
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulation of the isomerization of iminothioindoxyl switches by supramolecular confinement†
Daniel
Doellerer
 a,
Ann-Kathrin
Rückert
b,
Sandra
Doria
cd,
Michiel
Hilbers
e,
Nadja A.
Simeth
f,
Wybren Jan
Buma
eg,
Mariangela
Di Donato
*cd,
Ben L.
Feringa
*a,
Wiktor
Szymanski
*ah and
Stefano
Crespi
*b
a,
Ann-Kathrin
Rückert
b,
Sandra
Doria
cd,
Michiel
Hilbers
e,
Nadja A.
Simeth
f,
Wybren Jan
Buma
eg,
Mariangela
Di Donato
*cd,
Ben L.
Feringa
*a,
Wiktor
Szymanski
*ah and
Stefano
Crespi
*b
aStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl; w.c.szymanski@rug.nl
bDepartment of Chemistry – Ångström Laboratory, Uppsala University, Box 523, 751 20 Uppsala, Sweden. E-mail: stefano.crespi@kemi.uu.se
cICCOM-CNR, via Madonna del Piano 10, 50019 Sesto Fiorentino, Italy. E-mail: didonato@lens.unifi.it
dLaboratorio Europeo di Spettroscopia Non Lineare (LENS), via N. Carrara 1, 50019 Sesto Fiorentino, Italy
eVan 't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH, Amsterdam, The Netherlands
fInstitute for Organic and Biomolecular Chemistry, Department of Chemistry, University of Göttingen, Tammannstr. 2, 37077, Göttingen, Germany
gInstitute for Molecules and Materials, FELIX Laboratory, Radboud University, Toernooiveld 7c, 6525 ED, Nijmegen, The Netherlands
hMedical Imaging Center, University Medical Center Groningen, University of Groningen, 9713 GZ Groningen, The Netherlands
First published on 12th August 2024
Abstract
Here we present the formation of an iminothioindoxyl (ITI)⊂Cage complex that retains the photochemical properties of the photoswitch within a confined environment in water. At the same time, besides ultrafast switching inside the cage, the ITI photoswitch displays an intriguing bifurcation of the excited state isomerization pathway when encapsulated.
The study of supramolecular coordination complexes is a captivating and fast-growing area of chemistry and materials science.1,2 A characteristic feature of these complexes is that they comprise self-assembled architectures formed by non-covalent interactions between metal ions and organic ligands (Fig. 1A). The resulting structures are applied in distinct fields, including catalysis,3 sensing,4 drug delivery5 and nanotechnology.6
 | ||
| Fig. 1 (A) Structure of a flexible Pd2+ coordination cage. (B) Various photoswitches encapsulated in the Pd2+ cage: spiropyrans,7 azobenzenes8 and dihydropyrenes (DHP).9 (C) Photochemical isomerization of ITI.10 (D) This work: encapsulation of ITI photoswitch. | ||
Emerging targets within this context are embedded molecular photoswitches.11,12 These compounds can undergo discrete, reversible configurational changes upon stimulation by light at specific wavelengths, enabling external control over their chemical and physical properties.13 Displaying such switching behavior renders them suitable for a plethora of applications, ranging from smart materials14 and optoelectronics15 to photopharmacology.16,17 Well-known representatives of photoswitchable molecules are azobenzenes,18 stiff-stilbenes,19 spiropyrans,20 hydrazones,21 and diarylethenes,15 among others (Fig. 1B).
As there is an increasing demand for efficient and visible-light-responsive molecular photoswitches, a current focus is on combinations of key structural units. In this way hemithioindigo (HTI)-based molecular switches, consisting of a thioindigo and stilbene part,22 and iminothioindoxyl (ITI)-based systems, consisting of a thioindigo and azobenzene part,10 were recently developed. The latter molecules show activation with visible light and feature a large band separation of over 100 nm between the stable Z- and metastable E-isomer, along with short thermal half-lives of E-isomers (Fig. 1C).10,23
Drawing from Nature, which has fine-tuned retinal in the process of vision by confining this prototypical optical switch inside the protein rhodopsin,12,24 chemists have confined and encapsulated switching entities into artificial pockets.25–27 This process significantly altered the chemical properties of switches,12,28 including modifications of the reaction kinetics and isomer stability.29 Compared to studies in solution, the modification of the photochemical properties and excited state dynamics of photoswitches under confinement have been considerably less investigated.12,30,31 Ramamurthy and coworkers demonstrated the opening of new excited state dynamic pathways of azobenzenes by encapsulation.32 Recent efforts by the Klajn group7–9 (Fig. 1A and B) showed the switching in aqueous solution under confinement of otherwise water-insoluble azobenzenes.8 Furthermore, they demonstrated the ‘disequilibration’ of azobenzenes by triplet sensitization under confinement, distinctively favoring the formation of the Z-isomer over its E counterpart.11 It is showcasing how an artificial environment akin to rhodopsin could effectively modulate the potential energy surface of electronically excited states of photoswitches.
In this study, we illustrate the influence exerted by a metal organic cage on the properties of ITI photoswitches (Fig. 1C), which are rendered water-soluble when embedded in a hydrophobic pocket (Fig. 1D). Furthermore, investigation of the excited state dynamics of ITI under confinement reveals a slowdown and a remarkable bifurcation of the excited state process within the constrained spatial domain compared to organic solutions or polymeric media.
The photoactive molecule (ITI), ligand (1), and Pd-precursor (2) for the Cage were synthesized through modification of reported procedures (see ESI†). Pure ligand 1, formed by reacting imidazole with 1,3,5-tribromobenzene, employing K2CO3 as a base and CuSO4·5H2O as catalyst, was obtained through recrystallization from MeOH. Pd-precursor 2 was synthesized by reacting PdCl2 with tetramethylethylenediamine (TMEDA) in MeCN, yielding Pd[TMEDA]Cl2.33,34 The Pd-complex was then converted with AgNO3 in water as solvent and the aqueous solution of Pd-precursor 2 obtained pure after washing with chloroform. Next, 2 was dissolved in deionized H2O, added dropwise to 1 and filtered over cotton wool yielding the Cage. It is important to note that all reactions involving the Pd species were conducted under the exclusion of light. Modifications to the ITI synthesis were made as well,10 reducing its environmental impact by using p-toluenesulfonic acid for the ring closure of (phenylthio)acetic acid instead of AlCl3 and by performing the condensation reaction in toluene instead of benzene (see ESI† for detailed reaction conditions).
To study the response of the ITI photoswitch towards confinement and different viscous environments, ITI was encapsulated within the Cage. To prepare the ITI⊂Cage complex, an excess of ITI was added to an aqueous solution of Cage (1.00–1.45 mM) followed by stirring the solution for approximately 24 h. The remaining free ITI was removed by filtration over cotton wool, where encapsulation of ITI was indicated by the change of color to intense yellow. For a direct comparison of its behavior in different constrained environments, ITI was also embedded in a poly(methyl methacrylate) (PMMA) matrix by spin-coating the ITI-containing mixture onto a glass surface.
The successful formation of the inclusion complex ITI⊂Cage was confirmed by NMR and UV-Vis spectroscopies (NMR and UV-Vis Spectroscopy section in ESI,† Fig. S1, S3 and S4). A distinct absorption band around 420 nm, attributed to the ITI, was observed in the measured spectrum of the ITI⊂Cage compared to the Cage itself (see Fig. 2A). Molecular dynamics simulations of different conformations of ITI inside the Cage at the GFN-FF level confirm that the ITI⊂Cage is more energetically feasible than the bare switch dissolved in water (Fig. 2B and Molecular dynamics simulations section in ESI†).
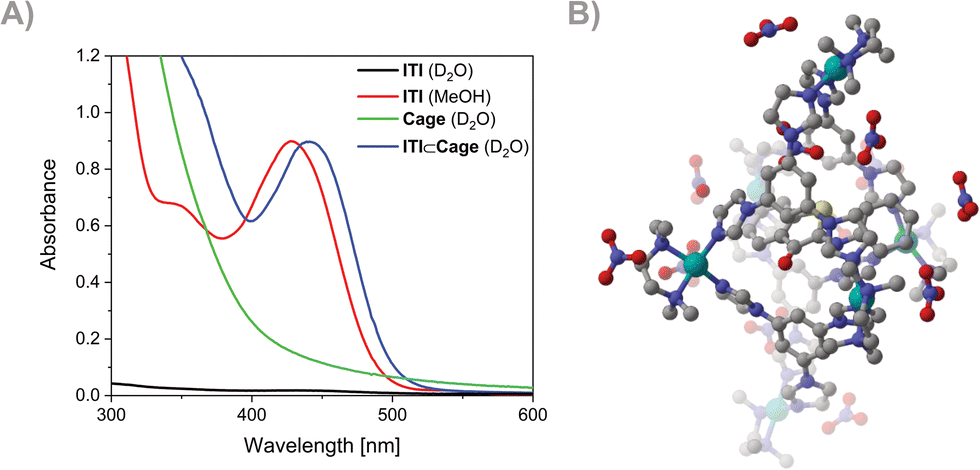 | ||
| Fig. 2 (A) UV-Vis spectroscopy of ITI in D2O (black), ITI in MeOH (c = ∼2.5 × 10−4 M, red), Cage in D2O (c = ∼1 × 10−3 M, green) and ITI⊂Cage in D2O (c = ∼9 × 10−4 M, blue) at 10 °C. (B) Optimized ITI⊂Cage at the GFN-FF level, with 12 NO3− counterions. | ||
After the successful preparation of the PMMA film and the ITI⊂Cage complex, transient absorption spectroscopy was performed to study the effect of encapsulation on the photodynamics of the switch and compare it to its behavior in solution (MeOH). The femtosecond transient spectroscopy experiments showed that in both constrained environments, the excited state dynamics upon irradiation at 400 nm qualitatively resembled the ones in solution.10 Indeed, instantaneously upon excitation, an intense excited state absorption band was observed, decaying on the sub-ps timescale, in support of the retention of an ultra-fast isomerization process in more constrained media (see Femtosecond TAS section in ESI†).
Global analysis of the transient absorption data (see ESI† for details) revealed that in PMMA, the intense excited state absorption band peaking at about 520 nm observed soon after excitation, decreased its intensity on a very fast 140 fs timescale (limit of time resolution of our setup). A second evolution associated decay spectrum (EADS) was distinguished, presenting a negative band around 425 nm, which was assigned to the ground state bleaching of the Z-isomeric form. In parallel with observations in solution (MeOH), the positive band around 475 nm also observed in the second spectral component, was assigned as a hot ground state absorption band of the photoproducts.10 Over time, the signal intensity diminished, and the positive band experienced a slight blue shift within roughly 11 ps, resulting from the vibrational cooling in the ground state (Fig. S7 in ESI;† the final spectral component, which represents the E/Z difference spectrum, persists longer than the timescale accessed with the setup). The spectra of the ITI⊂Cage complex exhibit features consistent with those observed in other organic solvents.10 Comparing the obtained results of the ITI⊂Cage complex with ITI in PMMA and methanol, a slower decay of the initial spectral component can be observed. Indeed, the kinetic analyses of ITI in MeOH and PMMA are almost superimposable, while the decay within the cage appears to be slower, however, all retrieved time constants from the performed global analysis are very similar. The main difference between the kinetics measured within Cage in comparison to the other media, resides in the relative weight of the first- and second-time component, being higher in comparison to MeOH and PMMA (Fig. S9 in ESI†). This finding suggests a specific interaction between the switch and metal–organic Cage in the excited state.
Studies on the effect of confinement on the photoswitching dynamics for some other systems,12 for example azobenzenes, suggest slower excited state dynamics under confinement than in solution.32 Those results were interpreted using a kinetic model considering the involvement of a distorted excited state configuration from which the molecule would isomerize on a slower timescale.32 When performing global analysis with a sequential scheme (see Fig. S8, ESI†), the main difference observed for ITI⊂Cage compared to ITI in PMMA and solvents is that only in the case of ITI⊂Cage the second EADS has residual intensity above 550 nm, while for all other cases, it goes almost to zero in the red region of the spectrum. This may suggest that the excited state population does not completely decay within the lifetime of the initial EADS and that a specific kinetic scheme is needed to better describe the spectral dynamics of ITI⊂Cage. We performed a target analysis on the transient absorption data to explore the feasibility of a bifurcation mechanism to the ITI⊂Cage complex, employing the kinetic scheme shown in Fig. 3A. According to this scheme, after the light pulse, Z-ITI⊂Cage populates an initial excited state (component 1, black), which branches, bringing the system both towards an intermediate excited state (component 2, red) or directly towards the conical intersection and the hot ground state (component 3, green) in 0.3 ps (k1 and k1′). The intermediate excited state then evolves towards the hot ground state but on a slower timescale (3.5 ps, k2). The different behavior observed in ITI⊂Cage underlines the effect of the supramolecular confinement of the Pd2+ cage in modifying the excited state dynamics of ITI (see Fig. 3B), compared to the dynamics in MeOH and PMMA. The dynamics in the ground state remain mostly unaltered: the hot ground state undergoes vibrational cooling throughout approximately 9 ps (k3), and the final component (component 4, blue), representing the E/Z difference spectrum and the population of E-ITI⊂Cage, decays on the timescale associated with thermal back-isomerization from E-to-Z (>1 ns, k4).
 | ||
| Fig. 3 (A) SADS (Species associated difference spectra) obtained from a global analysis of the transient absorption data performed by applying the kinetic scheme shown on the right part of the figure. (B) Pictorial representation of the excited state isomerization of the ITI⊂Cage complex. | ||
We further investigated the E-to-Z back-isomerization process, performing nanosecond transient absorption spectroscopy, showing that the ITI retained its thermal switching properties at room temperature under confinement. In particular, the E-ITI⊂Cage complex relaxes to the stable Z-form in 3 ms, slightly faster than the reported 18.5 ms in MeOH,10 showing an effect of the supramolecular confinement also on the ground state motion. Confining ITI in PMMA, on the other hand, has a more peculiar outcome. Similarly to azobenzene in polymeric matrices,35ITI showed biexponential kinetics (0.16 and 7.7 ms, see Fig. S6 in ESI†), reflecting the different nature of the surroundings in the glassy polymer. This observation directly contrasts with the result obtained when confining the switch in the metal–organic Cage. In particular, our studies suggest that ITI when confined within the Cage forms a single species, i.e., E-ITI⊂Cage (see Fig. 3B), via two different excited state pathways, due to the excited state stabilization by the cage. E-ITI⊂Cage will then revert back to its Z-form, within the supramolecular Cage. Interestingly, under these conditions the excited state dynamics seem to be affected by the surroundings more than the thermal isomerization. The non-covalent π interactions between the cage and the switch appear to be responsible for stabilizing the excited state, while the flexible nature of the supramolecular cavity exerts only a limited hindrance to its ground state motion. In contrast, the switch in PMMA experiences different degrees of spatial constraints, possibly due to the different porosities of the material, which have limited effect on the excited state, but considerably modify the ground state isomerization.
In summary, we have shown the formation of an ITI⊂Cage complex, wherein the hydrophobic cavity of the Cage is able to accommodate one equivalent of commonly water insoluble ITI switch. Our investigation using transient absorption spectroscopy showed that the ITI molecule retains its ultra-fast photochemical properties under confinement. Remarkably, the analysis of our results reveals that the decay from the electronically excited state of ITI confined within the Cage bifurcates, showcasing the interaction between the supramolecular cavity and the excited molecule. These results pave the way to further studies aimed to control the excited state dynamics and pathways of molecular switches by tailoring the supramolecular environment.
S. C. and B. L. F. conceived the project. D. D. synthesized, purified and characterized all molecules, carried out NMR and UV-Vis experiments. S. C., M. H., W. J. B., S. D. and M. D. D. carried out transient absorption spectroscopy measurements and related data processing. A.-K. R. performed computations, N. A. S. incorporated ITI in PMMA. D. D. and S. C. wrote the manuscript. M. D. D., B. L. F., W. S. and S. C. supervised the work. All authors discussed and commented on the manuscript. S. C. and B. L. F. acquired funding.
We thank Dr A. Ryabchun for his support in preparing the PMMA films. We gratefully acknowledge financial support from the Dutch Ministry of Education, Culture and Science (Bonus Incentive Scheme and Gravitation Program no: 024.001.035 to B. L. F.), the H2020 Excellent Science Marie Skłodowska-Curie Actions (Individual Fellowship 838280 to S. C.) and the Zernike Institute for Advanced Materials at the University of Groningen. S. C. thanks the Vetenskapsrådet (2021-05414 starting grant) and the Göran Gustafsson Foundations for financial support. The computations were enabled by resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) at the Tetralith cluster (NAISS 2023/5-413) partially funded by the Swedish Research Council through grant agreement no. 2022-06725.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Notes and references
- J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, Wiley-VCH, Weinheim, 1995 Search PubMed.
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed.
- R. Ham, C. J. Nielsen, S. Pullen and J. N. H. Reek, Chem. Rev., 2023, 123, 5225–5261 CrossRef CAS PubMed.
- S. Hiraoka and M. Fujita, J. Am. Chem. Soc., 1999, 121, 10239–10240 CrossRef CAS.
- B. Woods, D. Döllerer, B. Aikman, M. N. Wenzel, E. J. Sayers, F. E. Kühn, A. T. Jones and A. Casini, J. Inorg. Biochem., 2019, 199, 110781 CrossRef CAS PubMed.
- S. Datta, M. L. Saha and P. J. Stang, Acc. Chem. Res., 2018, 51, 2047–2063 CrossRef CAS PubMed.
- D. Samanta, D. Galaktionova, J. Gemen, L. J. W. Shimon, Y. Diskin-Posner, L. Avram, P. Král and R. Klajn, Nat. Commun., 2018, 9, 641 CrossRef PubMed.
- D. Samanta, J. Gemen, Z. Chu, Y. Diskin-Posner, L. J. W. Shimon and R. Klajn, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9379–9384 CrossRef CAS PubMed.
- M. Canton, A. B. Grommet, L. Pesce, J. Gemen, S. Li, Y. Diskin-Posner, A. Credi, G. M. Pavan, J. Andréasson and R. Klajn, J. Am. Chem. Soc., 2020, 142, 14557–14565 CrossRef CAS PubMed.
- M. W. H. Hoorens, M. Medved, A. D. Laurent, M. Di Donato, S. Fanetti, L. Slappendel, M. Hilbers, B. L. Feringa, W. Jan Buma and W. Szymanski, Nat. Commun., 2019, 10, 2390 CrossRef PubMed.
- J. Gemen, J. R. Church, T.-P. Ruoko, N. Durandin, M. J. Białek, M. Weißenfels, M. Feller, M. Kazes, M. Odaybat, V. A. Borin, R. Kalepu, Y. Diskin-Posner, D. Oron, M. J. Fuchter, A. Priimagi, I. Schapiro and R. Klajn, Science, 2023, 381, 1357–1363 CrossRef CAS PubMed.
- A. B. Grommet, L. M. Lee and R. Klajn, Acc. Chem. Res., 2020, 53, 2600–2610 CrossRef CAS PubMed.
- W. R. Browne and B. L. Feringa, Molecular Switches, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2011 Search PubMed.
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- J. Broichhagen, J. A. Frank and D. Trauner, Acc. Chem. Res., 2015, 48, 1947–1960 CrossRef CAS PubMed.
- P. Kobauri, F. J. Dekker, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 135, e202300681 CrossRef.
- Z. L. Pianowski, Molecular Photoswitches: Chemistry, Properties, and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2022 Search PubMed.
- F. Xu, J. Sheng, C. N. Stindt, S. Crespi, W. Danowski, M. F. Hilbers, W. J. Buma and B. L. Feringa, Chem. Sci., 2024, 15, 6763–6769 RSC.
- B. Seefeldt, R. Kasper, M. Beining, J. Mattay, J. Arden-Jacob, N. Kemnitzer, K. H. Drexhage, M. Heilemann and M. Sauer, Photochem. Photobiol. Sci., 2010, 9, 213–220 CrossRef CAS PubMed.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC.
- C. Petermayer and H. Dube, Acc. Chem. Res., 2018, 51, 1153–1163 CrossRef CAS PubMed.
- M. Medved, M. W. H. Hoorens, M. Di Donato, A. D. Laurent, J. Fan, M. Taddei, M. Hilbers, B. L. Feringa, W. J. Buma and W. Szymanski, Chem. Sci., 2021, 12, 4588–4598 RSC.
- X. Yang, M. Manathunga, S. Gozem, J. Léonard, T. Andruniów and M. Olivucci, Nat. Chem., 2022, 14, 441–449 CrossRef CAS PubMed.
- U. Akihiko, K. Takahashi and T. Osa, J. Chem. Soc., Chem. Commun., 1980, 837–838 Search PubMed.
- H. Dube, D. Ajami and J. Rebek, Angew. Chem., Int. Ed., 2010, 49, 3192–3195 CrossRef CAS PubMed.
- G. H. Clever, S. Tashiro and M. Shionoya, J. Am. Chem. Soc., 2010, 132, 9973–9975 CrossRef CAS PubMed.
- O. B. Berryman, H. Dube and J. Rebek, Isr. J. Chem., 2011, 51, 700–709 CrossRef CAS PubMed.
- A. B. Grommet, M. Feller and R. Klajn, Nat. Nanotechnol., 2020, 15, 256–271 CrossRef CAS PubMed.
- M. Petersen, B. Rasmussen, N. N. Andersen, S. P. A. Sauer, M. B. Nielsen, S. R. Beeren and M. Pittelkow, Chem. – Eur. J., 2017, 23, 17010–17016 CrossRef CAS PubMed.
- J. Sheng, J. Perego, S. Bracco, P. Cieciórski, W. Danowski, A. Comotti and B. L. Feringa, Angew. Chem., Int. Ed., 2024, e202404878 CAS.
- C. J. Otolski, A. M. Raj, V. Ramamurthy and C. G. Elles, Chem. Sci., 2020, 11, 9513–9523 RSC.
- R. P. King and S. L. Buchwald, Organometallics, 2019, 38, 3490–3493 CrossRef CAS.
- S. H. A. M. Leenders, R. Becker, T. Kumpulainen, B. de Bruin, T. Sawada, T. Kato, M. Fujita and J. N. H. Reek, Chem. – Eur. J., 2016, 22, 15468–15474 CrossRef CAS PubMed.
- M. Poutanen, O. Ikkala and A. Priimagi, Macromolecules, 2016, 49, 4095–4101 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02423k |
| This journal is © The Royal Society of Chemistry 2024 |