
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhenomenological observations of quinone-mediated zinc oxidation in an alkaline environment†
Christopher T.
Mallia
 ab and
Fikile R.
Brushett
*b
ab and
Fikile R.
Brushett
*b
aDepartment of Material Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: brushett@mit.edu
First published on 6th September 2024
Abstract
Redox-mediated electrochemistry is an area of growing interest, particularly in the context of energy storage. The development of such systems requires knowledge of underlying reaction mechanisms, which bear similarities to the processes that underpin corrosion and semiconductor electrochemistry. Herein we discuss an example system, quinone-mediated zinc oxidation in an alkaline environment, using knowledge from the corrosion and semiconductor fields to understand the phenomenological aspects of the reaction.
Widespread adoption of renewable electricity necessitates accompanying energy storage to address production intermittency and further development of sustainable processes that make use of electrical energy. To expand the portfolio of electrochemical technologies and methods which make direct use of electricity, redox-mediated reactions are of interest. Here, we define redox-mediated systems as any process in which electrochemically-generated reactants in a liquid phase transfer charge to and from a solid material, for the purposes of energy storage, conversion, or fabrication. Generically, they operate by using electricity to produce a dissolved reactant on an inert electrode, then transporting (e.g., fluid motion of electrolyte) that reactant to another solid species not physically in contact with an electrode, with which the reactant undergoes a chemical reaction. A finite electron potential difference is needed to drive the reaction between the solid material and the soluble redox mediator. This can be thought of as an equilibrium “overpotential”, which depends on the difference between the standard redox potentials for the solid and solution-phase species.
The redox-mediated format has found utility in a variety of innovative processes including biomass oxidation1 (to produce electricity), homogeneous chemical synthesis,2 and energy storage (the focus of this work).3 Example devices can be found in the literature which use redox-mediated reactions to store energy in configurations similar to metal–air batteries and fuel cells.1 Specifically within the energy storage context, the redox-mediated reaction is used to “target” an electrochemically-active material that stores charge, which is typically an insoluble and solid species, such as alkali-ion intercalating compounds.4 The high charge density of solid active materials allows such a system to surpass the energy density of traditional all-liquid redox flow batteries (RFBs), where the solubility of the dissolved redox couples governs the charge storage capacity, without sacrificing operational and scaling flexibility. However, as additional charge-transfer interfaces are introduced to the electrochemical system, redox-mediated reactions can be mechanistically complex, and likely involve convoluting factors (e.g., species transport to and from the surface, evolving surface and bulk chemistry of the solid) which complicate analysis and are difficult to control. Accordingly, we seek to understand how the interplay of reaction kinetics and transport phenomena may alter the macroscopic behavior of these novel electrochemical systems, and what lessons can be drawn from analogous phenomena in the corrosion and semiconductor fields. We note that the conclusions presented here remain specific to metallic substrates undergoing redox-mediated oxidation. The behavior of other materials (e.g. intercalation materials, polymers) are anticipated to be distinct.
While there are many possible redox-mediated chemistries, for the purposes of illustrating phenomenological behavior, we consider a reaction between organic mediators and a metal substrate. In a device analogue to a metal–air battery, a redox-mediated approach was previously explored within our group, where soluble redox species discharge a metallic zinc anode in a separate compartment (tank), resulting in a system where the capacity of the anodic half-cell could be mechanically increased by replacing the zinc metal in the external tank.5 This system used water-soluble quinone derivatives commonly employed as charge-storage materials in conventional RFBs,6,7 due to their standard redox potentials which are controllable through molecular engineering and sufficiently disparate from the zinc substrate as to induce a reaction. Zinc anodes are an ideal chemistry to explore for redox-mediated processes, as they are ubiquitous in primary alkaline “dry-cells”, providing a well-developed research area from which to draw information. Additionally, zinc and its associated compounds are relatively non-toxic, naturally-abundant, and low cost compared to many other chemistries of comparable electrochemical performance. The study primarily focused on proof-of-concept device performance over extended operation, and post-cycling characterization to verify the occurrence of the predicted reactions. However, further experiments conducted within our group on this particular system demonstrated that there remained fundamental questions on the interaction between the organic redox-mediators and zinc anode, which may impact device performance and longevity. The phenomenological consequences of some of these studies are discussed here. Fig. 1 schematically demonstrates the potential scale associated with the reactions involved in this half-cell.
 | ||
| Fig. 1 Electrode potential scale showing the location of the approximate redox potentials both versus a standard reference (E0) and absolute electron energy (EF), for both the zinc/zincate couple and quinone redox couples selected as mediators. Equilibrium overpotentials between the liquid/solid components are indicated on the right-hand side. Please see ESI† for complete compound names and discussion of reactions. | ||
Zinc anodes are often used in highly alkaline (pH ≥ 14) electrolytes to provide ionic conductivity and to minimize parasitic hydrogen evolution.8 In high-pH systems, metallic zinc is anticipated to oxidize according to a 2-electron reaction (Scheme S1, ESI†). We explored several quinone derivatives as candidate redox-mediators, identical to the prior work.5–7,9 A discussion of cell performance for these molecules in similar operating circumstances can be found elsewhere.5 We elected to use an alkaline electrolyte (1 M KOH) in which these quinone derivatives are soluble and kinetically facile. All tested mediators are believed to undergo 2-electron reduction reactions,10 resulting in soluble and moderately stable dianion species which appear chemically reversible on inert electrodes (e.g., platinum).6
From an energetic perspective, all redox-mediators possess a characteristic reduction potential (measured as the half-wave potential of their cyclic voltammograms) more positive than that of the zinc electrode in these conditions (Fig. 1). This difference indicates the driving force for the reduction of the soluble species through chemical reaction with the metallic zinc surface. Accordingly, we posit that the rate of reduction for each species is directly linked to the oxidation behavior of the zinc. In these circumstances, oxidation of zinc bears mechanistic resemblance to corrosion, where liquid electrolyte can oxidize bare metals in destructive ways, and, therefore, we posit the mediator mimics a corrosive agent. Thus, the faradaic corrosion current is expected to be related to the reduction rate of selected mediator.
Zinc and zinc alloys have been extensively studied in corrosion literature, providing approximate rates of reaction in electrolytes free of redox mediators.8,11–13 The effects of particular oxidants on pure zinc metal, as in the case of redox-mediators, have not been as well characterized, obfuscating comparison with prior literature. Ongoing work in our group seeks to use corrosion rate measurement techniques such as linear polarization in the presence of redox-mediators, to quantify the effectiveness of different mediators. In our present results, macroelectrode and microelectrode voltammetry indicated that all species were incompletely reduced in the presence of zinc metal, consistent with previously reported results.5 While the details of the reaction mechanisms and efficiencies will be the subject of a future publication, here we highlight phenomenologically important aspects of the process.
To explore the zinc surface evolution on a shorter experimental timescale, disk electrodes (2-mm diameter) were fabricated in-house, and used to define the surface area available for reaction (Fig. 2). In all cases (supporting electrolyte and with different mediators), pitting occurred after less than 1 h, regardless of mediator presence or specific chemistry, on the surface of the mirror-polished zinc electrode. Zinc is known to be unstable towards aqueous electrolytes at nearly all pH values, and the alkaline electrolyte used here is anticipated to corrode zinc, in parallel with the desired reaction of mediator reduction. The consistency in the macroscopic appearance of pitting, both with and without mediator, indicates this early stage of surface oxidation is relatively unaffected in morphological development by the mediator, which only appears to affect the rate of metal dissolution. These visual characteristics were common for all tested mediators, but the time to achieve the same physical appearance varied, suggesting some mediators reacted quicker than others. Furthermore, crossed-polarizer optical microscopy evinced that during this early reaction period, and simultaneously with the appearance of pits, the underlying metallic grain structure revealed, suggesting amorphous surface layers had been removed consistent with etching. Subsequent saturation of the solution near the electrode with zincate (Zn(OH)42−), a corrosion species, causes precipitation of several insoluble products that deposit on the surface, as well as grow from the underlying metal. This is reflected in the surface after 24 h of exposure as a thin, microscopically uniform coating on the base metal at the solid–electrolyte interface. Again, this visual result was observed for all mediator species, but at slightly different timescales, appearing in as little as 1 h to several, but faster than in the supporting electrolyte alone. In some cases (particularly for 2-hydroxy 1,4-naphthoquinone, which possesses the largest overpotential), it appeared that the mediator had accelerated corrosion to the point of causing recessing of the electrode surface, visible to the naked eye. In reaction cases with the mediator, discoloration of the surface oxide indicated the presence of an unidentified compound, but colors were often similar to that of the solution.
 | ||
| Fig. 2 Optical microscopy shows different stages in Zn electrode morphology evolution. The surface is mechanically polished, but retains micron-scale scratches/mechanical inclusions (a). Upon brief (ca. 1 h) exposure to alkaline electrolyte (1 M KOH, with or without mediator) the surface layer is microscopically etched (b), revealing more subsurface features. Upon imaging in crossed polarizers (at the same time), (c), the underlying grain structure is revealed. (d) Finally, upon extended (ca. 24 h) exposure to alkaline electrolyte, the surface is covered in a non-metallic layer of corrosion products. | ||
Anticipated corrosion products for the zinc anode are complicated and remain an area of active research in alkaline environments without redox mediators.11 What is understood thermodynamically is the prevalence of zinc oxide (ZnO) as a stable product, as well as several other hydroxy species that can be amorphous or crystalline.14 Thermodynamic data available in the literature indicates that all the reported hydroxy species are anticipated to decompose to the oxide species at room temperature, but the kinetics of the transformation are not well understood.15 Thus, we conclude that the interfaces created in these experiments likely are composed of multiple species. This chemical heterogeneity will be the subject of future study. Additional discussion of Zn corrosion is available in the ESI.†
The morphological evolution of the zinc surface indicates that the zinc oxidation, and subsequent mediator reduction occurring on the surface are unlikely to be mechanistically identical for all mediators, but have little effect on the morphology on short (<1 h) timescales. For additional schematic illustration, Fig. S1 (ESI†) depicts the temporal evolution of the surface structure and anticipated electrochemical response. In all cases, the initial stages bear the hallmarks of pitting and grain boundary corrosion, as would be anticipated from classic understandings of metallic dissolution. In later stages, there is evidence that anisotropic dissolution occurs filling the resultant voids with corrosion products. This latter observation spurred the question: would the deposited corrosion product passivate the surface and prevent further reaction with the redox-mediators? Thus, we elected to study the corrosion behavior at longer timescales, by using an electrode allowed to corrode in supporting electrolyte over 24 h (Fig. 2d). While this does not fully capture the early mechanism of surface corrosion brought on by the mediator + electrolyte combination, it does create a “model” electrode surface that simulated a zinc anode upon extended exposure to the electrolyte used in experiments without the additional complexity of the redox-mediator. Upon observation of this more extensively corroded surface both optically and electrochemically, it became clear that it was covered, likely with a combination of amorphous and crystalline corrosion products such as zinc hydroxides (ZnOHx) and ZnO. This observation was supported by the slight plateau in the open-circuit potential (OCP) relative to the bare metal in contact with alkaline electrolyte, and additionally with a dramatic change in the impedance spectra. In the latter experiment, the interfacial resistance appeared to decrease over a 24 h period, consistent with an increase in surface area during transient corrosion, but eventually plateaued to a consistent value, indicating passivation.
Due to natural non-stoichiometry with respect to the base compound, ZnO in the corrosion layer is expected to be n-type semiconducting, not electronically insulating.16,17 Therefore, to confirm that the produced surface layer was semiconducting, we performed the Mott–Schottky analysis on the corroded electrode in the presence of the redox-mediators. As shown in Fig. 3, the produced layer appears to possess a space-charge layer capacitance that changes with applied bias. While Mott–Schottky analysis is known to be insufficient to quantitatively characterize chemically heterogeneous, polycrystalline, and composite semiconductor materials,18 it is still expected to present qualitative evidence of the flat-band potential, a key indicator that the surface possesses mobile solid charge carriers in the oxide layer.19 Additionally, despite the qualitative nature of the measurement, the procedure is well-suited to the experimental configuration of the corroded electrode in an ionically-conductive electrolyte containing soluble mediators. The estimated value of the flat band potential observed in these experiments is ca. −1.2 V (vs. Hg/HgO), but without precise knowledge of the oxide free-carrier concentration and double-layer capacitance, differentiating the contributions of the space-charge and Helmholtz layers is difficult.18 This quantitatively results in the flat-band potential being predicted to be more negative than in reality, because, for an n-type semiconductor, the Helmholtz capacitance will shift the intercept to more negative potentials, owing to a finite potential drop across the double-layer.18
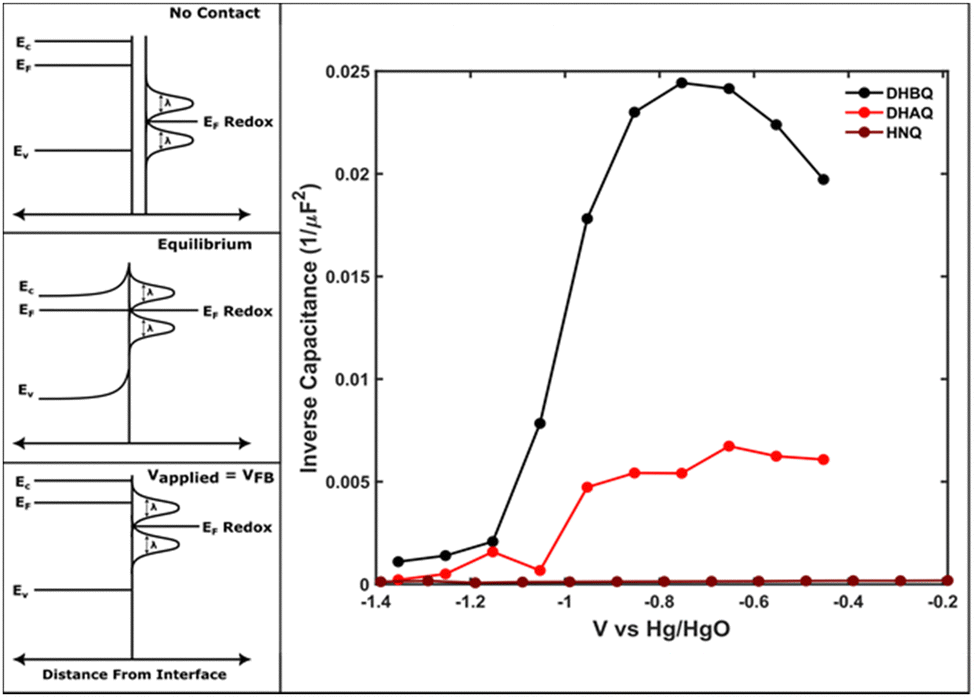 | ||
| Fig. 3 Semiconducting electrode–electrolyte interface, and the basis for the Mott–Schottky analysis (exaggerated for clarity). Upon contact, the semiconductor and electrolyte equilibrate in electron energies, and at the flat band potential (VFB) the solid returns to the equilibrium (bulk) band structure. This flat band potential (VFB) is quantitatively the x-intercept of the inverse capacitance with applied bias. | ||
In light of these combined observations, we posit that the redox-mediated reaction process transitions from a region of transient corrosion, to one where corrosion and semiconducting electrochemistry occur in parallel (Fig. S1, ESI†). Likely at timescales longer than presented here, as the corrosion layer densifies and mass transport of the redox-mediator to and from the metal subsurface is hindered, the interface reaction will be dominated by semiconductor electrochemistry (if any reaction occurs at all). The semiconducting nature of the interface presents an opportunity for refinement, as the theories of Marcus and Gerischer indicate that the electrochemical reaction rate is intimately related to the solid charge transport characteristics and solution electrochemical redox properties.20
While we emphasize that the initial results shown here do not constitute a quantitative description of the redox-mediated reaction kinetics, they do shed light on the nature of the phenomena present at these dynamic interfaces, and further demonstrate the research areas and associated methods to characterize these reactions in greater detail. With the discovery that early times are dominated by pitting corrosion, we anticipate that corrosion rate measurements coupled with electron microscopy and X-ray techniques will elucidate the product morphology and evolution rate (and dependence on mediator). The presence of both semiconducting and corroding interfaces in parallel poses an interesting problem for analytically predicting behavior of these materials in full systems. As practical embodiments are likely to attempt to maximize both power and energy densities, these experiments indicate that rate of reaction might be heterogeneous across timescales of system operation, and potentially spatially, and that multiple modes of reaction will need consideration in design. Finally, the production of a semiconducting layer upon extended reaction is an interesting outcome. While such phenomena may present additional complexity for energy storage, it invites the possibility for these reactions to be used for intentional production of semiconducting interfaces, for example in a fabrication setting. Whereas conventional high-vacuum deposition techniques can already produce well-defined interfacial materials and structures, they are often prohibitively expensive and can stifle research advancements. Creating such materials using low-temperature benchtop methods via electrochemistry,14,21 and coupling to regenerable reactants such as redox-mediators, the barrier to entry for studying systems produced in this way is dropped significantly.
The authors thank Prof. Carl Thompson, Nicholas Matteucci II, Alexander Quinn, and Trent Weiss for insightful discussions during preparation of this work. C. T. Mallia gratefully acknowledges support under and awarded by the Department of Defense, Office of Naval Research, through the National Defense Science and Engineering Graduate Fellowship (2020–2023).
Data availability
Data for this article, including electrochemical impedance spectroscopy and photographic images are available at redox-mediated zinc oxidation, DOI: 10.17605/OSF.IO/Z6EB9.Conflicts of interest
The authors declare no conflicts of interest in the work presented.References
- C. W. Anson and S. S. Stahl, Chem. Rev., 2020, 120(8), 3749–3786 CrossRef.
- F. Wang, et al. , Joule, 2021, 5(1), 149–165 CrossRef.
- Q. Wang, S. M. Zakeeruddin, D. Wang, I. Exnar and M. Grätzel, Angew. Chem., Int. Ed., 2006, 45(48), 8197–8200 CrossRef.
- Q. Huang and Q. Wang, ChemPlusChem, 2015, 80(2), 312–322 CrossRef.
- A. M. Fenton, et al., ACS Omega, 2022, acsomega.2c05798 Search PubMed.
- Z. Yang, et al., Adv. Energy Mater., 2018, 8(8), 1702056 CrossRef.
- L. Tong, et al., ACS Energy Lett., 2019, 4(8), 1880–1887 CrossRef CAS.
- M. Bockelmann, et al., J. Electrochem. Soc., 2019, 166(6), A1132–A1139 CrossRef CAS.
- K. Lin, et al., Science, 2015, 349(6255), 1529–1532 CrossRef CAS.
- M. T. Huynh, et al., J. Am. Chem. Soc., 2016, 138(49), 15903–15910 CrossRef CAS PubMed.
- B. Beverskog and I. Puigdomenech, Corros. Sci., 1997, 39(1), 107–114 CrossRef CAS.
- Y. Ein-Eli, M. Auinat and D. Starosvetsky, J. Power Sources, 2003, 114(2), 330–337 CrossRef CAS.
- A.-R. El-Sayed, H. S. Mohran and H. M. Abd El-Lateef, Metall. Mater. Trans. A, 2012, 43(2), 619–632 CrossRef CAS.
- C. Woll, Prog. Surf. Sci., 2007, 82(2–3), 55–120 CrossRef.
- C. Debiemme-Chouvy and J. Vedel, J. Electrochem. Soc., 1991, 138(9), 2538–2542 CrossRef CAS.
- O. Fruhwirth, G. W. Herzog and J. Poulios, Surf. Technol., 1980, 11(4), 259–267 CrossRef CAS.
- L. Schmidt-Mende and J. L. MacManus-Driscoll, Mater. Today, 2007, 10(5), 40–48 CrossRef CAS.
- A. Hankin, et al. , J. Mater. Chem. A, 2019, 7(45), 26162–26176 RSC.
- K. Gelderman, L. Lee and S. W. Donne, J. Chem. Educ., 2007, 84(4), 685 CrossRef CAS.
- R. Memming Electron Transfer Theories, Semiconductor Electrochemistry, Wiley-VCH Verlag GmbH & Co. KgaA, Weinheim, Germany, 2015, pp. 127–168 Search PubMed.
- A. Moezzi, A. M. McDonagh and M. B. Cortie, Chem. Eng. J., 2012, 185–186, 1–22 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02746a |
| This journal is © The Royal Society of Chemistry 2024 |