
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulating the photolysis of aryl azides in a supramolecular host to develop photoactivatable fluorophores†
Xujun
Qiu‡
a,
Eric
Pohl‡
b,
André
Jung
 c,
Qianyu
Cai
a,
Haopu
Su
a,
Olaf
Fuhr
de,
Ute
Schepers
*ab and
Stefan
Bräse
*ac
c,
Qianyu
Cai
a,
Haopu
Su
a,
Olaf
Fuhr
de,
Ute
Schepers
*ab and
Stefan
Bräse
*ac
aInstitute of Organic Chemistry (IOC), Karlsruhe Institute of Technology (KIT), Kaiserstrasse 12, 76131 Karlsruhe, Germany. E-mail: ute.schepers@kit.edu; braese@kit.edu
bInstitute of Functional Interfaces (IFG), Karlsruhe Institute of Technology (KIT), Kaiserstrasse 12, 76131 Karlsruhe, Germany
cInstitute of Biological and Chemical Systems - Functional Molecular Systems (IBCS-FMS), Karlsruhe Institute of Technology (KIT), Kaiserstrasse 12, 76131 Karlsruhe, Germany
dInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Kaiserstrasse 12, 76131 Karlsruhe, Germany
eKarlsruhe Nano Micro Facility (KNMFi), Karlsruhe Institute of Technology (KIT), Kaiserstrasse 12, 76131 Karlsruhe, Germany
First published on 3rd October 2024
Abstract
Photolysis of aryl azides is a convenient method to approach more functionalized systems in chemical biology. Here, we present a set of photoactivatable aryl azides that undergo controlled reaction pathways within the cucurbit[7]uril (CB7) cavity upon photolysis. The fluorescence turn-on process is utilized for bioimaging.
Photoconvertible molecules which can undergo precisely directed photoreactions, significantly enhancing the emission from a weak or non-fluorescent state, are highly demanded.1,2 This ‘turn-on’ process can be utilized for tracking the biological activity and real-time localization imaging.3,4 Organic azides have shown their versatile applications in chemical biology, as they can be easily functionalized using various methods.5,6 Photolysis of aryl azides is of crucial importance in chemical biology, with applications like light-induced protein labeling7,8 and RNA photo-crosslinking.9 Nevertheless, applications of aryl azides in living cells as photoactivatable probes are seldomly documented.10–13 One possibility is the generation of highly toxic singlet nitrene during photolysis of aryl azides, reducing cell viability. Additionally, singlet nitrenes may undergo uncontrollable photoreaction pathways, leading to undesirable products.14
To overcome these challenges, we previously developed a novel method to selectively control the aryl azide photoreaction pathway to a carboline with dramatically enhanced fluorescence by implementing the photoreaction within the cavity of a macrocyclic molecule, cucurbit[7]uril (CB7).15
In this study, we designed and synthesized three aryl azides with a push–pull system. We demonstrated control over the photoreaction within the cavity of CB7, transforming low emission aryl azides into highly fluorescent carbolines. Time-dependent density functional theory (TD-DFT) calculations were used to study the mechanism of this ‘turn-on’ process. Additionally, we successfully applied the photoactivation process to living cells for optical imaging (Scheme 1).
 | ||
| Scheme 1 Illustration of photoactivatable aryl azides’ ‘turn-on’ emission in CB7 (left) and structures of azides and carbolines (right). | ||
We applied various electron-donating groups to the designed azides to modulate intramolecular charge transfer (ICT) and to adjust their absorption properties.16 The syntheses and structures of aryl azides Az-1, Az-2, and Az-3 are shown in Fig. 1, with full characterization in the ESI† (Fig. S1–S11 and Tables S1, S2). Photolysis of these azides was initially investigated in water using 405 nm LEDs (M405L4 – 405 nm, 1000 mW (Min) Mounted LED, 1000 mA, THORLABS) at room temperature. As indicated by the NMR spectra (Fig. S12, S14 and S16, ESI†), new peaks appeared after photolysis of Az-1, Az-2, and Az-3. Liquid chromatography-mass spectrometry (LC-MS) analysis of the reaction mixtures of Az-1 and Az-2 (Fig. S13 and S15, ESI†) revealed product masses of 348.23 g mol−1 for Az-1 and 350.27 g mol−1 for Az-2. Compared to the starting masses of Az-1 (376.26 g mol−1) and Az-2 (378.28 g mol−1), these new masses indicate the formation of carboline products through photoreaction. This can be attributed to the highly hydrophobic aromatic ring conjugation in Az-1 and Az-2, which promotes the molecule aggregation in water, forming noncovalent stacked systems.17,18 These systems prevent water from attacking, leading to intramolecular C–H amination. The crystal packing structures of Az-1 and Az-2 further support this hypothesis (Fig. S4 and S8, ESI†).
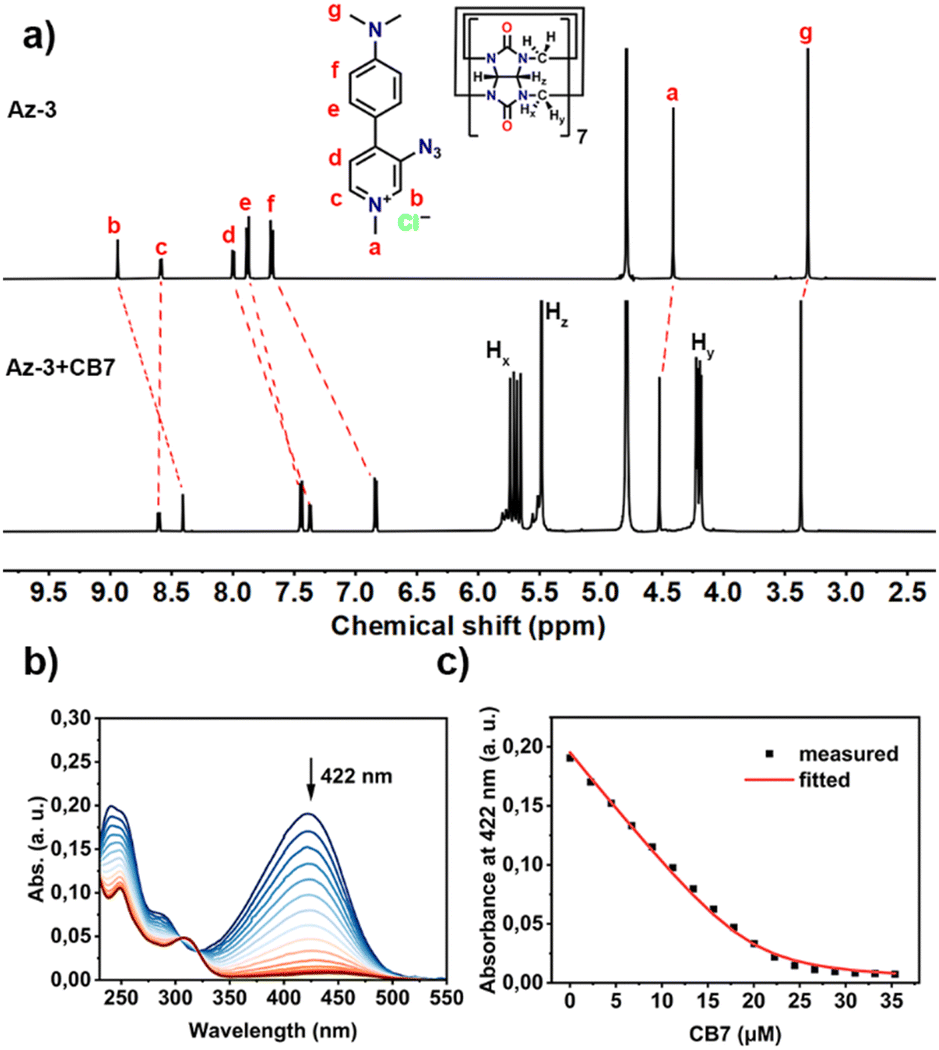 | ||
Fig. 1 (a) 1H NMR spectra (500 MHz, D2O) of Az-3 (0.5 mM) and Az-3 + CB7 (0.5 mM each); (b) UV-vis titration spectra of Az-3 (20 μM) upon addition of CB7 (0–35 μM) in water at 298 K, and (c) the binding isotherm fitted with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest binding model. :1 host–guest binding model. | ||
In contrast, Az-3, which has a smaller aromatic conjugate, was found to form various products after irradiation. This was evidenced by the presence of methyl groups in the resulting NMR spectra (Fig. S16, ESI†). LC–MS also revealed that the photolysis of Az-3 in water produces a mixture of photoproducts with different molecular masses (Fig. S17, ESI†). The photoreaction processes were further monitored using UV-vis and emission spectra (Fig. S18–S20, ESI†), which suggest the reaction completes at 420 s for Az-1, 240 s for Az-2 and 180 s for Az-3.
We then investigated the host–guest interaction of the synthesized aryl azides with CB7 using NMR techniques. As shown in Fig. S21–S23 (ESI†), the addition of CB7 to the Az-1 solution resulted in significant proton shifts, whereas signals d and e exhibited an upfield shift, suggesting the deep encapsulation of azido moieties within CB7 cavities. The formation of a 1:1 complex of Az-1 with CB7 was also observed by MALDI-TOF-MS (Fig. S24, ESI†), which showed a peak of m/z 1510.21 corresponding to [Az-1+CB7-Cl]+ ions. The deep encapsulation within the CB7 cavity was also observed for Az-2 and Az-3 (Fig. 1(a) and Fig. S25–S32, ESI†).
The binding constants of the aryl azides and CB7 were determined by UV-vis titration (Fig. S33–S38, ESI†). The addition of CB7 to aryl azides resulted in a decrease in absorption intensity at 368 nm for Az-1 (Fig. S33, ESI†), 422 nm for both Az-2 (Fig. S35, ESI†) and Az-3 (Fig. 1(b) and Fig. S37, ESI†). The binding constants were obtained by non-linear fitting, which were calculated to be (3.79 ± 0.18) × 105 M−1 for Az-1 (Fig. S34, ESI†), (1.76 ± 0.05) × 105 M−1 for Az-2 (Fig. S36, ESI†), and (1.40 ± 0.03) × 106 M−1 for Az-3 (Fig. 1(c) and Fig. S38, ESI†). These values are comparable to those of previously studied aryl azides.15
Furthermore, the photoreaction of aryl azides within the CB7 cavities was performed under the same conditions as in water. As shown in Fig. S39–S56 (ESI†), the formation of carbolines through tuned intramolecular C–H amination within the CB7 cavities was observed. The resulting carbolines were characterized as shown in Fig. S42–S44 (ESI†) for Cb-1, Fig. S48–S50 (ESI†) for Cb-2, and Fig. S54–S56 (ESI†) for Cb-3. LC–MS revealed molecular masses of 348.23 g mol−1 for Cb-1 (Fig. S41, ESI†), 350.27 g mol−1 for Cb-2 (Fig. S47, ESI†), and 226.23 g mol−1 for Cb-3 (Fig. S53, ESI†). The reaction progress was also tracked by UV-vis and emission spectroscopy, which suggest the carbolines formed within 240 s for Cb-1 (Fig. S57, ESI†), 100 s for Cb-2 (Fig. S58, ESI†), and 180 s for Cb-3 (Fig. S59, ESI†).
Interestingly, the emission spectra of the photoreaction products showed substantial increases in intensity. For Cb-1·CB7, the emission intensity at λ539nm increased 16-fold compared to the weak emission of Az-1·CB7 (Fig. 2(a)). Cb-2·CB7 exhibited a 49-fold enhancement at λ556nm, while Cb-3·CB7 showed a 60-fold enhancement at λ480nm compared to the barely fluorescent Az-2·CB7 (Fig. 2(b)) and Az-3·CB7 (Fig. 2(c)). The photophysical properties of the carbolines are shown in Table S3 (ESI†), with photoreaction quantum yields of 16.3%, 31.4%, and 25.6% for Az-1, Az-2, and Az-3 in CB7, respectively.
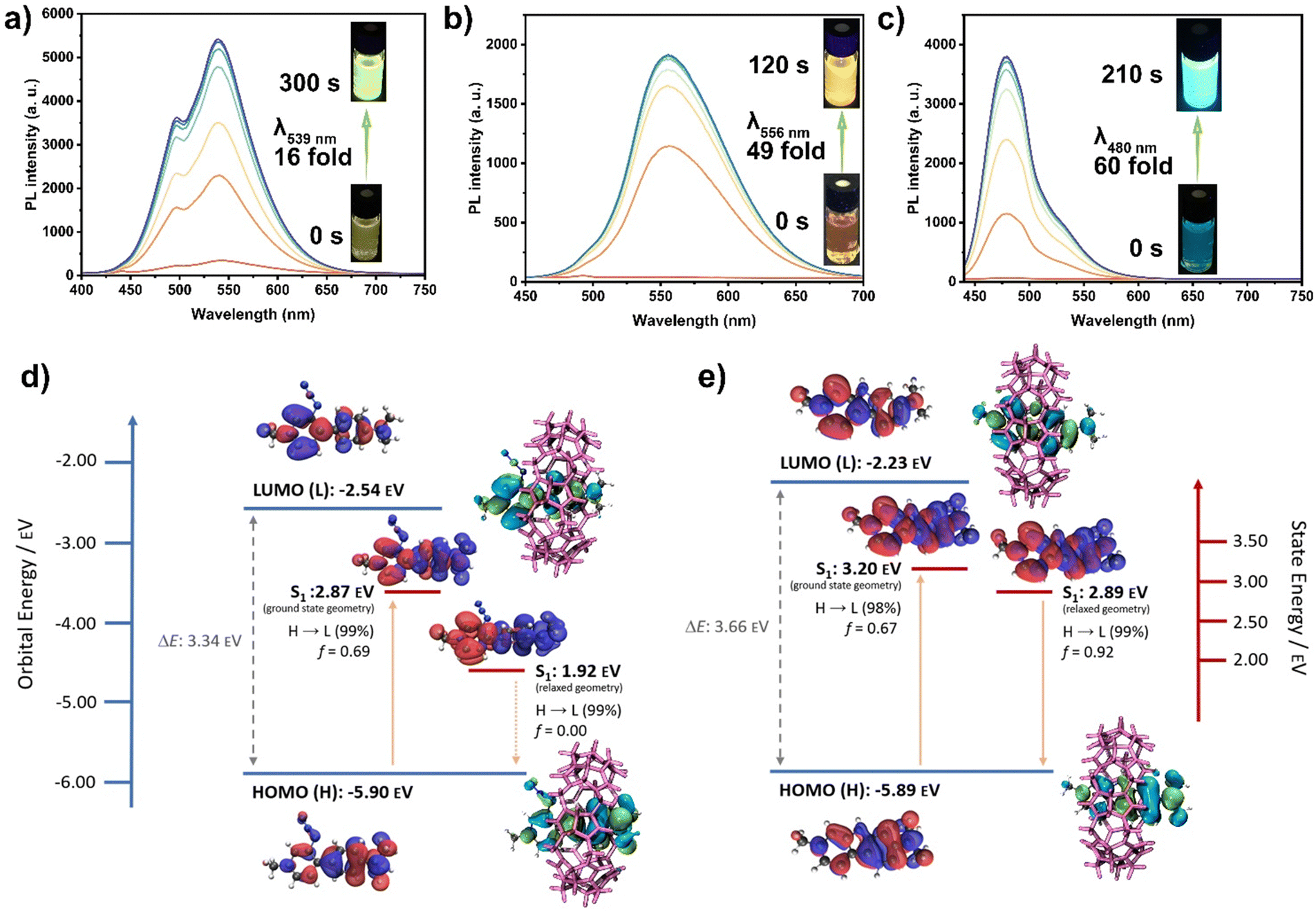 | ||
| Fig. 2 Photoluminescence (PL) spectra of photolysis of (a) Az-1·CB7; (b) Az-2·CB7; and (c) Az-3·CB7 after different reaction times; (d) and (e) computed molecular orbitals (HOMO/LUMO) and difference density plots of the first excited singlet state at the ground state and relaxed S1 geometry of Az-3 and Cb-3, calculated at the (TD-)DFT PBE0/6-31+G(d,p)//B3LYP/6-31G(d) level of theory in water (PCM). | ||
To better understand the nature of the fluorescence enhancement upon carboline formation, particularly the cyclization of Az-3 to Cb-3 within CB7, we performed DFT calculations (Fig. 2(d), (e) and Fig. S62, S63, ESI†). The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) orbital energies of azides and carbolines were compared, which are shown in Fig. 2(d), (e) and Fig. S62 (ESI†). The calculated results reveal similar bandgaps of carbolines and corresponding azides. For Az-3·CB7 and Cb-3·CB7 (Fig. 2(d) and (e)), hole and electron wavefunctions are distributed within the whole molecule at the ground state geometry. At the relaxed excited state geometry, hole and electron wavefunctions exhibit near-complete separation, which likely causes the S1 state of Az-3·CB7 to become non-emissive. This is further supported by the near-zero oscillator strength observed in the system. In general, the state energies of the excited singlet states S1 for azides were found to be lower than the corresponding carbolines. Additionally, absorption and emission spectra were simulated (Fig. S63, ESI†), revealing acceptable agreement with experimental results. The non-emissive nature of Az-1 and Az-2 could not be described via TD-DFT (Fig. S62, ESI†). However, the shortcomings of TD-DFT concerning the accurate description of (long-range) CT states are well known.19,20
Considering these findings, we explored this fluorescence photoactivation process for live cell imaging. Emission experiments performed in cell media showed enhanced fluorescence intensity for azide–CB7 complexes compared to their free forms (Fig. S64, ESI†). Following the photoreaction, emission intensities increased for both unbound and CB7-complexed carbolines (Fig. S65, ESI†). Interestingly, the PL intensities of Cb-1 and Cb-2 were higher than those observed within the CB7 cavity (Fig. S65, ESI†), likely due to large aromatic structures fitting into protein pockets in serum and reducing water quenching. In contrast, CB7 effectively shields the smaller Cb-3 from water-based quenching. To verify this hypothesis, we tested the photophysical properties of Cb-1, Cb-2, Cb-3, and their CB7 complexes in the presence of bovine serum albumin (BSA), the most abundant protein in HeLa cell medium21 and varying concentrations of fetal bovine serum (FBS) (Fig. S66–S83, ESI†).
We evaluated the cytotoxicity of aryl azides, carboline products, and their CB7 complexes on HeLa cells at different concentrations (50 μM, 5 μM, and 0.5 μM) (Fig. S67, ESI†). Both aryl azides and carbolines exhibit similarly low cytotoxicity (LC50 > 50 μM). We then implemented azide–CB7 complexes in living cells, followed by photoactivation. As shown in Fig. 3, fluorescence intensity increased after irradiation, indicating intracellular carboline formation. Photoreaction without CB7 led to faster reaction times and reduced PL for azides, highlighting the role of CB7 in enhancing cellular uptake and directing the photoreaction (Fig. S68, ESI†). Additionally, we preactivated azides·CB7 to carbolines in cell media, prior to the uptake for imaging (Fig. S69a, ESI†). As shown in Fig. S69b (ESI†), carboline–CB7 complexes Cb-1·CB7 and Cb-2·CB7 were accumulated within endosomal vesicles, while Cb-3·CB7 was predominantly found in mitochondria. We cultivated cells with carboline–CB7 complexes under light exposure to assess long-term effects and biocompatibility, revealing no significant cytotoxicity (Fig. S84, ESI†).
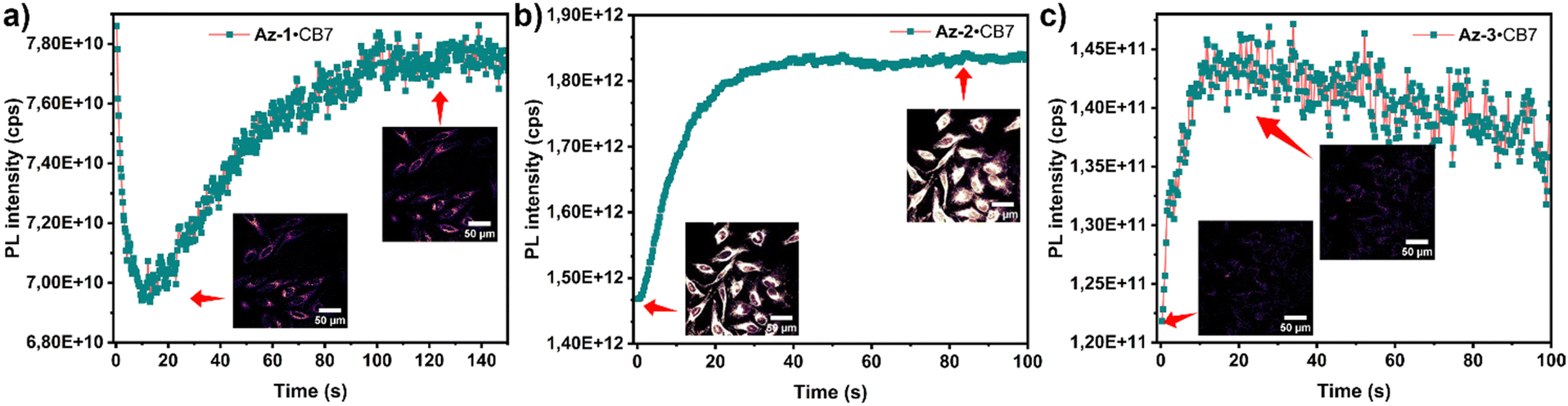 | ||
| Fig. 3 PL intensity and confocal imaging of (a) Az-1·CB7; (b) Az-2·CB7; and (c) Az-3·CB7 upon reaction in HeLa cells. | ||
In summary, we developed a novel approach for developing photoactivatable fluorescent probes utilizing the CB7 host to control the photoreaction pathway of aryl azides into carbolines, resulting in a significantly enhanced fluorescence intensity. DFT calculations elucidated the fluorescence ‘turn-on’ mechanism. Besides, we successfully applied this photoactivation process in live cell imaging, highlighting the potential of controlling aryl azide photolysis within supramolecular hosts for biochemical applications.
The supports from China Scholarship Council, bwHPC, Deutsche Forschungsgemeinschaft (DFG) under Germany's Excellence Strategy-3DMM2O-EXC-2082/1-390761711, and INST 40/575-1 FUGG (Justus 2 cluster) are gratefully acknowledged. Prof. H.-A. Wagenknecht (IOC, KIT) and Dr F. Biedermann (INT, KIT) are gratefully acknowledged for access to photophysics equipment used in this study.
Data availability
Data for this article, including NMR and IR, are available at Chemotion Repository at https://www.chemotion-repository.net/welcomevia the DOI in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- X. Gu, E. Zhao, T. Zhao, M. Kang, C. Gui, J. W. Y. Lam, S. Du, M. M. T. Loy and B. Z. Tang, Adv. Mater., 2016, 28, 5064–5071 CrossRef CAS PubMed.
- Q. Gong, X. Zhang, W. Li, X. Guo, Q. Wu, C. Yu, L. Jiao, Y. Xiao and E. Hao, J. Am. Chem. Soc., 2022, 144, 21992–21999 CrossRef CAS PubMed.
- C. Xu, H. Zou, L. Hu, H. Shen, H. H. Y. Sung, H. Feng, R. T. K. Kwok, J. W. Y. Lam, L. Zheng and B. Z. Tang, ACS Mater. Lett., 2022, 4, 1831–1839 CrossRef CAS.
- K. Kikuchi, L. D. Adair, J. Lin, E. J. New and A. Kaur, Angew. Chem., Int. Ed., 2023, 62, e202204745 CrossRef CAS PubMed.
- C. Bednarek, I. Wehl, N. Jung, U. Schepers and S. Bräse, Chem. Rev., 2020, 120, 4301–4354 CrossRef CAS PubMed.
- N. Z. Fantoni, A. H. El-Sagheer and T. Brown, Chem. Rev., 2021, 121, 7122–7154 CrossRef CAS PubMed.
- Y. Zhang, J. Tan and Y. Chen, Chem. Commun., 2023, 59, 2413–2420 RSC.
- N. E. S. Tay, K. A. Ryu, J. L. Weber, A. K. Olow, D. C. Cabanero, D. R. Reichman, R. C. Oslund, O. O. Fadeyi and T. Rovis, Nat. Chem., 2023, 15, 101–109 CrossRef CAS PubMed.
- K. L. Buchmueller, B. T. Hill, M. S. Platz and K. M. Weeks, J. Am. Chem. Soc., 2003, 125, 10850–10861 CrossRef CAS PubMed.
- S. J. Lord, N. R. Conley, H.-L. D. Lee, R. Samuel, N. Liu, R. J. Twieg and W. E. Moerner, J. Am. Chem. Soc., 2008, 130, 9204–9205 CrossRef CAS PubMed.
- A. V. Anzalone, Z. Chen and V. W. Cornish, Chem. Commun., 2016, 52, 9442–9445 RSC.
- S. H. Liyanage, N. G. H. Raviranga, J. G. Ryan, S. S. Shell, O. Ramström, R. Kalscheuer and M. Yan, JACS Au, 2023, 3, 1017–1028 CrossRef CAS PubMed.
- X. Qiu, E. Pohl, Q. Cai, J. Seibert, Y. Li, S. Leopold, O. Fuhr, M. A. R. Meier, U. Schepers and S. Bräse, Adv. Funct. Mater., 2024, 2401938 CrossRef.
- M.-L. Tsao, N. Gritsan, T. R. James, M. S. Platz, D. A. Hrovat and W. T. Borden, J. Am. Chem. Soc., 2003, 125, 9343–9358 CrossRef CAS PubMed.
- X. Qiu, Y. Wang, S. Leopold, S. Lebedkin, U. Schepers, M. M. Kappes, F. Biedermann and S. Bräse, Small, 2024, 20, 2307318 CrossRef CAS PubMed.
- A. Slama-Schwok, M. Blanchard-Desce and J. M. Lehn, J. Phys. Chem., 1990, 94, 3894–3902 CrossRef CAS.
- K. Leduskrasts, A. Kinens and E. Suna, Chem. Commun., 2019, 55, 12663–12666 RSC.
- H. Park, G. Niu, C. Wu, C. Park, H. Liu, H. Park, R. T. K. Kwok, J. Zhang, B. He and B. Z. Tang, Chem. Sci., 2022, 13, 2965–2970 RSC.
- M. Campetella, F. Maschietto, M. J. Frisch, G. Scalmani, I. Ciofini and C. Adamo, J. Comput. Chem., 2017, 38, 2151–2156 CrossRef CAS PubMed.
- T. Froitzheim, S. Grimme and J.-M. Mewes, J. Chem. Theory Comput., 2022, 18, 7702–7713 CrossRef CAS PubMed.
- S. Curry, H. Mandelkow, P. Brick and N. Franks, Nat. Struct. Mol. Biol., 1998, 5, 827–835 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2351099 and 2351100. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc03907f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |