
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAuto tandem triple cascade organocatalysis: access to bis-lactone and butenolide derivatives†
Stéphane
Wittmann
a,
Elodie
Deschamps
a,
Chloée
Bournaud
a,
Regis
Guillot
 a,
Jean-François
Brière
b,
Giang
Vo-Thanh
*a and
Martial
Toffano
*a
a,
Jean-François
Brière
b,
Giang
Vo-Thanh
*a and
Martial
Toffano
*a
aInstitut de Chimie Moléculaire et des Matériaux d’Orsay, CNRS-UMR-8182, Université Paris-Saclay, Bât. H. Moissan. 19 avenue des sciences, 91400 Orsay, France
bNormandie Univ, UNIROUEN, INSA Rouen, CNRS, COBRA, 76000 Rouen, France
First published on 7th August 2024
Abstract
The synthesis of bis-lactone and butenolide derivatives was described using alkylidene Meldrum's acid as nucleophiles. The process operates in a triple cascade through an auto tandem catalysis promoted by DBU.
Dimeric butenolides and bis-lactones are found in many natural products and possess interesting biological properties. (−)-Lindenanolide F is a dimeric butenolide, which was isolated by Fujiwara and co-workers in 2002 from the root of Lindera chunii, a plant mostly found in Asia.1 It is frequently used in traditional Chinese medicine to improve the wind-cold-dampness arthralgia syndrome. (−)-Salprzelactone is a seco-norabietane diterpenoid containing a bis-lactone motif, which was isolated by Wu and co-workers in 2013 from the root of Salvia przewalskii, a plant native to China.2 It showed stronger antibacterial activity against A. aerogenes than streptomycin, acheomycin and ampicillin. Its total synthesis was first described by Zhai and co-workers in 2017.3 Bielschowskysin is a diterpene whose structure was reported by Rodriquez and co-workers in 2004.4 It was isolated from the Caribbean gorgonian octocoral Pseudopterogorgia kallos and was reported to exhibit antiplasmodial activity and selective in vitro cytotoxicity against small-cell lung and renal cancer cell lines (Fig. 1).
 | ||
| Fig. 1 Natural products possessing a butenolide and saturated and unsaturated bis-lactone motif. | ||
Few approaches towards the synthesis of dimeric butenolides and bis-lactone units have been described in the literature.5,6 In 2013, Sulikowski and co-workers reported the total synthesis of bielschowskysin in which the formation of the saturated and unsaturated bis-lactone unit was created by means of an intramolecular [2+2]-photocycloaddition upon irradiation of a substrate containing two butenolide moieties.7 More recently, Shenvi et al. described a stereo and heteroselective butenolide coupling leading to the formation of expected product in good to excellent yields.8 It should be noted that most of these methods required the coupling of two lactones or butanolide moieties that were constructed beforehand (Scheme 1).
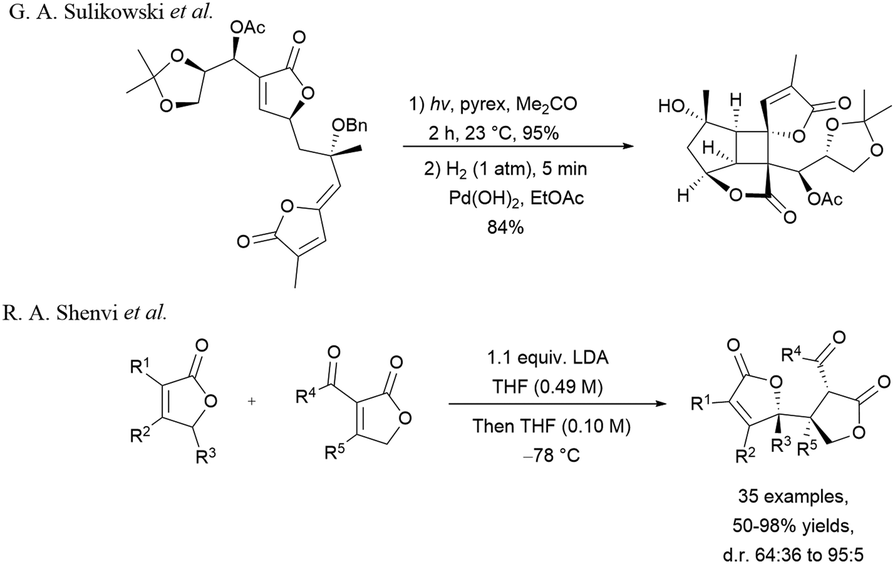 | ||
| Scheme 1 Approaches towards the synthesis of saturated and unsaturated bis-lactone motif. | ||
Although access to saturated and unsaturated lactones has been developed, the discovery of new strategies is still of great interest. Producing more complex structures while conserving energy, resources and minimizing waste requires new solutions. However, these solutions must remain simple, elegant and efficient in order to remain viable. With this in mind, tandem and multi-catalysis strategies remain one of the best solutions to access rapidly to complex organic structures from simple building blocks while minimizing steps.9 Among these strategies, auto tandem catalysis (ATC) appears to be a very interesting option, since the same catalyst promotes at least several different chemical transformations in one-pot. The main advantages of ATC consist in both the multiple role of a single catalyst, while overcoming the challenging compatibility between reagents in order to achieve an economical step-economy domino process.10 We have recently discovered a new reactivity of ketone-derived alkylidene Meldrum's acid derivatives for the enantioselective synthesis of 5,6- or 3,6-dihydropyrane-2-one with very high ee (Scheme 2).10a,11 The selectivity for the conjugated (path a) or non-conjugated dihydropyrane-2-one (path b), is controlled by the nature of the catalyst. The more basic cinchona catalyst induced a second catalytic cycle resulting in an auto tandem process to form preferentially conjugated-dihydropyrane-2-one, starting from the non-conjugated one.
 | ||
| Scheme 2 Previous and present works. | ||
We report herein a triple cascade ATC process leading to the formation of bis-lactone 3aa from alkylidene derived from Meldrum's acid 1a and furandione 2a as electrophiles.
In the course of our study, besides the formation of unprecedented 5,6-dihydropyranone 4aa from benzylidene Meldrum's acid 1a and dihydro-4,4-dimethyl-2,3-furandione 2a, we were surprised to also observe the formation of bis-lactone byproduct 3aa (33% isolated yield) when the reaction was carried out in the presence of DBU as catalyst in THF (Scheme 3). The structure of the bis-lactone 3aa was carefully characterized by NMR, MS analysis methods and confirmed by X-ray diffraction analysis. It is noteworthy to mention that this finding is extremely excited not only because structure of 3aa is found within bioactive molecules, but also its synthesis has not been well developed in literature as previously mentioned.
 | ||
| Scheme 3 Base catalysed formation of bis-lactone 3aa. | ||
In the first investigation, various parameters were screened, such as solvent, catalyst loading, base, temperature and concentration (see ESI† for details). It was determined that a total conversion and the best yields of bis-lactone 3aa (77%) as a major product (3aa/4aa = 98/2) were obtained in toluene at 70 °C and at 0.1 M concentration. This relatively low concentration and moderate temperature is necessary to limit the formation of oligomers promoting selectivity for the bis-lactone 3aa.12
With the optimal conditions in hand, we then investigated the substrate scope by varying the aryl group R on the alkylidene derivatives 1a (Table 1).
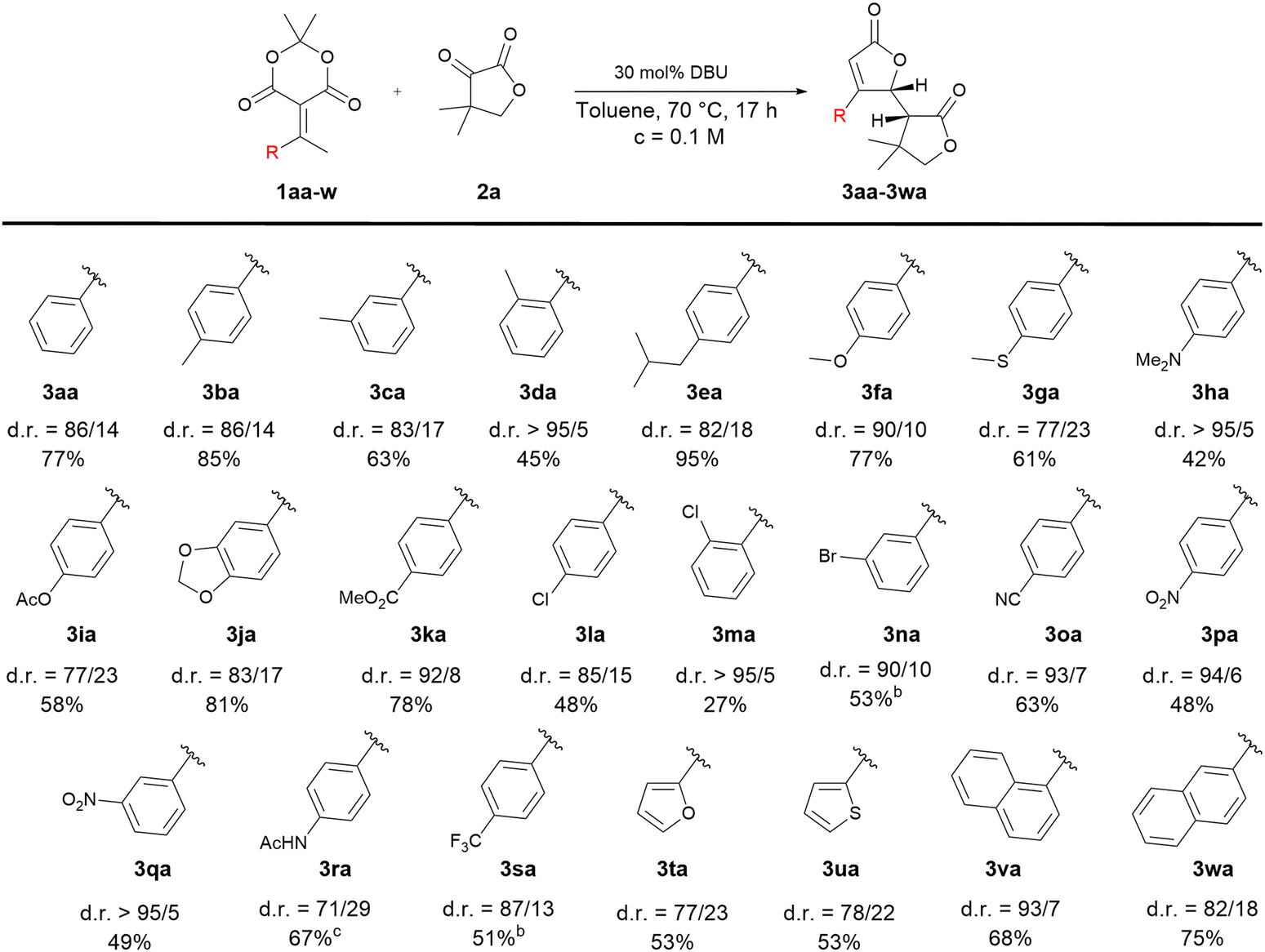
In most cases, a complete formation of the bis-lactone 3aa–3waversus the conjugated spiro-compound 4aa–4wa was observed. Only in the case of 3ha, the spiro-compound was obtained alongside the bis-lactone in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Various substituents on the aromatic ring seemed to be tolerated for this transformation. Substrates with alkyl substituents and electron-donating substituents led to the corresponding bis-lactones in good to excellent yields (3ba, 3ca, 3ea, 3fa, 3ga and 3ja). Acetate, ester, cyano and amino substituents were also well tolerated (3ia, 3ka, 3oa, 3ha and 3ra). However, moderate yields were obtained with substrates containing halogens, nitro and CF3 substituents (3la, 3na, 3pa, 3qa and 3sa). In these cases, the lower yields might be due to the lower reactivity of the alkylidene Meldrum's acid derivatives or the intermediates formed might be more prone to polymerization (vide infra). In other cases, the low yields obtained could be explained by the low solubility of the starting materials in the solvent. Substrates containing ortho-substituted aryl groups (3da and 3ma) proved to be more challenging as the bis-lactones were obtained in 45% and 27% respectively. Finally, naphthyl groups as well as furan and thiophene moieties were also well tolerated (3ta–3wa), albeit with moderate yields. Generally speaking, all the products were obtained in moderate to good diastereoselectivity with ratios ranging from 71/29 to 95/5, but in the majority of cases, the diastereoselectivity was greater than 82/18. We assume that the diastereo-determining step is the ring closure of intermediate 6aa (Scheme 6). It is controlled by both steric (see 3ba, 3ca, 3da and 3va, 3wa) and electronic effects (see 3ha, 3ma, 3na, 3oa, 3pa, 3qp) where ortho-substitution or electron-withdrawing group increase the diastereoselectivity up to 95/5 d.r. Moreover, the relative configuration of the major diastereomer 3aa was (R, S) determined by X-ray diffraction analysis of its single crystal, which was obtained by recrystallization (see ESI†). Next, the scope of electrophiles was investigated under standard reaction conditions (Table 2).
:1 ratio. Various substituents on the aromatic ring seemed to be tolerated for this transformation. Substrates with alkyl substituents and electron-donating substituents led to the corresponding bis-lactones in good to excellent yields (3ba, 3ca, 3ea, 3fa, 3ga and 3ja). Acetate, ester, cyano and amino substituents were also well tolerated (3ia, 3ka, 3oa, 3ha and 3ra). However, moderate yields were obtained with substrates containing halogens, nitro and CF3 substituents (3la, 3na, 3pa, 3qa and 3sa). In these cases, the lower yields might be due to the lower reactivity of the alkylidene Meldrum's acid derivatives or the intermediates formed might be more prone to polymerization (vide infra). In other cases, the low yields obtained could be explained by the low solubility of the starting materials in the solvent. Substrates containing ortho-substituted aryl groups (3da and 3ma) proved to be more challenging as the bis-lactones were obtained in 45% and 27% respectively. Finally, naphthyl groups as well as furan and thiophene moieties were also well tolerated (3ta–3wa), albeit with moderate yields. Generally speaking, all the products were obtained in moderate to good diastereoselectivity with ratios ranging from 71/29 to 95/5, but in the majority of cases, the diastereoselectivity was greater than 82/18. We assume that the diastereo-determining step is the ring closure of intermediate 6aa (Scheme 6). It is controlled by both steric (see 3ba, 3ca, 3da and 3va, 3wa) and electronic effects (see 3ha, 3ma, 3na, 3oa, 3pa, 3qp) where ortho-substitution or electron-withdrawing group increase the diastereoselectivity up to 95/5 d.r. Moreover, the relative configuration of the major diastereomer 3aa was (R, S) determined by X-ray diffraction analysis of its single crystal, which was obtained by recrystallization (see ESI†). Next, the scope of electrophiles was investigated under standard reaction conditions (Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Yieldb (%) | |||
| Products | 3 (d.r.) | 4 | |||
| a Reaction was performed with 0.5 mmol of benzylidene 1a, 0.525 mmol of electrophile and 0.15 mmol of DBU in 5 mL of dry toluene at 70 °C for 17 h. b Isolated yields. c Determined by 1H NMR on crude product. | |||||
| 1 | 2b |

|
3ab | 32 (67/33) | 41 |
| 2 | 2c |

|
3ac | 18 (71/29) | 31 |
| 3 | 2d |

|
3ad | 59 (nd) | 0 |
| 4 | 2e |

|
3ae | 52 (87/13) | 24 |
| 5 | 2f |

|
3af | 60 (82/18) | 12 |
| 6 | 2g |

|
3ag | 43 (82/18) | 22 |
| 7 | 2h |

|
3ah | 25 (80/20) | 30 |
| 8 | 2i |

|
3ai | 58 (89/11) | 12 |

Finding suitable electrophiles for this transformation proved to be challenging due to the presence of spirolactone 4.13 Thus, 4-cyanobenzoylformate led to the formation of the bis-lactone 3ab in 32% yield (entry 1). Using N-methylisatin as an electrophile gave the desired product 3ac in only 18% yield (entry 2). In a course of our studies, a new furandione was synthesized serving as an electrophile for this reaction, and the expected bis-lactone 3ad was isolated in 59% yield without any trace of spiro-compound 4ad (entry 3). Interestingly, fluorinated ketones offer interesting fluorinated butenolides. Thus, trifluoroacetophenone led to the formation of butenolide 3ae in 52% yield (d.r. 87/13), alongside the spiro-compound 4ae in 24% yield. Surprisingly, we were unable to increase the 3ae/4ae ratio despite a longer reaction time and/or the addition of more catalyst. This phenomenon was observed for all trifluoromethyl derivatives. It seems that electron-withdrawing substituent favors the formation of butenolide 3, whereas electron-donating group offers lower yields (compare 3ah–3agvs.3af–3ai). In any case, the diastereoselectivity still remains stable for 3. In order to get insight into the mechanism, the reaction was carried out starting from enantioenriched dihydropyranone 4aa (99%ee) with DBU under the standard reaction conditions. The expected bis-lactone 3aa was isolated as the sole product (61% yield with 84/16 d.r.) as a racemic form (Scheme 4). This result clearly proves that dihydropyranone 4aa is an intermediate leading to the formation of the bis-lactone 3aa. Unfortunately, this transformation is not stereoselective. Therefore, we assume a possible rearrangement involving the achiral transition state intermediate 6aa (Scheme 6).
 | ||
| Scheme 4 Mechanistical study from enantiopure dihydrolactone 4aa. | ||
Based on our previous works on the synthesis of dihydropyranones,10a,11 we postulated a base-catalyzed mechanism for the formation of bis-lactones from alkylidene Meldrum's acid derivatives. This sequence proceeds through a triple cascade auto tandem catalysis promoted by the Brønsted base. Dihydropyranone 4aa was obtained following two catalytic cycles in accordance with our previous work for the asymmetric synthesis of dihydropyranones (Scheme 5).10a
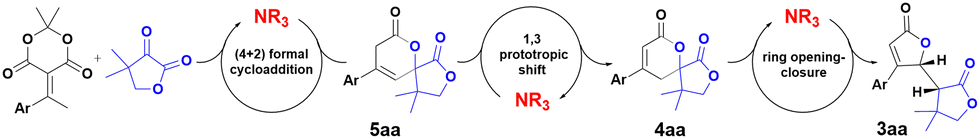 | ||
| Scheme 5 Triple auto tandem cascade. | ||
The first cycle leads to the non-conjugated dihydropyranone 5aa from 1a. The 1,3-prototropic shift process gives the conjugated spirolactone 4aa in a second catalytic cycle promoted by the same base. The last catalytic cycle consists in the deprotonation of the conjugated dihydropyranone 4aa, leading to the ring opening and the formation of the achiral diene intermediate 6aa (Scheme 6). The diene would undergo intramolecular 1,4-addition ring closure to form the desired bis-lactone 3aa. The presence of significant amount of oligomer in the case of electron-withdrawing substituted alkylidene 1l, 1n, 1p, 1q or 1s is probably enhanced by the presence of this highly reactive intermediate 6.
 | ||
| Scheme 6 Proposed mechanism in a triple cascade auto-tandem process. | ||
In conclusion, we have developed a new method for the synthesis of bis-lactones and fluorinated butenolides from alkylidene Meldrum's acid derivatives with satisfactory overall yields through the one-pot triple steps. We have also demonstrated that this transformation occurs through a DBU-catalyzed auto tandem catalysis in a triple-cascade process, including a rearrangement of a dihydropyranone intermediate via a ring-opening step.
We are grateful to the Charm3At-SynOrg Interlabex for a post-doctoral fellowship to S. Wittmann, the University Paris-Saclay for an apprenticeship grant to E. Deschamps and the CNRS (UMR 8182) for financial supports.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- C. F. Zhang, N. Nakamura, S. Tewtrakul, M. Hattori, Q. S. Sun, Z. T. Wang and T. Fujiwara, Chem. Pharm. Bull., 2002, 50, 1195 CrossRef CAS PubMed.
- H.-L. Jiang, X.-Z. Wang, J. Xiao, X.-H. Luo, X.-J. Yao, Y.-Y. Zhao, Y.-J. Chen, P. Crew and Q.-X. Wu, Tetrahedron, 2013, 69, 6687 CrossRef CAS.
- B. Ma, H. Zheng, Y. Li, H. Yang, C. Tao, B. Cheng and H. Zhai, Tetrahedron Lett., 2017, 58, 1775 CrossRef CAS.
- J. Marrero, A. D. Rodriguez, P. Baran, R. G. Raptis, J. A. Sanchez, E. Ortega-Barria and T. L. Capson, Org. Lett., 2004, 6, 1661 CrossRef CAS PubMed.
- S. K. Bagal, R. M. Adlington, R. A. B. Brown and J. E. Baldwin, Tetrahedron Lett., 2005, 46, 4633 CrossRef CAS.
- J. Langer, M. Gärtner, H. Görls and D. Walther, Synthesis, 2006, 2697 CAS.
- S. D. Townsend and G. A. Sulikowski, Org. Lett., 2013, 15, 5096 CrossRef CAS PubMed.
- B. J. Huffman, S. Chen, J. L. Schwarz, R. E. Plata, E. N. Chin, L. L. Lairson, K. N. Houk and R. A. Shenvi, Nat. Chem., 2020, 12, 310 CrossRef CAS PubMed.
- (a) D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365 CrossRef CAS; (b) N. T. Patil, V. S. Shinde and B. Gajula, Org. Biomol. Chem., 2012, 10, 211 RSC; (c) S. M. Inamdar, V. S. Shinde and N. T. Patil, Org. Biomol. Chem., 2015, 13, 8116 RSC; (d) T. L. Lohr and T. J. Marks, Nat. Chem., 2015, 7, 477 CrossRef CAS PubMed; (e) J. Zhou, Multicatalyst System in Asymmetric Catalysis, Wiley, 2015 Search PubMed; (f) A. Galván, F. J. Fañanás and F. Rodríguez, Eur. J. Inorg. Chem., 2016, 1306 CrossRef; (g) G. Szőllősi, Catal. Sci. Technol., 2018, 8, 389 RSC; (h) J. F. Campos and S. Berteina-Raboin, Catalysts, 2020, 10, 631 CrossRef CAS; (i) S. P. Sancheti, U. Urvashi, M. P. Shah and N. T. Patil, ACS Catal., 2020, 10, 3462 CrossRef CAS; (j) R. Calmanti, M. Selva and A. Perosa, Green Chem., 2021, 23, 1921 RSC; (k) S. Martínez, L. Veth, B. Lainer and P. Dydio, ACS Catal., 2021, 11, 3891 CrossRef; (l) X. Xiao, B.-X. Shao, Y.-J. Lu, Q.-Q. Cao, C.-N. Xia and F.-E. Chen, Adv. Synth. Catal., 2021, 363, 352 CrossRef.
- For recent exemple of ATC see: (a) M. Toffano, R. Guillot, C. Bournaud, J. Brière and G. Vo-Thanh, Adv. Synth. Catal., 2021, 363(18), 4452 CrossRef CAS and references herein; (b) J. R. Alexander, V. I. Shchepetkina, K. S. Stankevich, R. J. Benedict, S. P. Bernhard, R. J. Dreiling and M. J. Cook, Org. Lett., 2021, 23, 559 CrossRef CAS PubMed; (c) J. T. Maddigan-Wyatt, M. T. Blyth, J. Ametovski, M. L. Coote, J. F. Hooper and D. W. Lupton, Chem. – Eur. J., 2021, 27, 16232 CrossRef CAS PubMed; (d) J.-X. Zhu, Z.-C. Chen, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2022, 61, e202200880 CrossRef CAS PubMed; (e) Z.-L. He, Y. Zhang, Z.-C. Chen, W. Du and Y.-C. Chen, Org. Lett., 2022, 24, 6326 CrossRef CAS PubMed.
- S. Wittmann, T. Martzel, C. T. P. Truong, M. Toffano, S. Oudeyer, R. Guillot, C. Bournaud, V. Gandon, J.-F. Brière and G. Vo-Thanh, Angew. Chem., Int. Ed., 2021, 60, 11110 CrossRef CAS PubMed.
- An insoluble paste is formed during the reaction. NMR analysis identify it, as an oligomer. The exact structure of this compound was not identified.
- Benzoylformate derivatives, acenaphthoquinone, phenanthrenequinone as well as various ketoester derivatives also mostly led to the formation of spirolactone 4 as the sole product (See ESI†).
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, NMR spectra and crystallographic data. CCDC 2350164. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc03029j |
| This journal is © The Royal Society of Chemistry 2024 |