Wang, Y. G. Huang and M. Hong, J. Am. Chem. Soc., 2021, 143, 10920–10929 CrossRef PubMed.
- A. Sikder, C. Esen and R. K. O'Reilly, Acc. Chem. Res., 2022, 55, 1609–1619 CrossRef PubMed.
- N. B. Leontis and E. Westhof, RNA, 2001, 7, 499–512 CrossRef PubMed.
- G. Das and R. H. D. Lyngdoh, J. Biomol. Struct. Dyn., 2013, 32, 1500–1520 CrossRef PubMed.
- F. V. Murphy and V. Ramakrishnan, Nat. Struct. Mol. Biol., 2004, 11, 1251–1252 CrossRef PubMed.
- G. Varani and W. H. McClain, EMBO Rep., 2000, 1, 18–23 CrossRef PubMed.
- O. Schrader, Nucleic Acids Res., 2003, 31, 988–998 CrossRef PubMed.
- X. Zhong, X. Tao, J. Stombaugh, N. Leontis and B. Ding, EMBO J., 2007, 26, 3836–3846 CrossRef PubMed.
- Q. M. Qiu, P. Zhou, L. Gu, L. Hao, M. Liu and H. Li, Chem. – Eur. J., 2017, 23, 7201–7206 CrossRef PubMed.
- T. Hagen, A. Laski, A. Brummer, A. Pruska, V. Schlosser, A. Clery, F. H. Allain, R. Zenobi, S. Bergmann and J. Hall, J. Am. Chem. Soc., 2021, 143, 15120–15130 CrossRef PubMed.
- Z. Li, W. Song, Y. Zhu, L. Yan, X. Zhong, M. Zhang and H. Li, Chem. – Eur. J., 2023, 29, e202203979 CrossRef PubMed.
- J. Sheng, A. Larsen, B. D. Heuberger, J. C. Blain and J. W. Szostak, J. Am. Chem. Soc., 2014, 136, 13916–13924 CrossRef PubMed.
- S. E. Lietzke, C. L. Barnes, J. A. Berglund and C. E. Kundrot, Structure, 1996, 4, 917–930 CrossRef PubMed.
- M. C. Wahl, S. T. Rao and M. Sundaralingam, Nat. Struct. Biol., 1996, 3, 24–31 CrossRef PubMed.
- C. R. Groom and J. C. Cole, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 240–245 CrossRef.
- R. B. Martin, Acc. Chem. Res., 1985, 18, 32–38 CrossRef.
- T. Sawada, M. Yoshizawa, S. Sato and M. Fujita, Nat. Chem., 2009, 1, 53–56 CrossRef CAS PubMed.
- J. A. Thomas, Nat. Chem., 2009, 1, 25–26 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- P.-S. Ganesh, S.-Y. Kim, D.-S. Choi, S. Kaya, G. Serdaroğlu, G. Shimoga, E.-J. Shin and S.-H. Lee, J. Anal. Sci. Technol., 2021, 12, 47 CrossRef CAS.
- F. Šebesta and J. V. Burda, Chem. – Eur. J., 2015, 22, 1037–1047 CrossRef PubMed.
- E. van Lenthe, R. van Leeuwen, E. J. Baerends and J. G. Snijders, Int. J. Quantum Chem., 1996, 57, 281–293 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. Zhang and T. Lu, Phys. Chem. Chem. Phys., 2021, 23, 20323–20328 RSC.
- R. M. Smith, A. E. Martell and Y. Chen, Pure Appl. Chem., 1991, 63, 1015–1080 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- K. E. Connell, T. Kurucsev and B. Norden, Aust. J. Chem., 1988, 41, 1509 CrossRef CAS.
- J. Novotna, I. Goncharova and M. Urbanova, Chirality, 2012, 24, 432–438 CrossRef CAS PubMed.
- J. Pandit, T. P. Seshadri and M. A. Viswamitra, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 342–345 CrossRef.
- P. Zhou, J.-F. Yao, C.-F. Sheng and H. Li, CrystEngComm, 2013, 15, 8430–8436 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General produces, and crystallographic details. CCDC 2288977–2288981. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce01056b |
| This journal is © The Royal Society of Chemistry 2024 |