
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled hydrolysis of AlMe3 to tetramethylalumoxane and a new look at incipient adducts with water†‡
Krzesimir
Korona
 a,
Iwona
Justyniak
*b,
James
Pogrebetsky
b,
Marta
Lemieszka
a,
Piotr
Bernatowicz
b,
Antoni
Pietrzykowski
a,
Adam
Kubas
*b and
Janusz
Lewiński
*ab
a,
Iwona
Justyniak
*b,
James
Pogrebetsky
b,
Marta
Lemieszka
a,
Piotr
Bernatowicz
b,
Antoni
Pietrzykowski
a,
Adam
Kubas
*b and
Janusz
Lewiński
*ab
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: janusz.lewinski@pw.edu.pl
bInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: ijustyniak@ichf.edu.pl; akubas@ichf.edu.pl
First published on 7th August 2024
Abstract
We present an efficient route to tetramethylalumoxane by the controlled hydrolysis of AlMe3 in the presence of pyridine. The AlMe3(pyr) hydrolysis by 0.5 and 1 equiv. of H2O has been followed with real-time 1H NMR. Based on high-level quantum-chemical calculations, we conclude that hypervalent, pentacoordinate aluminium species are critical in the first steps of hydrolysis.
Alumoxanes belong to the family of organometallics comprising an Al–O–Al unit with pendant organic substituents.1,2 The chemistry of alumoxanes has a rich history; initial studies trace back to the end of the 1950s.1 They have attracted attention as highly active catalysts in the polymerisation of a wide range of organic monomers.2,3 In particular, methylalumoxane (MAO) is an effective cocatalyst in olefin polymerisation.4–6 Interestingly, many structural models have been proposed for MAO,7–15 and only very recently, its molecular structure was crystallographically documented as a discrete sheet cluster.16
The hydrolytic approach seems conceptually the simplest way to alumoxanes. However, all attempts for the initial three decades have focused on hydrolysis reactions involving homoleptic short-chain-alkyl aluminates.1,17 Although various methods of introducing water have been used to lower the rate of the hydrolysis reaction (e.g., using hydrated salts18 or the dropwise addition of water at low temperatures18,19), complexity and limited stability of the resulting species have prevented their detailed structural investigations. Moreover, the hydrolysis of short-chain-alkyl homoleptic aluminates has been monitored using variable-temperature 1H NMR spectroscopy,18–21 mass spectrometry,22,23 and the identity of intermediates has been supported by quantum-chemical calculations.22–27 Simultaneously, various alternative non-hydrolytic approaches to alumoxanes have also been used, such as the decomposition of aluminium alkoxides,28 reactions of AlR3 compounds with carboxylic acids11,29–31 or R2BOH,32–35 and other methods.36–40 Strikingly, only the phthalic acid alkylation has paved the way for isolating the tetraramethylalumoxane moieties arranged as nodes in the bipyridine-linked polymeric chains.30 A milestone in the hydrolytic synthesis of molecular alkylalumoxanes has been achieved by Barron et al., employing sterically demanding tert-butylaluminium compounds. The authors have isolated and structurally characterised the first tetraraalkylalumoxane, [tBu2AlOAltBu2]2, its adduct with pyridine (py), and a series of tert-butylaluminium hydroxides, (oxide)hydroxides and [(tBuAlO)n] cages.41–46
Further studies on the controlled hydrolysis of alkylaluminium compounds are highly desired and may lead to the accelerated development of well-defined alumoxanes. In the course of our systematic studies on the controlled hydrolysis of various organometallics to desired molecular complexes and functional materials,47–50 herein we present an efficient route to a well-defined tetramethylalumoxane by the controlled hydrolysis of AlMe3 in the presence of pyridine derivatives as strong coordinating ligands. To gain additional insight into the AlMe3 hydrolysis, we have examined the reaction system by variable-temperature 1H NMR spectroscopy along with theoretical calculations of the incipient AlMe3·H2O adducts in the presence of donor molecules.
Homoleptic aluminium alkyls readily form four-coordinate Lewis acid–base adducts with neutral donor ligands, which reduces their reactivity.51 Thus, we chose a series of [AlMe3L] adducts as model precursors, where L = THF, pyrrolidine, pyridine (py) and p-dimethylaminopyridine (dmap). The controlled hydrolysis of [AlMe3(L)] (1, L = py, dmap) by 0.5 equiv. of H2O was performed in a THF/hexane mixture, starting at −78 °C to ambient temperature (Fig. 1). Subsequent crystallisation led to the colourless crystals of tetrametylalumoxane adducts [AlMe2(L)(μ3-O)AlMe2]2 with py (3) and dmap (4), isolated in high yields. In contrast, using THF or pyrrolidine as donor ligands, analogous reactions were unselective and led to viscous mixtures of products according to 1H NMR spectra (Fig. S10 and S11, ESI‡). Compounds 3 and 4 are isostructural and crystallise as a dimer with Ci symmetry based on an essentially planar [Al4(μ3-O)2] core (Fig. 2 and Fig. S23, ESI‡). The Al centres are four-coordinate, adopting distorted tetrahedral geometries, and the py/dmap ligands are coordinated to the exocyclic Al1 centres. Similarly to other reported [Al4(μ3-O)2] cores,30,41 the exocyclic Al–O bonds are slightly shorter than the endocyclic ones (by ca. 0.05–0.1 Å). Both compounds are essentially isostructural to the asymmetric unit of a tetramethylalumoxane-based coordination polymer [AlMe2(μ3-O)AlMe2(1,2-bis(4-pirydyl)ethane)]n.30 The 1H NMR spectra of 3 and 4 confirm the presence of two distinct methyl groups and py/dmap moiety in the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio (Fig. S6 and S8, ESI‡). Using 1D transient NOE and 1H–13C HSQC experiments for 3, we assigned the 1H/13C NMR chemical shifts at −0.79/−9.8 ppm to the exocyclic Al-bonded methyl groups and the resonances at −1.05/−7.6 ppm to the endocyclic Al-bonded methyl moieties (Fig. S6, S7, S11 and S12 and Table S2, ESI‡). Despite two independent Al centres in 3 and 4, we have observed only one broad resonance in the 27Al spectrum at ca. 150 ppm, typical for the four-coordinate Al centres.52 The dimeric form of 3 and 4 in THF solution was proved by DOSY (Table S1 and Fig. S14 and S15, ESI‡) and ESI-MS (Fig. S18 and S19, ESI‡).
:2:1 ratio (Fig. S6 and S8, ESI‡). Using 1D transient NOE and 1H–13C HSQC experiments for 3, we assigned the 1H/13C NMR chemical shifts at −0.79/−9.8 ppm to the exocyclic Al-bonded methyl groups and the resonances at −1.05/−7.6 ppm to the endocyclic Al-bonded methyl moieties (Fig. S6, S7, S11 and S12 and Table S2, ESI‡). Despite two independent Al centres in 3 and 4, we have observed only one broad resonance in the 27Al spectrum at ca. 150 ppm, typical for the four-coordinate Al centres.52 The dimeric form of 3 and 4 in THF solution was proved by DOSY (Table S1 and Fig. S14 and S15, ESI‡) and ESI-MS (Fig. S18 and S19, ESI‡).
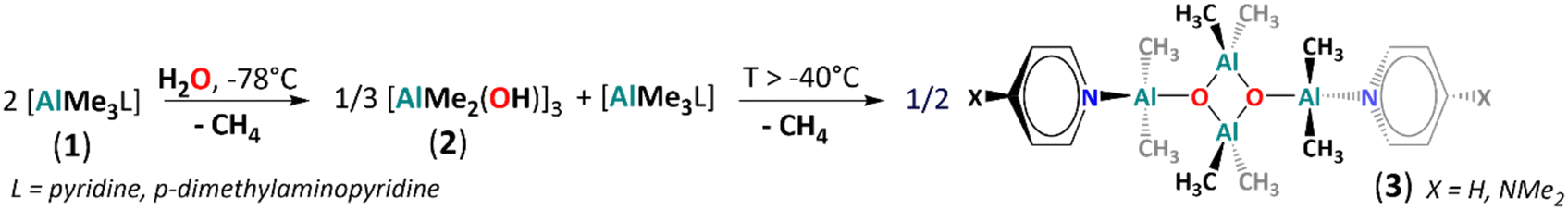 | ||
| Fig. 1 Synthesis of [AlMe2(pyr)(μ3-O)AlMe2]2 (3) and [AlMe2(dmap)(μ3-O)AlMe2]2 (4). | ||
 | ||
| Fig. 2 The molecular structure of [AlMe2(pyr)(μ3-O)AlMe2]2 (3). Hydrogen atoms were omitted for clarity. | ||
To gain a deeper understanding of the AlMe3 hydrolysis, we examined the title reaction system (with py) by variable-temperature 1H NMR experiments. The 1H NMR-tracked hydrolysis of 1 was carried out with one and two equiv. of H2O. (Fig. 3) The reaction progress was monitored from 193 K to 298 K with a temperature interval of 10 K. The parent solution of 1 in THF-d8 exhibits a single resonance of the Al–Me protons at −0.87 ppm, and its chemical shift is only slightly different from that for a solution of AlMe3 in THF-d8 (−0.95 ppm, Fig. S1 and S2, ESI‡), suggesting a dynamic equilibrium between 1 and a [AlMe3(THF-d8)] adduct. For the reaction at 0.5 equiv. of H2O per 1, new signals characteristic of Al–Me protons (−0.95 ppm) and an Al–OH proton (7.98 ppm) immediately appeared in the spectrum after the addition of H2O at 193 K. Their integrals ratio of 6:1 indicates the formation of a [AlMe2OH] (2) species with concomitant liberation of methane (signal at 0.18 ppm). At this temperature, the formed hydroxide compound 2 does not interact with the excessive amount of 1, which chemical shift remains intact. Notably, no signals indicating the presence of a labile AlMe3·OH2 adduct were observed. In the temperature range of 193–233 K, the reaction system is intact, and only the –OH proton resonance gradually shifts to 7.50 ppm at 233 K. Above 233 K, the resonances of the final product 3 appear and gradually grow with temperature gain to 298 K with simultaneous expiring of the signals from 1 and 3. Notably, the Al–Me resonance of 2 vanishes faster than that from 1, suggesting a side reaction of 2 at 298 K.
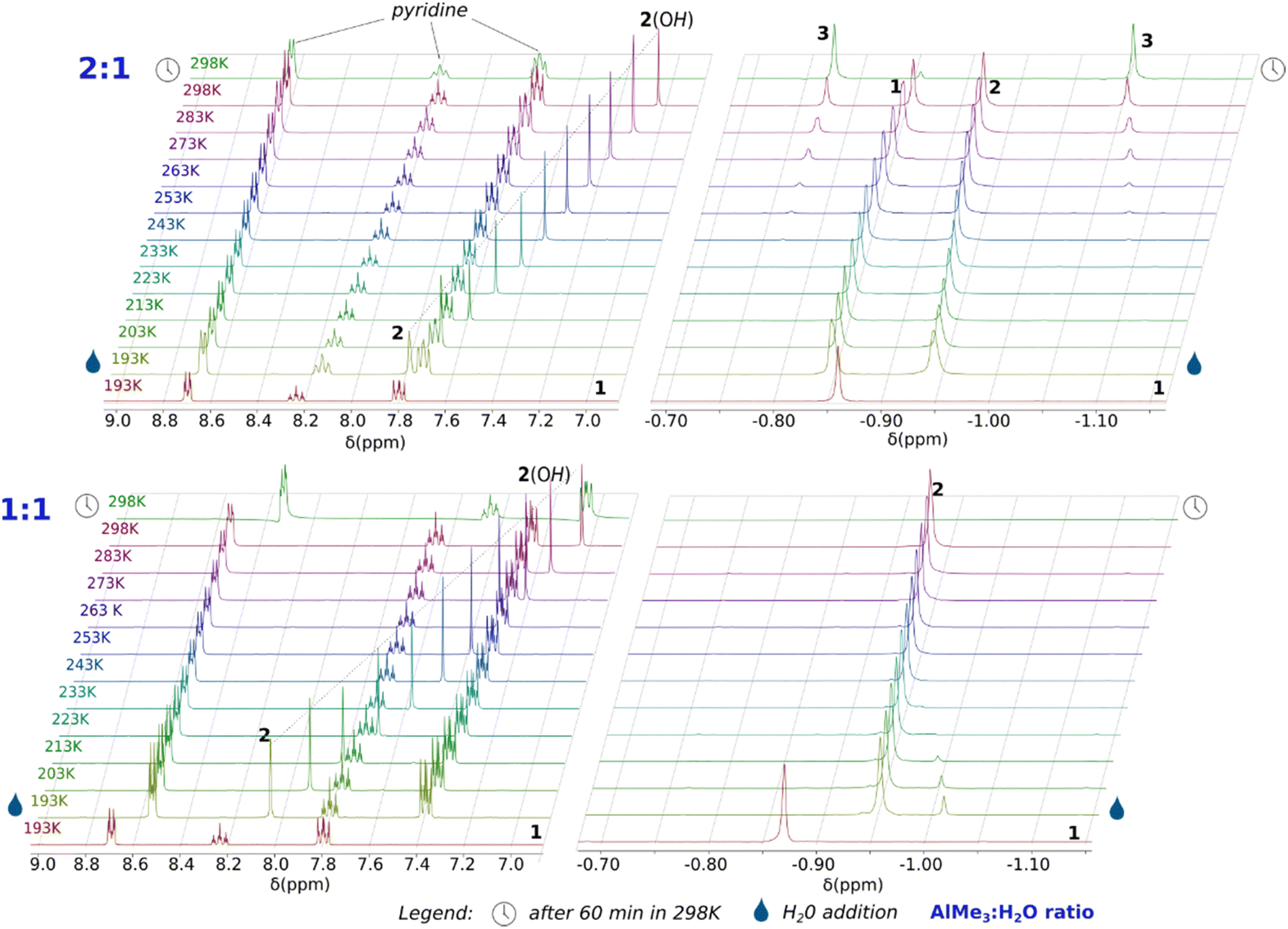 | ||
| Fig. 3 Variable-temperature NMR spectra for the reaction of 1 with H2O in 2:1 (top) and 1:1 (down) stoichiometries. | ||
In the case of the equimolar AlMe3/H2O/py reaction system, the 1H NMR spectra demonstrate that the addition of H2O to a THF-d8 solution of 1 at 193 K affords effectively the hydroxide compound 2 with concomitant disappearance of 1. Interestingly, initially, the low-intensity resonance at −1.03 was observed at 193 K, which could be related to the formation of an unstable adduct of AlMe3 with H2O; this signal relatively quickly dissipates as a result of leaving the sample at this temperature (for detailed discussion vide infra). Notably, no new signals were observed with the temperature gain. Instead, a gradual expiration of the resonances of 2 takes place at 298 K due to an oligomerisation of the hydroxide species to an amorphous solid (Fig. S12, ESI‡).
In order to determine the nature of the hydroxide, DOSY NMR experiments were performed. The estimated molecular weight indicates values corresponding to the formation of OH-bridged trimer solvated by three molecules of THF-d8 or pyridine (M ≈ 460 g mol−1, Table S1, ESI‡). In turn, the 27Al NMR spectrum of 2 shows a signal at 146 ppm, suggesting the four-coordinate Al centre.52 Thus, the neutral donor molecules are outside the first coordination sphere and likely solvate the trimer via hydrogen bonding. The formation of trimeric dialklylaluminium hydroxide species solvated by neutral donor molecules was previously anticipated by Pasynkiewicz.1 Moreover, such type of adducts featuring the bulky tBu groups have been isolated in crystal form and structurally characterised by Barron for [Al(tBu)2(μ-OH)]3·2L (L = THF, MeCN).41
While our results corroborate to some degree with previous experimental1,20,21 and computational23–29 investigations of the initial steps of AlMe3 hydrolysis that the character of an initial AlMe3/H2O adduct is unclear as the presence of both donor solvent molecules and pyridine (as a strong Lewis base) in the first coordination sphere should be considered. To determine the character of this unstable intermediate state, we performed state-of-the-art quantum-chemical calculations, including the possibility of the formation of hypervalent five-coordinate adducts,51,53 which have not been considered before. The energies reported below have been obtained at the domain-based local pair natural orbital coupled cluster method with singles, doubles, and iterative triples (DLPNO-CCSD(T1))54–56 and include estimates for complete PNO space,57 complete basis set,58 zero-point energy, and solvent effects (THF). Details of calculations can be found in the ESI,‡ where the comparison with density functional theory results is provided for completeness. First, we have considered the binding energies of water, THF, and pyridine to AlMe3. Here, the energies of −16.8, −23.3, and −26.5 kcal mol−1 have been found (Table S7, ESI‡). According to our expectation, the four-coordinate AlMe3·py adduct is the most stable, and in the presence of THF and water, one can consider an equilibrium presented in Fig. 4.
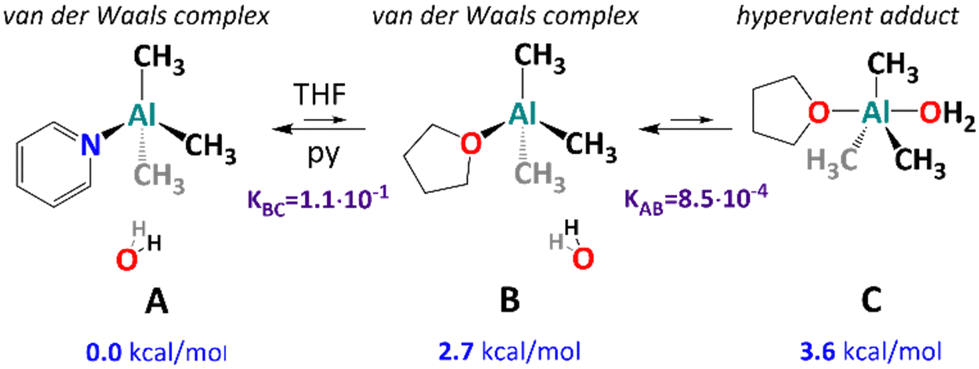 | ||
| Fig. 4 Computationally modelled incipient complexes of AlMe3 with H2O in the presence of neutral donor ligands. | ||
For the py adduct, the H2O molecule remains outside the first coordination sphere (structure A in Fig. 4; see also Fig. S24, ESI‡), and no stable hypervalent minimum has been located. The replacement of py with a THF molecule (structure B) leads to slight energy elevation (2.7 kcal mol−1), which corresponds to an equilibrium constant of 8.5 × 10−4 at 195 K (Tables S8 and S9, ESI‡), and the process has a negligible activation energy of 3.3 kcal mol−1 (Table S8, ESI‡). Interestingly, a van der Waals complex B remains in equilibrium with a hypervalent complex C involving H2O in the first coordination sphere (the Al–O distance equals 2.34 Å, see Fig. S24, ESI‡). The energy difference between the van der Waals complex B and the five-coordinate complex C is small enough (0.9 kcal mol−1) to result in a relatively large equilibrium constant of 1.1 × 10−1 (as shown in several works,13,22–27 the quantum-chemical modelling of AlMe3 hydrolysis is highly challenging, and further steps of the hydrolysis process deserve advanced calculations and will be published due course). It is worth noting that similarly to the A → B step, the B → C step features rather a small activation barrier of 6.3 kcal mol−1. Therefore, the whole reaction process A → B → C is under thermodynamic control.
In summary, we developed the efficient synthesis of tetramethylalumoxane by the controlled hydrolysis of AlMe3 in the presence of pyridines as strong coordinating ligands. Moreover, using both high-level quantum-chemical calculations and variable temperature 1H NMR studies, we provided a new look at incipient complexes of AlMe3 with H2O in the presence of neutral donor ligands and revealed the immediate formation of the quasi-stable trimeric dimethylaluminium hydroxide species, respectively. Our studies contribute to extensive research on the organoaluminium compounds’ hydrolysis and should be helpful in a rational design of studies on alumoxanes reactivity and their further stepwise transformations.
We acknowledge financial support from the National Science Centre, Poland, grant OPUS 2020/37/B/ST4/03310.
Data availability
The data supporting this article was included as part of the ESI.‡Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Pasynkiewicz, Polyhedron, 1990, 9, 429–453 CrossRef CAS.
- B. Wu, C. J. Harlan, R. W. Lenz and A. R. Barron, Macromolecules, 1997, 30, 316–318 CrossRef CAS.
- Z. Florjańczyk, A. Plichta and M. Sobczak, Polymer, 2006, 47, 1081–1090 CrossRef.
- E. Zurek and T. Ziegler, Prog. Polym. Sci., 2004, 29, 107–148 CrossRef CAS.
- M. Bochmann, Organometallics, 2010, 29, 4711–4740 CrossRef CAS.
- W. Kaminsky, Macromolecules, 2012, 45, 3289–3297 CrossRef CAS.
- H. Sinn, Macromol. Symp., 1995, 97, 27–52 CrossRef CAS.
- E. Zurek, T. K. Woo, T. K. Firman and T. Ziegler, Inorg. Chem., 2001, 40, 361–370 CrossRef CAS PubMed.
- Z. Boudene, T. De Bruin, H. Toulhoat and P. Raybaud, Organometallics, 2012, 31, 8312–8322 CrossRef CAS.
- Z. Falls, N. Tymińska and E. Zurek, Macromolecules, 2014, 47, 8556–8569 CrossRef CAS.
- A. F. R. Kilpatrick, J.-C. Buffet, P. Nørby, N. H. Rees, N. P. Funnell, S. Sripothongnak and D. O’Hare, Chem. Mater., 2016, 28, 7444–7450 CrossRef CAS.
- M. Linnolahti and S. Collins, ChemPhysChem, 2017, 18, 3369–3374 CrossRef CAS PubMed.
- S. Collins, G. Hasan, A. Joshi, J. S. McIndoe and M. Linnolahti, ChemPhysChem, 2021, 22, 1326–1335 CrossRef CAS PubMed.
- A. Joshi, S. Collins, M. Linnolahti, H. S. Zijlstra, E. Liles and J. S. McIndoe, Chem. – Eur. J., 2021, 27, 8753–8763 CrossRef CAS PubMed.
- S. Collins and M. Linnolahti, ChemPhysChem, 2023, 24, e202300342 CrossRef CAS PubMed.
- L. Luo, J. M. Younker and A. V. Zabula, Science, 2024, 384, 1424–1428 CrossRef PubMed.
- H. S. Zijlstra and S. Harder, Eur. J. Inorg. Chem., 2015, 19–43 CrossRef CAS.
- A. Storr, K. Jones and A. W. Laubengayer, J. Am. Chem. Soc., 1968, 90, 3173–3177 CrossRef CAS.
- M. Bolesławski and J. Serwatowski, J. Organomet. Chem., 1983, 254, 159–166 CrossRef.
- M. Bolesławski and J. Serwatowski, J. Organomet. Chem., 1983, 255, 269–278 CrossRef.
- S. Pasynkiewicz, A. Sadownik and A. Kunicki, J. Organomet. Chem., 1977, 124, 265–269 CrossRef CAS.
- A. Joshi, H. S. Zijlstra, E. Liles, C. Concepcion, M. Linnolahti and J. S. McIndoe, Chem. Sci., 2021, 12, 546–551 RSC.
- S. Collins, A. Joshi and M. Linnolahti, Chem. – Eur. J., 2021, 27, 15460–15471 CrossRef CAS PubMed.
- L. Negureanu, R. W. Hall, L. G. Butler and L. A. Simeral, J. Am. Chem. Soc., 2006, 128, 16816–16826 CrossRef CAS PubMed.
- R. Glaser and X. Sun, J. Am. Chem. Soc., 2011, 133, 13323–13336 CrossRef CAS PubMed.
- M. Linnolahti, A. Laine and T. A. Pakkanen, Chem. – Eur. J., 2013, 19, 7133–7142 CrossRef CAS PubMed.
- J. T. Hirvi, M. Bochmann, J. R. Severn and M. Linnolahti, ChemPhysChem, 2014, 15, 2732–2742 CrossRef CAS PubMed.
- S. J. Obrey, S. G. Bott and A. R. Barron, Organometallics, 2001, 20, 5162–5170 CrossRef CAS.
- J. Lewiński, I. Justyniak, J. Zachara and E. Tratkiewicz, Organometallics, 2003, 22, 4151–4157 CrossRef.
- J. Lewiński, W. Bury, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2006, 45, 2872–2875 CrossRef PubMed.
- A. Pietrzykowski, I. Justyniak, V. Szejko, T. Skrok, T. Radzymiński, K. Suwińska and J. Lewiński, Chem. – Eur. J., 2024, e202402021 CrossRef PubMed.
- L. Synoradzki, M. Bolesławski and J. Lewínski, J. Organomet. Chem., 1985, 284, 1–4 CrossRef CAS.
- R. Anulewicz-Ostrowska, S. Luliński, J. Serwatowski and K. Suwińska, Inorg. Chem., 2000, 39, 5763–5767 CrossRef CAS PubMed.
- V. C. Gibson, S. Mastroianni, A. J. P. White and D. J. Williams, Inorg. Chem., 2001, 40, 826–827 CrossRef CAS.
- R. Tanaka, T. Hirose, Y. Nakayama and T. Shiono, Polym. J., 2016, 48, 67–71 CrossRef CAS.
- R. J. Wehmschulte and P. P. Power, J. Am. Chem. Soc., 1997, 119, 8387–8388 CrossRef CAS.
- Y. V. Kissin and A. J. Brandolini, Macromolecules, 2003, 36, 18–26 CrossRef CAS.
- F.-J. Wu, L. S. Simeral, A. A. Mrse, J. L. Eilertsen, L. Negureanu, Z. Gan, F. R. Fronczek, R. W. Hall and L. G. Butler, Inorg. Chem., 2007, 46, 44–47 CrossRef CAS PubMed.
- Y. Yang, H. Zhu, H. W. Roesky, Z. Yang, G. Tan, H. Li, M. John and R. Herbst-Irmer, Chem. – Eur. J., 2010, 16, 12530–12533 CrossRef CAS PubMed.
- Y. Yang, H. Li, C. Wang and H. W. Roesky, Inorg. Chem., 2012, 51, 2204–2211 CrossRef CAS PubMed.
- M. R. Mason, J. M. Smith, S. G. Bott and A. R. Barron, J. Am. Chem. Soc., 1993, 115, 4971–4984 CrossRef CAS.
- C. J. Harlan, M. R. Mason and A. R. Barron, Organometallics, 1994, 13, 2957–2969 CrossRef CAS.
- C. C. Landry, C. J. Harlan, S. G. Bott and A. R. Barron, Angew. Chem., Int. Ed. Engl., 1995, 34, 1201–1202 CrossRef CAS.
- C. J. Harlan, E. G. Gillan, S. G. Bott and A. R. Barron, Organometallics, 1996, 15, 5479–5488 CrossRef CAS.
- C. J. Harlan, S. G. Bott, B. Wu, R. W. Lenz and A. R. Barron, Chem. Commun., 1997, 22, 2183–2184 RSC.
- C. N. McMahon and A. R. Barron, J. Chem. Soc., Dalton Trans., 1998, 3703–3704 RSC.
- W. Bury, E. Krajewska, M. Dutkiewicz, K. Sokołowski, I. Justyniak, Z. Kaszkur, K. J. Kurzydłowski, T. Płociński and J. Lewiński, Chem. Commun., 2011, 47, 5467–5469 RSC.
- W. Bury, E. Chwojnowska, I. Justyniak, J. Lewiński, A. Affek, E. Zygadło-Monikowska, J. Bąk and Z. Florjańczyk, Inorg. Chem., 2012, 51, 737–745 CrossRef CAS PubMed.
- M. Terlecki, S. Badoni, M. K. Leszczyński, S. Gierlotka, I. Justyniak, H. Okuno, M. Wolska-Pietkiewicz, D. Lee, G. De Paëpe and J. Lewiński, Adv. Funct. Mater., 2021, 31, 2105318 CrossRef CAS.
- V. Gupta, I. Justyniak, E. Chwojnowska, V. Szejko and J. Lewiński, Inorg. Chem., 2023, 62, 16274–16279 CrossRef CAS PubMed.
- J. Lewiński and A. E. H. Wheatley, in Modern Organoaluminum Reagents, ed. S. Woodward and S. Dagorne, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, vol. 41, pp. 1–58 Search PubMed.
- J. Lewiński and D. Kubicki, Encyclopedia of Spectroscopy and Spectrometry, Elsevier, 2017, pp. 318–329 Search PubMed.
- G. A. Papoian and R. Hoffmann, Angew. Chem., Int. Ed., 2000, 39, 2408–2448 CrossRef.
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 CrossRef PubMed.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed.
- Y. Guo, C. Riplinger, D. G. Liakos, U. Becker, M. Saitow and F. Neese, J. Chem. Phys., 2020, 152, 024116 CrossRef CAS PubMed.
- J. Pogrebetsky, A. Siklitskaya and A. Kubas, J. Chem. Theory Comput., 2023, 19, 4023–4032 CrossRef CAS PubMed.
- J. Šponer and P. Hobza, J. Phys. Chem. A, 2000, 104, 4592–4597 CrossRef.
Footnotes |
| † Dedicated to the memory of Prof. Stanisław Pasynkiewicz and his invaluable contribution to organoaluminium chemistry. |
| ‡ Electronic supplementary information (ESI) available: Experimental section, XRD, NMR, IR, data, computational details. CCDC 2352082 and 2369858. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc03672g |
| This journal is © The Royal Society of Chemistry 2024 |